
Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drosophila Strains and Genetics
2.2. Immunohistochemistry
2.3. Preparation of Tissue Sections for Electron and Light Microscopy
2.4. Live Imaging of Embryos
3. Results
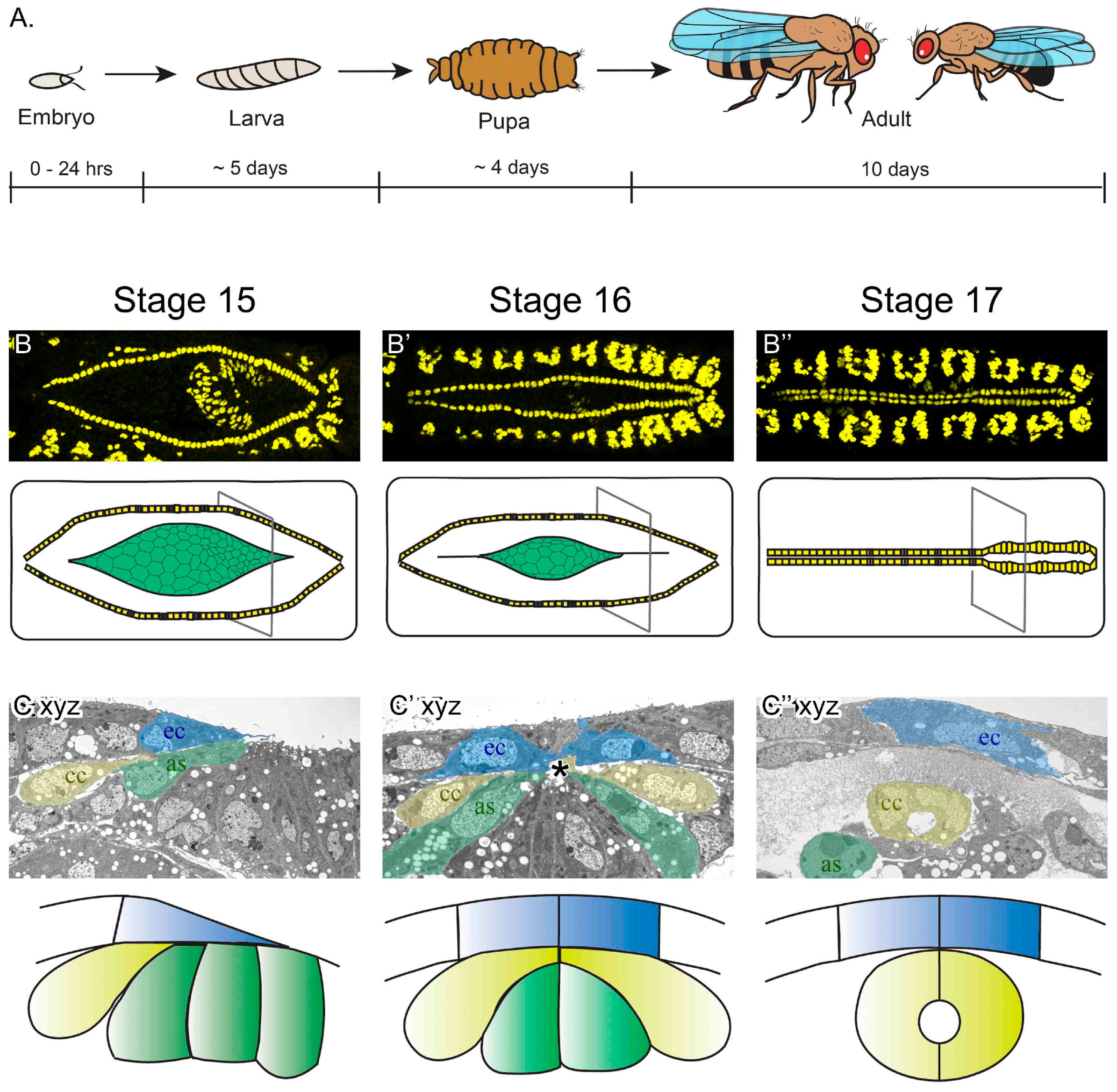
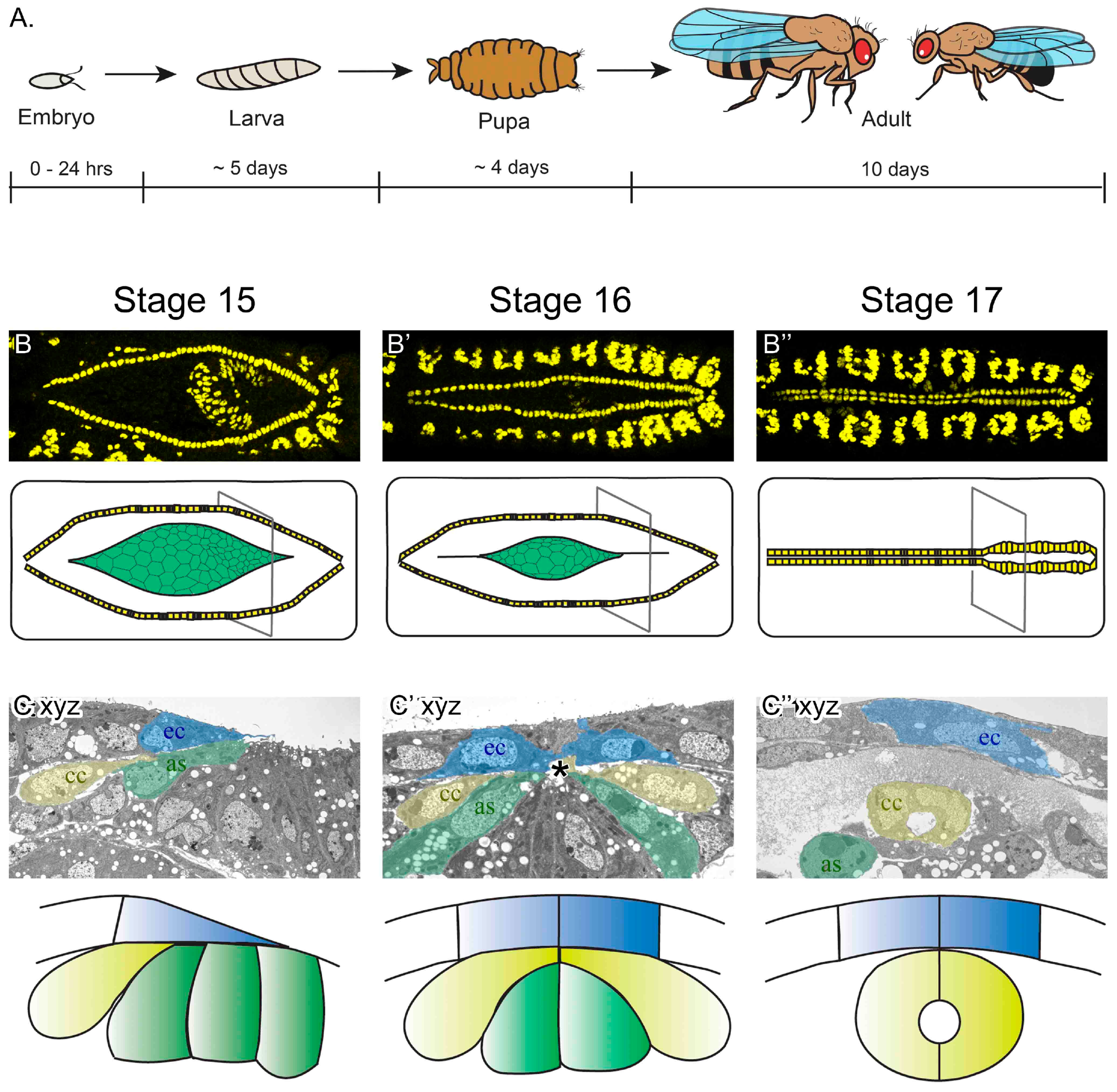
3.1. Key Steps in Drosophila Melanogaster Embryonic Heart Development
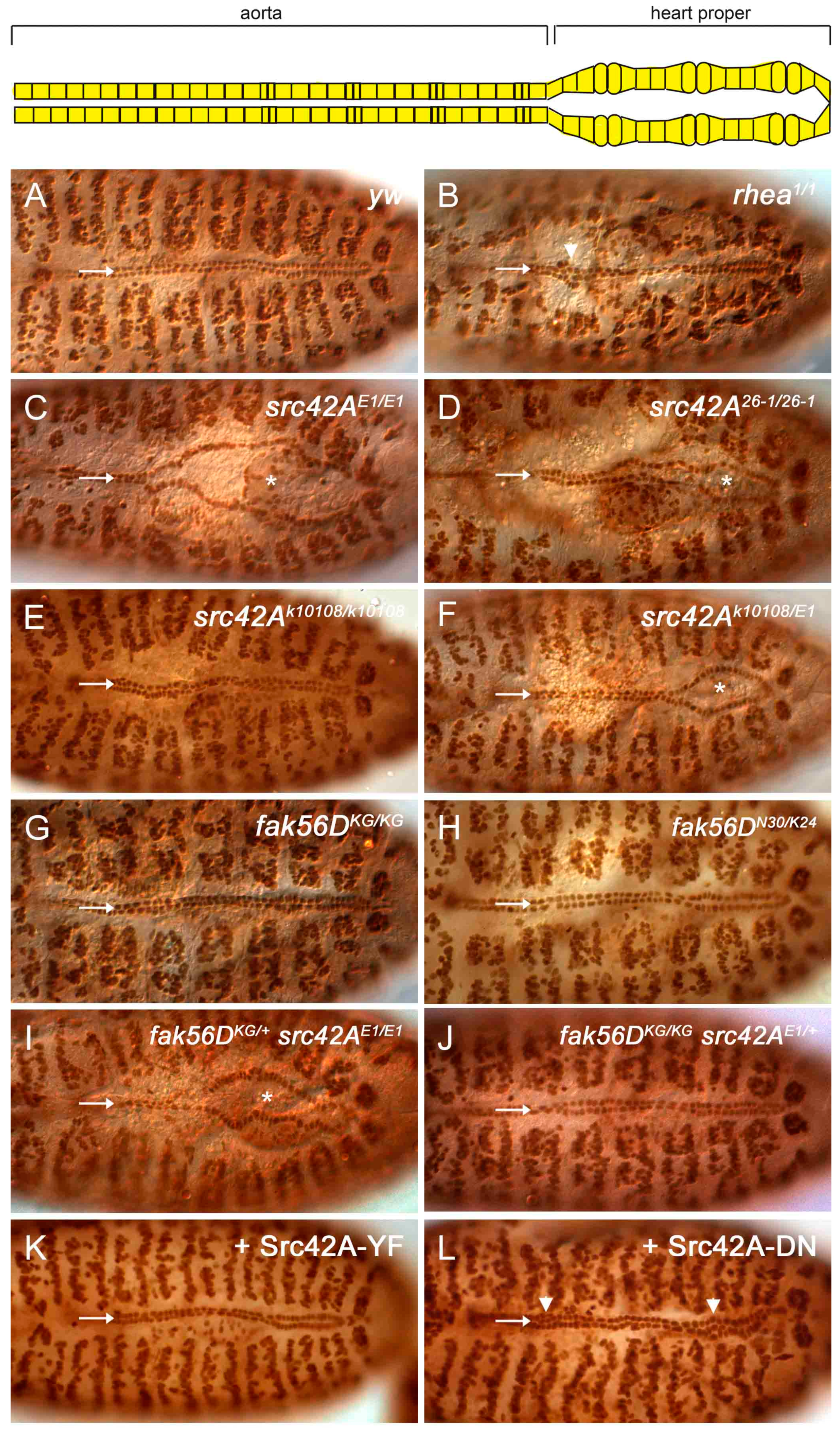
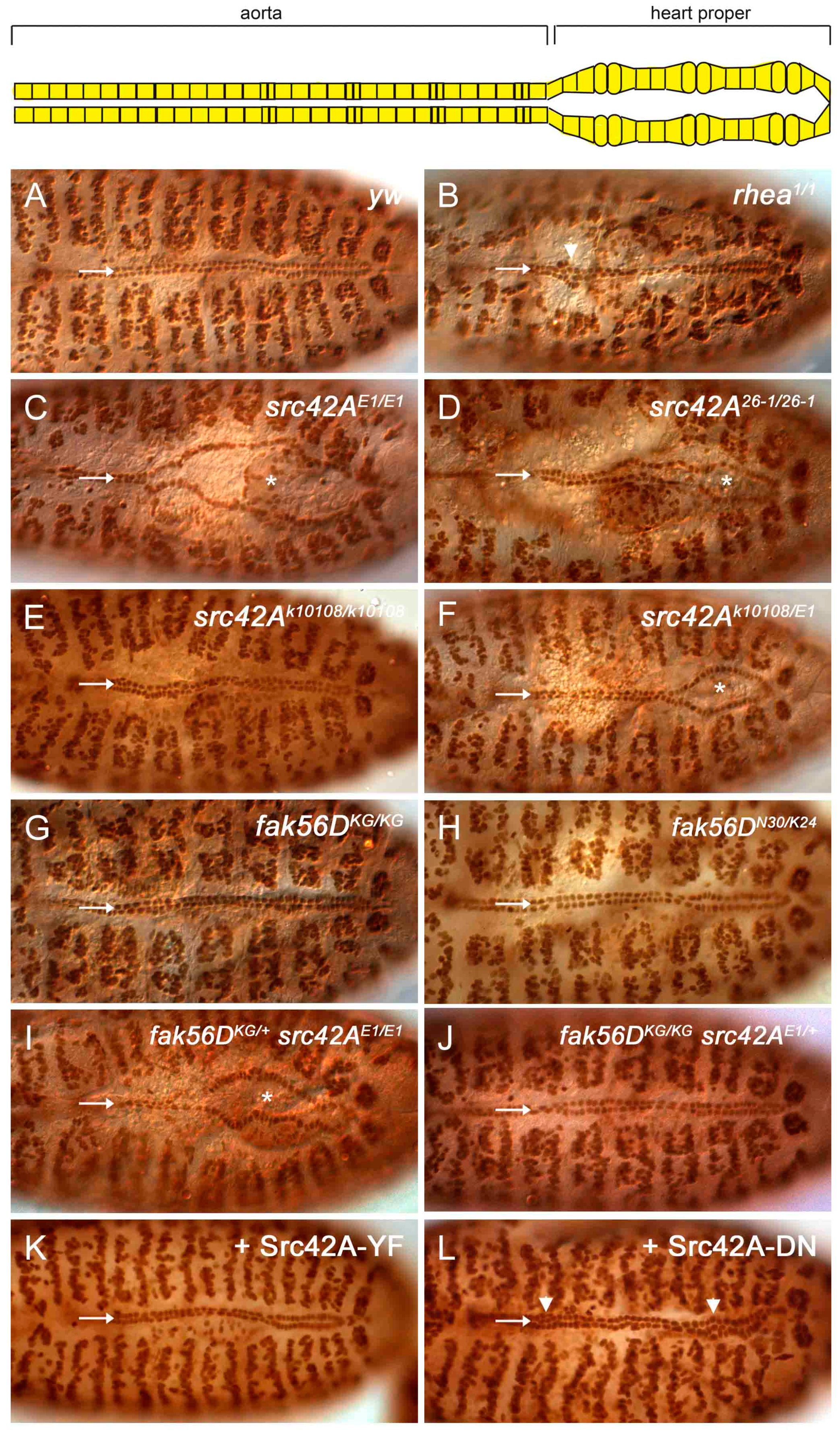
3.2. Src42A Is Required for Drosophila Heart Development
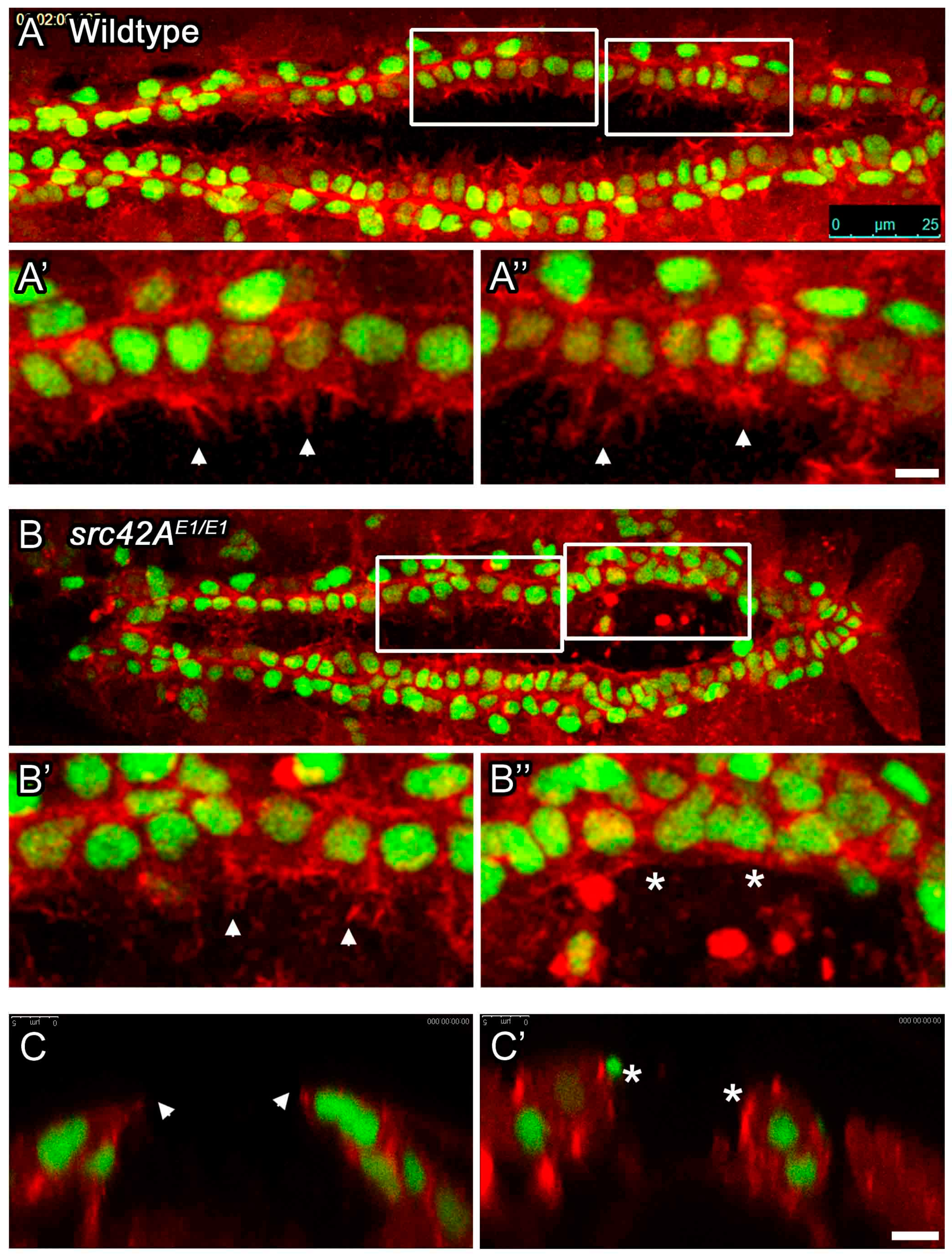
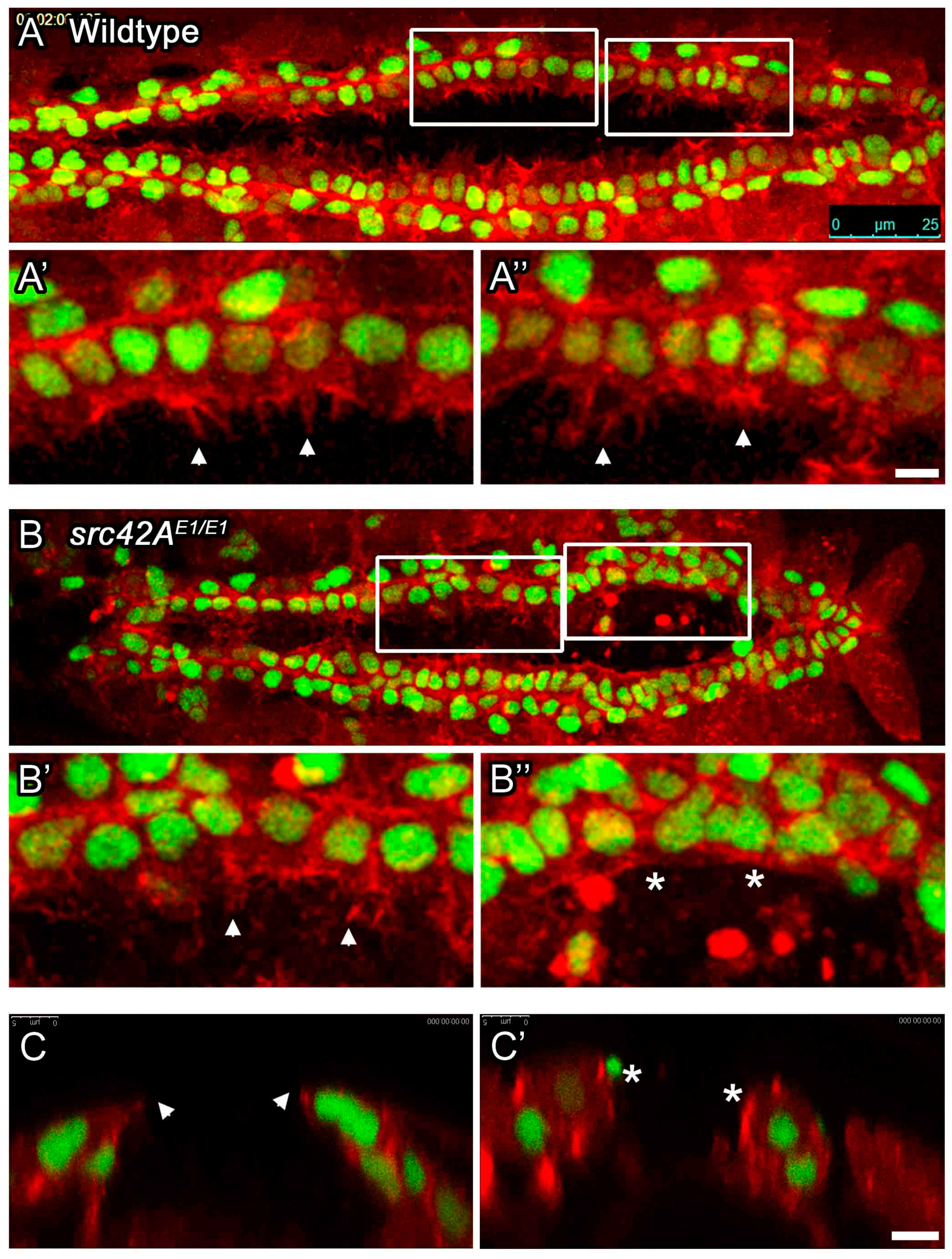
3.3. Src42A Is Required for Cell Migratory Dynamics in the Posterior Heart
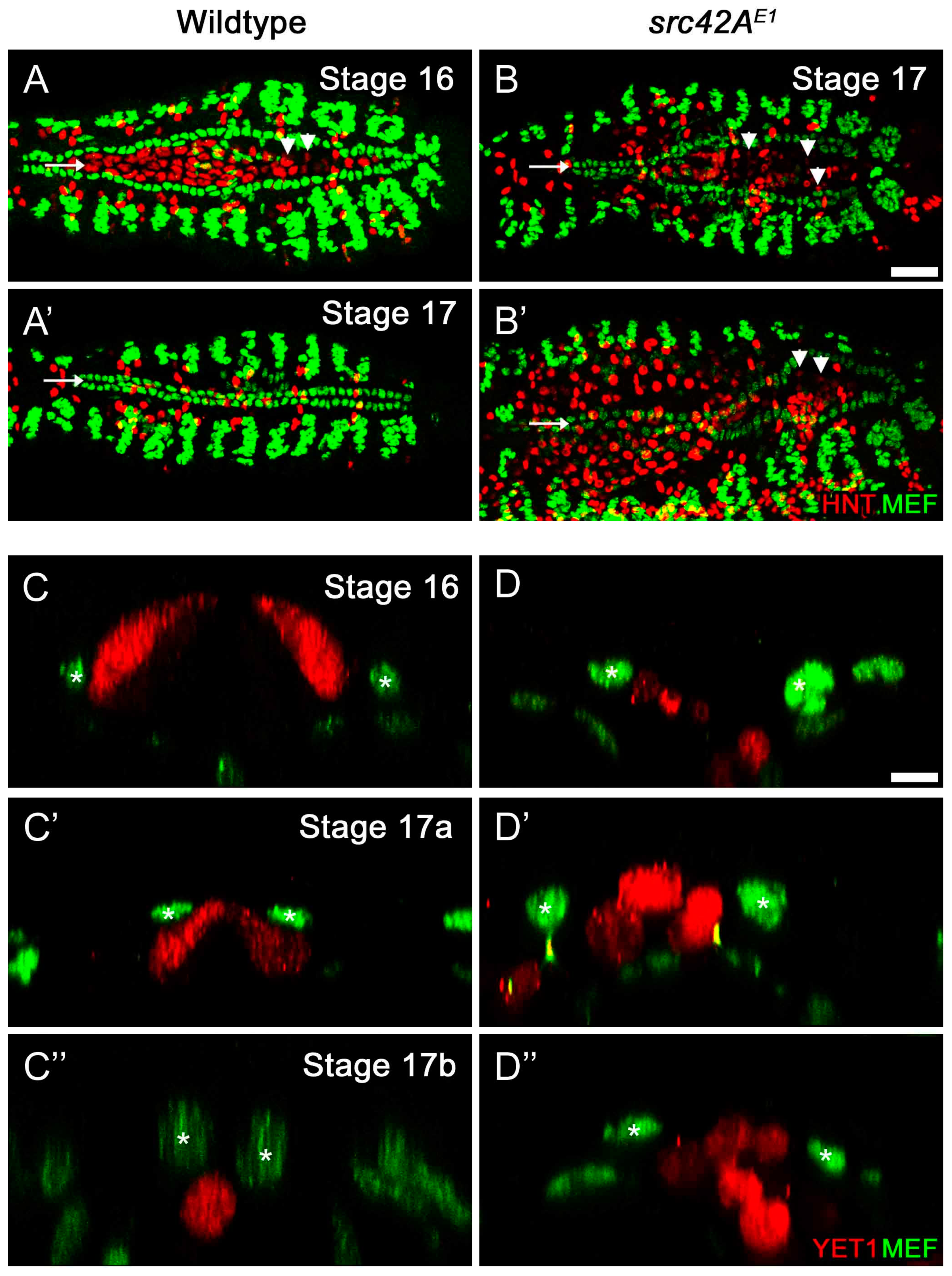
3.4. Heart-Specific Modification of Src42A Function
3.5. Delayed Migration in src42A Mutants Correlates with Stalled Internalization of the Amnioserosa
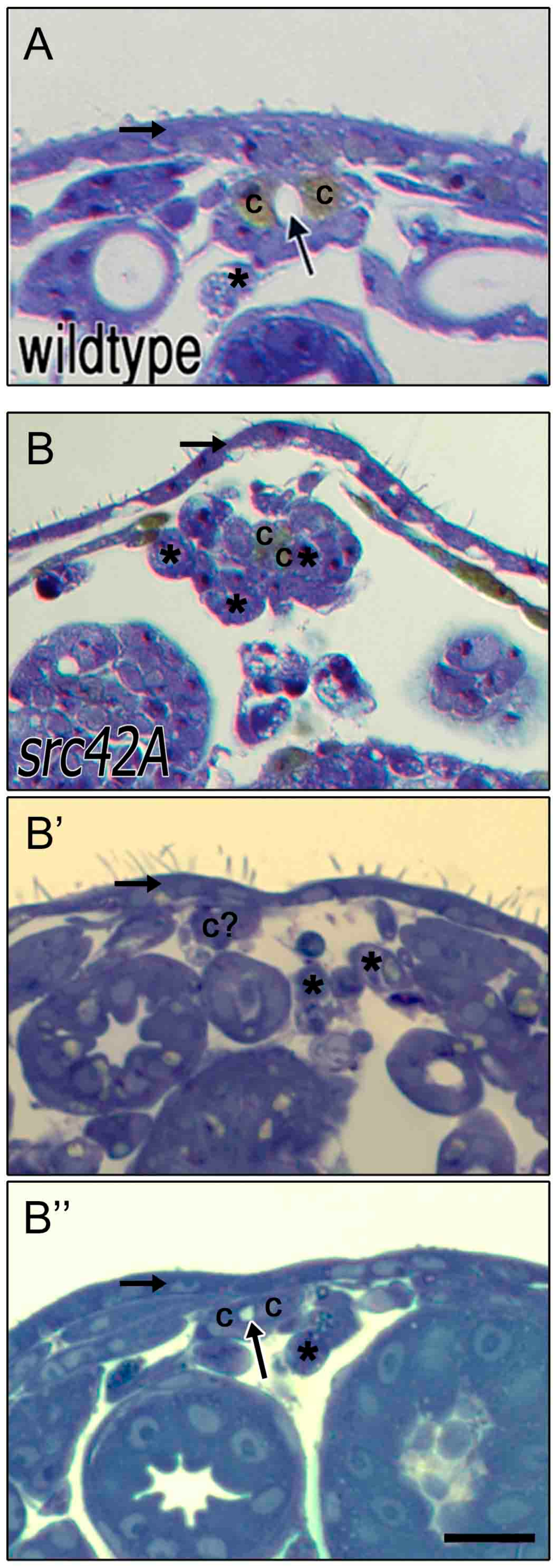
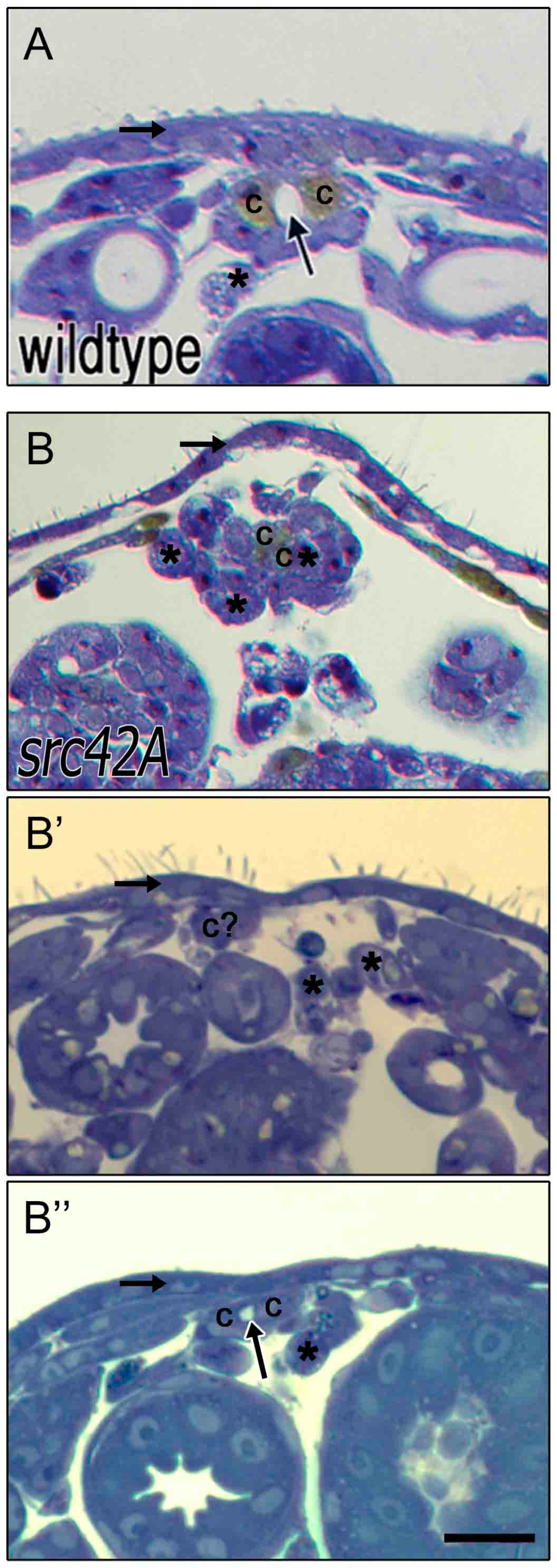
3.6. Src42A Is Not Essential for Development of A Lumen
4. Discussion
4.1. Genetic Analysis to Map Protein Networks Underlying Heart Development
4.2. Detailed Cell Biological Analysis Suggests that Src42A Functions in An Integrin-Independent manner
4.3. A Role for Transient Tissues in Proper Heart Development
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scansen, B.A.; Schneider, M.; Bonagura, J.D. Sequential segmental classification of feline congenital heart disease. J. Vet. Cardiol. 2015, 17, S10–S52. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M.A.; Sisson, D.D. Evaluation of canine congenital heart disease using an echocardiographic algorithm. J. Am. Anim. Hosp. Assoc. 2001, 37, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Buergelt, C.D. Equine cardiovascular pathology: An overview. Anim. Health Res. Rev. 2003, 4, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Buczinski, S.; Rezakhani, A.; Boerboom, D. Heart disease in cattle: Diagnosis, therapeutic approaches and prognosis. Vet. J. 2010, 184, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Ohwada, K.; Murakami, T. Morphologies of 469 cases of congenital heart diseases in cattle. J. Jpn. Vet. Med. Assoc. 2000, 53, 205–209. [Google Scholar] [CrossRef]
- MacDonald, K.A. Congenital heart diseases of puppies and kittens. Vet. Clin. N. Am. Small Anim. Pract. 2006, 36, 503–531. [Google Scholar] [CrossRef] [PubMed]
- Meurs, K.M. Genetics of cardiac disease in the small animal patient. Vet. Clin. N. Am. Small Anim. Pract. 2010, 40, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Andelfinger, G.; Wright, K.N.; Lee, H.S.; Siemens, L.M.; Benson, D.W. Canine tricuspid valve malformation, a model of human Ebstein anomaly, maps to dog chromosome 9. J. Med. Genet. 2003, 40, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, N.; Kumagai, D.; Matsumoto, K.; Inokuma, H.; Furuoka, H.; Kobayashi, Y. Detection of the nonsense mutation of OPA3 gene in Holstein Friesian cattle with dilated cardiomyopathy in Japan. J. Vet. Med. Sci. 2015, 77, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Pfister, R.; Acksteiner, C.; Baumgarth, J.; Burst, V.; Geissler, H.J.; Margulies, K.B.; Houser, S.; Bloch, W.; Flesch, M. Loss of beta1D-integrin function in human ischemic cardiomyopathy. Basic Res. Cardiol. 2007, 102, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Fedak, P.W.M.; Moravec, C.S.; McCarthy, P.M.; Altamentova, S.M.; Wong, A.P.; Skrtic, M.; Verma, S.; Weisel, R.D.; Li, R.-K. Altered expression of disintegrin metalloproteinases and their inhibitor in human dilated cardiomyopathy. Circulation 2006, 113, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Fedak, P.W.M.; Dai, X.; Du, C.; Zhou, Y.-Q.; Henkelman, M.; Mongroo, P.S.; Lau, A.; Yamabi, H.; Hinek, A.; et al. Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation 2006, 114, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. In vivo functions of integrins: Lessons from null mutations in mice. Matrix Biol. 2000, 19, 203–209. [Google Scholar] [CrossRef]
- Fässler, R.; Meyer, M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995, 9, 1896–1908. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.E.; Sutherland, A.E.; Klimanskaya, I.V.; Andrieux, A.; Meneses, J.; Pedersen, R.A.; Damsky, C.H. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995, 9, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Valencik, M.L.; Keller, R.S.; Loftus, J.C.; McDonald, J.A. A lethal perinatal cardiac phenotype resulting from altered integrin function in cardiomyocytes. J. Card. Fail. 2002, 8, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Keller, R.S.; Shai, S.Y.; Babbitt, C.J.; Pham, C.G.; Solaro, R.J.; Valencik, M.L.; Loftus, J.C.; Ross, R.S. Disruption of integrin function in the murine myocardium leads to perinatal lethality, fibrosis, and abnormal cardiac performance. Am. J. Pathol. 2001, 158, 1079–1090. [Google Scholar] [CrossRef]
- Shai, S.-Y.; Harpf, A.E.; Babbitt, C.J.; Jordan, M.C.; Fishbein, M.C.; Chen, J.; Omura, M.; Leil, T.A.; Becker, K.D.; Jiang, M.; et al. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ. Res. 2002, 90, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.L.; Yang, J.T.; Hynes, R.O. Mesodermal defects and cranial neural crest apoptosis in alpha5 integrin-null embryos. Development 1997, 124, 4309–4319. [Google Scholar] [PubMed]
- Yang, J.T.; Rayburn, H.; Hynes, R.O. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development 1993, 119, 1093–1105. [Google Scholar] [PubMed]
- Yang, J.T.; Rayburn, H.; Hynes, R.O. Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development 1995, 121, 549–560. [Google Scholar] [PubMed]
- Sujkowski, A.; Wessells, R. Drosophila models of cardiac aging and disease. In Life Extension; Springer: Cham, Switzerland, 2015; pp. 127–150. [Google Scholar]
- Bier, E.; Bodmer, R. Drosophila, an emerging model for cardiac disease. Gene 2004, 342, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Markwald, R.; Eisenberg, C.; Eisenberg, L.; Trusk, T.; Sugi, Y. Epithelial-mesenchymal transformations in early avian heart development. Acta Anatomica 1996, 156, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Medioni, C.; Sénatore, S.; Salmand, P.-A.; Lalevée, N.; Perrin, L.; Sémériva, M. The fabulous destiny of the Drosophila heart. Curr. Opin. Genet. Dev. 2009, 19, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.; Jacobs, J.R. Dissecting the role of the extracellular matrix in heart disease: Lessons from the Drosophila genetic model. Vet. Sci. 2017, 4, 24. [Google Scholar] [CrossRef]
- Vanderploeg, J.; Vazquez Paz, L.L.; MacMullin, A.; Jacobs, J.R. Integrins are required for cardioblast polarisation in Drosophila. BMC Dev. Biol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Morse, E.M.; Brahme, N.N.; Calderwood, D.A. Integrin cytoplasmic tail interactions. Biochemistry 2014, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Vanderploeg, J.; Jacobs, J.R. Talin is required to position and expand the luminal domain of the Drosophila heart tube. Dev. Biol. 2015, 405, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Raza, Q.; Jacobs, J.R. Guidance signalling regulates leading edge behaviour during collective cell migration of cardiac cells in Drosophila. Dev. Biol. 2016, 419, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Chen, Y.-C.; Kuo, C.-T.; Wenshin Yu, H.; Chen, Y.-Q.; Chiou, A.; Kuo, J.-C. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Kratimenos, P.; Koutroulis, I.; Marconi, D.; Syriopoulou, V.; Delivoria-Papadopoulos, M.; Chrousos, G.P.; Theocharis, S. Multi-targeted molecular therapeutic approach in aggressive neuroblastoma: The effect of Focal Adhesion Kinase-Src-Paxillin system. Expert Opin. Ther. Targets 2014, 18, 1395–1406. [Google Scholar] [PubMed]
- Wu, Y.; Zhang, K.; Seong, J.; Fan, J.; Chien, S.; Wang, Y.; Lu, S. In-situ coupling between kinase activities and protein dynamics within single focal adhesions. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Willey, C.D.; Balasubramanian, S.; Rodríguez Rosas, M.C.; Ross, R.S.; Kuppuswamy, D. Focal complex formation in adult cardiomyocytes is accompanied by the activation of beta3 integrin and c-Src. J. Mol. Cell. Cardiol. 2003, 35, 671–683. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, D.; Wang, X.; Wan, J.; Liu, C.; Zhang, H. Kindlin-2 phosphorylation by Src at Y193 enhances Src activity and is involved in Migfilin recruitment to the focal adhesions. FEBS Lett. 2015, 589, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Tu, Y.; Guan, J.-L.; Xiao, G.; Wu, C. Kindlin-2 tyrosine phosphorylation and interaction with Src serve as a regulatable switch in the integrin outside-in signaling circuit. J. Biol. Chem. 2014, 289, 31001–31013. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gong, H.; Jiang, G.; Ye, Y.; Wu, J.; You, J.; Zhang, G.; Sun, A.; Komuro, I.; Ge, J.; et al. Src is required for mechanical stretch-induced cardiomyocyte hypertrophy through angiotensin II type 1 receptor-dependent β-arrestin2 pathways. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Takahashi, F.; Ui-Tei, K.; Kojima, T.; Saigo, K. Requirements of genetic interactions between Src42A, armadillo and shotgun, a gene encoding E-cadherin, for normal development in Drosophila. Development 2005, 132, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, L.G.; Stewart, R.A.; Li, D.-M.; Xu, T. Drosophila Src-family kinases function with Csk to regulate cell proliferation and apoptosis. Oncogene 2004, 23, 4754–4762. [Google Scholar] [CrossRef] [PubMed]
- Ranganayakulu, G.; Schulz, R.A.; Olson, E.N. Wingless signaling induces nautilus expression in the ventral mesoderm of the Drosophila embryo. Dev. Biol. 1996, 176, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, N.; McMillan, S.C.; Chaudhary, R.; Mok, J.; Reed, B.H. Autophagy promotes caspase-dependent cell death during Drosophila development. Autophagy 2009, 5, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Millard, T.H.; Martin, P. Dynamic analysis of filopodial interactions during the zippering phase of Drosophila dorsal closure. Development 2008, 135, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Tokusumi, T.; Russell, M.; Gajewski, K.; Fossett, N.; Schulz, R.A. U-shaped protein domains required for repression of cardiac gene expression in Drosophila. Differentiation 2007, 75, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.H. Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. Methods Cell Biol. 1994, 44, 445–487. [Google Scholar] [PubMed]
- MacMullin, A.; Jacobs, J.R. Slit coordinates cardiac morphogenesis in Drosophila. Dev. Biol. 2006, 293, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Reed, B.H.; McMillan, S.C.; Chaudhary, R. The preparation of Drosophila embryos for live-imaging using the hanging drop protocol. J. Vis. Exp. 2009, 25. [Google Scholar] [CrossRef] [PubMed]
- Muliyil, S.; Krishnakumar, P.; Narasimha, M. Spatial, temporal and molecular hierarchies in the link between death, delamination and dorsal closure. Development 2011, 138, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.L.; Rebay, I.; Hynes, R.O. Expression of DFak56, a Drosophila homolog of vertebrate focal adhesion kinase, supports a role in cell migration in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 14978–14983. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, J.; Sawamoto, K.; Okabe, M.; Takagi, Y.; Tezuka, T.; Yoshikawa, S.; Ryo, H.; Okano, H.; Yamamoto, T. Cloning and characterization of Dfak56, a homolog of focal adhesion kinase, in Drosophila melanogaster. J. Biol. Chem. 1999, 274, 29196–29201. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Fessler, L.I.; Edeen, P.T.; Madigan, S.J.; McKeown, M.; Hunter, T. DFak56 is a novel Drosophila melanogaster focal adhesion kinase. J. Biol. Chem. 1999, 274, 35621–35629. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.-I.; Kao, H.-H.; Grabbe, C.; Lee, Y.-T.; Ghose, A.; Lai, T.-T.; Peng, K.-P.; Van Vactor, D.; Palmer, R.H.; Chen, R.-H.; et al. Fak56 functions downstream of integrin alphaPS3betanu and suppresses MAPK activation in neuromuscular junction growth. Neural Dev. 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Grabbe, C.; Zervas, C.G.; Hunter, T.; Brown, N.H.; Palmer, R.H. Focal adhesion kinase is not required for integrin function or viability in Drosophila. Development 2004, 131, 5795–5805. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Bershadsky, A.; Pankov, R.; Yamada, K.M. Transmembrane crosstalk between the extracellular matrix—Cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2001, 2, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.A.; Brand, A.H. The GAL4 system: A versatile system for the expression of genes. Methods Mol. Biol. 2008, 420, 79–95. [Google Scholar] [PubMed]
- Takahashi, F.; Endo, S.; Kojima, T.; Saigo, K. Regulation of cell-cell contacts in developing Drosophila eyes by Dsrc41, a new, close relative of vertebrate c-src. Genes Dev. 1996, 10, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Serrels, A.; Miller, D.; Timpson, P.; Serrels, B.; Frame, M.C.; Brunton, V.G. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: Effects on E-cadherin dynamics. Cancer Res. 2010, 70, 9413–9422. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.W.; McLean, G.W.; Wyke, A.W.; Paraskeva, C.; Parkinson, E.K.; Frame, M.C.; Brunton, V.G. The catalytic activity of the Src family kinases is required to disrupt cadherin-dependent cell-cell contacts. Mol. Biol. Cell 2000, 11, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Tateno, M.; Nishida, Y.; Adachi-Yamada, T. Regulation of JNK by Src during Drosophila development. Science 2000, 287, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Raza, Q.S.; Vanderploeg, J.; Jacobs, J.R. Matrix Metalloproteinases are required for membrane motility and lumenogenesis during Drosophila heart development. PLOS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Harpaz, N.; Ordan, E.; Ocorr, K.; Bodmer, R.; Volk, T. Multiplexin promotes heart but not aorta morphogenesis by polarized enhancement of slit/robo activity at the heart lumen. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Hyun, C.; Park, I.-C. Congenital heart diseases in small animals: Part II. Potential genetic aetiologies based on human genetic studies. Vet. J. 2006, 171, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Bodmer, R. Probing the polygenic basis of cardiomyopathies in Drosophila. J. Cell. Mol. Med. 2012, 16, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Hashima, J.N.; Rogers, V.; Langley, S.M.; Ashraf, M.; Sahn, D.J.; Ohtonen, P.; Davis, L.E.; Hohimer, A.R.; Rasanen, J. Fetal ventricular interactions and wall mechanics during ductus arteriosus occlusion in a sheep model. Ultrasound Med. Biol. 2015, 41, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Hofstadler, G.; Tulzer, G.; Altmann, R.; Schmitt, K.; Danford, D.; Huhta, J.C. Spontaneous closure of the human fetal ductus arteriosus—A cause of fetal congestive heart failure. Am. J. Obstet. Gynecol. 1996, 174, 879–883. [Google Scholar] [CrossRef]
- Weichert, J.; Hartge, D.R.; Axt-Fliedner, R. The fetal ductus arteriosus and its abnormalities—A review. Congenit. Heart Dis. 2010, 5, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Tananari, Y.; Maeno, Y.; Takagishi, T.; Sasaguri, Y.; Morimatsu, M.; Kato, H. Role of apoptosis in the closure of neonatal ductus arteriosus. Jpn. Circ. J. 2000, 64, 684–688. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanderploeg, J.; Jacobs, J.R. Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila. Vet. Sci. 2017, 4, 23. https://doi.org/10.3390/vetsci4020023
Vanderploeg J, Jacobs JR. Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila. Veterinary Sciences. 2017; 4(2):23. https://doi.org/10.3390/vetsci4020023
Chicago/Turabian StyleVanderploeg, Jessica, and J. Roger Jacobs. 2017. "Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila" Veterinary Sciences 4, no. 2: 23. https://doi.org/10.3390/vetsci4020023
APA StyleVanderploeg, J., & Jacobs, J. R. (2017). Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila. Veterinary Sciences, 4(2), 23. https://doi.org/10.3390/vetsci4020023