
Beta Adrenergic Signaling: A Targetable Regulator of Angiosarcoma and Hemangiosarcoma

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Beta Adrenergic Signaling and Cancer
2.1. Inhibition of Beta Adrenergic Signaling and Therapeutic Responses
2.2. Beta Adrenergic Signaling and the Regulation of Tumor Cell Biology
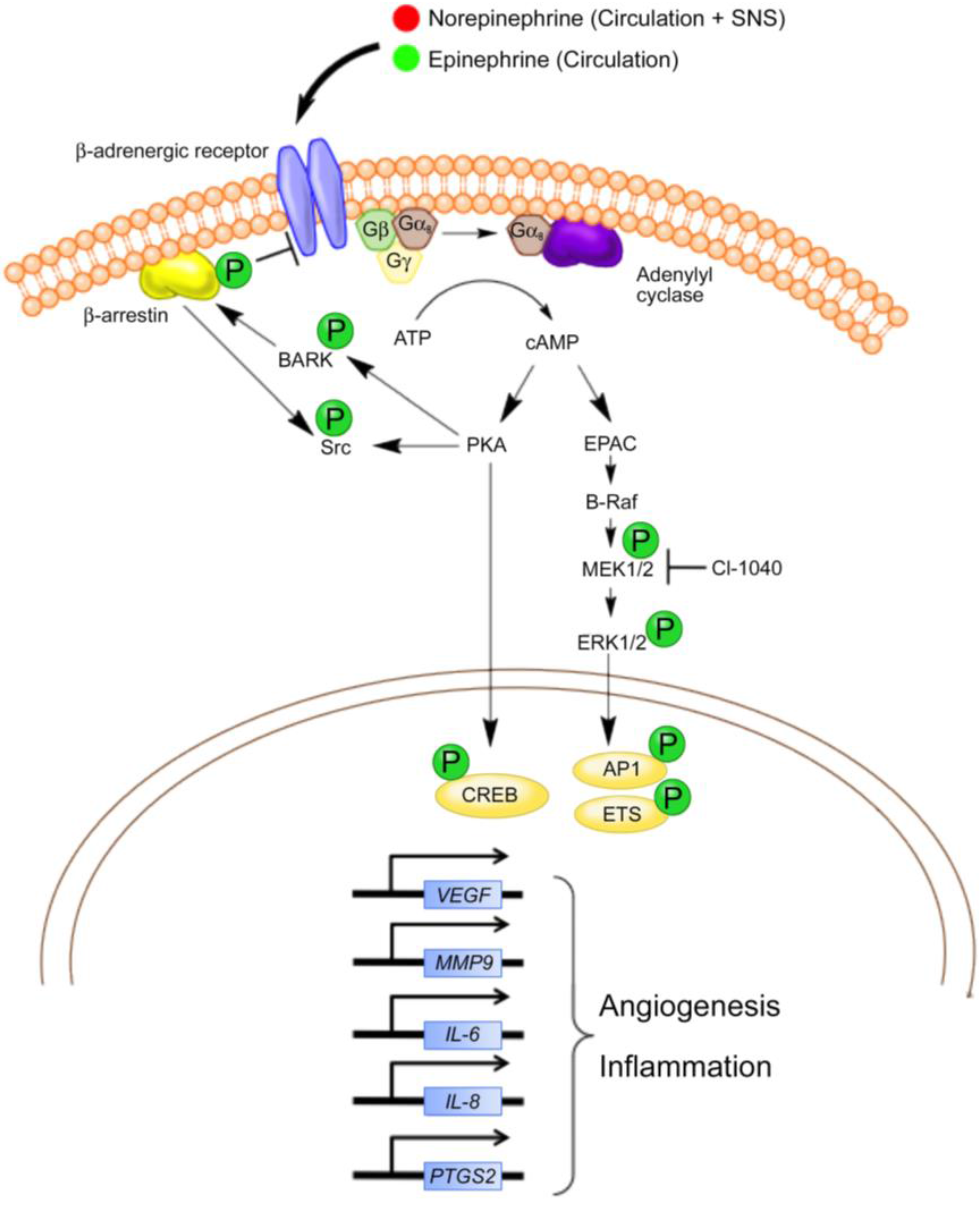
3. Inhibition of Beta Adrenergic Signaling in Vascular Tumors
3.1. Beta Blockade of Benign Vascular Tumors: Studies in Infantile Hemangiomas Point the Way
3.2. Beta Blockade of Malignant Vascular Tumors
4. Beta Adrenergic Regulation of the Hematopoietic and Tumor Cell Microenvironments
4.1. The β-AR-CXCR4-CXCL12 Signaling Axis
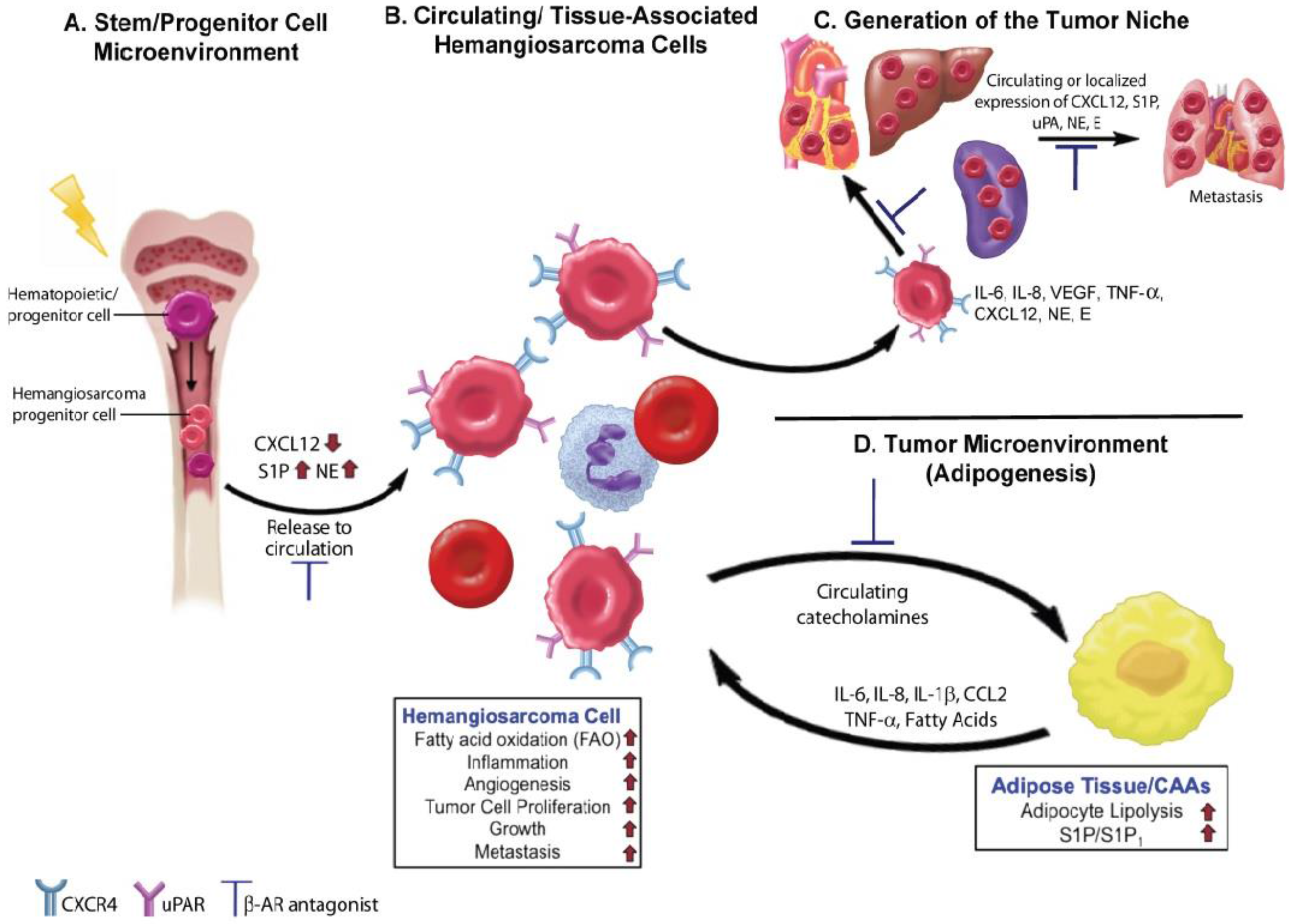
4.2. Angiogenesis and Inflammation
4.3. Lipid Metabolism
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Coindre, J.M.; Terrier, P.; Guillou, L.; Le Doussal, V.; Collin, F.; Ranchere, D.; Sastre, X.; Vilain, M.O.; Bonichon, F.; N'Guyen Bui, B. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: A study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001, 91, 1914–1926. [Google Scholar] [CrossRef]
- Lurkin, A.; Ducimetiere, F.; Vince, D.R.; Decouvelaere, A.V.; Cellier, D.; Gilly, F.N.; Salameire, D.; Biron, P.; de Laroche, G.; Blay, J.Y.; et al. Epidemiological evaluation of concordance between initial diagnosis and central pathology review in a comprehensive and prospective series of sarcoma patients in the Rhone-Alpes region. BMC Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Penel, N.; Mallet, Y.; Robin, Y.M.; Fournier, C.; Grosjean, J.; Ceugnart, L.; Clisant, S.; Lefebvre, J.L. Prognostic factors for adult sarcomas of head and neck. Int. J. Oral Maxillofac. Surg. 2008, 37, 428–432. [Google Scholar] [CrossRef]
- Fury, M.G.; Antonescu, C.R.; van Zee, K.J.; Brennan, M.F.; Maki, R.G. A 14-year retrospective review of angiosarcoma: Clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer J. 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.W.; Goldblum, J.R. Malignant vascular neoplasms. In Enzinger and Weiss’s Soft Tissue Tumors, 4th ed.; Mosby: St. Louis, MO, USA, 2011; pp. 917–954. [Google Scholar]
- Cohen, S.M.; Storer, R.D.; Criswell, K.A.; Doerrer, N.G.; Dellarco, V.L.; Pegg, D.G.; Wojcinski, Z.W.; Malarkey, D.E.; Jacobs, A.C.; Klaunig, J.E.; et al. Hemangiosarcoma in rodents: Mode-of-action evaluation and human relevance. Toxicol. Sci. 2009, 111, 4–18. [Google Scholar] [CrossRef]
- Janigan, D.T.; Husain, A.; Robinson, N.A. Cardiac angiosarcomas: A review and a case report. Cancer 1986, 57, 852–859. [Google Scholar] [CrossRef]
- Smith, V.C.; Eisenberg, B.L.; McDonald, E.C. Primary splenic angiosarcoma. Case report and literature review. Cancer 1985, 55, 1625–1627. [Google Scholar] [CrossRef]
- Koch, M.; Nielsen, G.P.; Yoon, S.S. Malignant tumors of blood vessels: Angiosarcomas, hemangioendotheliomas, and hemangioperictyomas. J. Surg. Oncol. 2008, 97, 321–329. [Google Scholar] [CrossRef]
- Priester, W.A.; McKay, F.W. The occurrence of tumors in domestic animals. Natl. Cancer Inst. Monogr. 1980, 54, 1–210. [Google Scholar] [PubMed]
- Clifford, C.A.; Mackin, A.J.; Henry, C.J. Treatment of canine hemangiosarcoma: 2000 and beyond. J. Vet. Intern. Med. 2000, 14, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Hargis, A.M.; Ihrke, P.J.; Spangler, W.L.; Stannard, A.A. A retrospective clinicopathologic study of 212 dogs with cutaneous hemangiomas and hemangiosarcomas. Vet. Pathol. 1992, 29, 316–328. [Google Scholar] [CrossRef]
- Bergman, P. Hemangiosarcoma. In Textbook of Veterinary Internal Medicine, 7th ed.; Ettinger, S., Feldman, E., Eds.; Elsevier: St. Louis, MO, USA, 2010; pp. 2175–2180. [Google Scholar]
- Schultheiss, P.C. A retrospective study of visceral and nonvisceral hemangiosarcoma and hemangiomas in domestic animals. J. Vet. Diagn. Invest. 2004, 16, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Thamm, D. Hemangiosarcoma. In Small Animal Clinical Oncology, 4th ed.; Withrow, S., Vail, D., Eds.; WB Saunders Elsevier: St. Louis, MO, USA, 2007; pp. 785–795. [Google Scholar]
- Kim, J.H.; Graef, A.J.; Dickerson, E.B.; Modiano, J.F. Pathobiology of hemangiosarcoma in dogs: Research advances and future perspectives. Vet. Sci. (Submitted).
- Italiano, A.; Cioffi, A.; Penel, N.; Levra, M.G.; Delcambre, C.; Kalbacher, E.; Chevreau, C.; Bertucci, F.; Isambert, N.; Blay, J.Y.; et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2011, 118, 3330–3336. [Google Scholar] [CrossRef] [PubMed]
- Penel, N.; Italiano, A.; Ray-Coquard, I.; Chaigneau, L.; Delcambre, C.; Robin, Y.M.; Bui, B.; Bertucci, F.; Isambert, N.; Cupissol, D.; et al. Metastatic angiosarcomas: Doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve the outcome. Ann. Oncol. 2011, 23, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.J.; Hosoya, K.; Lara-Garcia, A.; Kisseberth, W.; Couto, G. VAC protocol for treatment of dogs with stage III hemangiosarcoma. J. Am. Anim. Hosp. Assoc. 2013, 49, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Finotello, R.; Stefanello, D.; Zini, E.; Marconato, L. Comparison of doxorubicin-cyclophosphamide with doxorubicin-dacarbazine for the adjuvant treatment of canine hemangiosarcoma. Vet. Comp. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dervisis, N.G.; Dominguez, P.A.; Newman, R.G.; Cadile, C.D.; Kitchell, B.E. Treatment with DAV for advanced-stage hemangiosarcoma in dogs. J. Am. Anim. Hosp. Assoc. 2011, 47, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.A.; Mullin, C.M.; de Lorimier, L.P.; Burgess, K.E.; Risbon, R.E.; Fred, R.M., 3rd; Drobatz, K.; Clifford, C.A. Doxorubicin and deracoxib adjuvant therapy for canine splenic hemangiosarcoma: A pilot study. Can. Vet. J. 2013, 54, 237–242. [Google Scholar] [PubMed]
- Mullin, C.M.; Arkans, M.A.; Sammarco, C.D.; Vail, D.M.; Britton, B.M.; Vickery, K.R.; Risbon, R.E.; Lachowicz, J.; Burgess, K.E.; Manley, C.A.; et al. Doxorubicin chemotherapy for presumptive cardiac hemangiosarcoma in dogs. Vet. Comp. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.E.; Rassnick, K.M.; Northrup, N.C.; Kristal, O.; Chretin, J.D.; Cotter, S.M.; Kintzer, P.; Frimberger, A.E.; Morrison-Collister, K.E.; Wood, C.A.; et al. Treatment of vascular and soft-tissue sarcomas in dogs using an alternating protocol of ifosfamide and doxorubicin. Vet. Comp. Oncol. 2003, 1, 171–179. [Google Scholar] [PubMed]
- Sorenmo, K.; Duda, L.; Barber, L.; Cronin, K.; Sammarco, C.; Usborne, A.; Goldschmidt, M.; Shofer, F. Canine hemangiosarcoma treated with standard chemotherapy and minocycline. J. Vet. Intern. Med. 2000, 14, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Sorenmo, K.; Samluk, M.; Clifford, C.; Baez, J.; Barrett, J.S.; Poppenga, R.; Overley, B.; Skorupski, K.; Oberthaler, K.; van Winkle, T.; et al. Clinical and pharmacokinetic characteristics of intracavitary administration of pegylated liposomal encapsulated doxorubicin in dogs with splenic hemangiosarcoma. J. Vet. Intern. Med. 2007, 21, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Sorenmo, K.U.; Baez, J.L.; Clifford, C.A.; Mauldin, E.; Overley, B.; Skorupski, K.; Bachman, R.; Samluk, M.; Shofer, F. Efficacy and toxicity of a dose-intensified doxorubicin protocol in canine hemangiosarcoma. J. Vet. Intern. Med. 2004, 18, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Teske, E.; Rutteman, G.R.; Kirpenstein, J.; Hirschberger, J. A randomized controlled study into the efficacy and toxicity of pegylated liposome encapsulated doxorubicin as an adjuvant therapy in dogs with splenic haemangiosarcoma. Vet. Comp. Oncol. 2011, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.L.; Rook, K.A.; Clifford, C.A.; Gregor, T.P.; Sorenmo, K.U. Efficacy of doxorubicin-based chemotherapy for non-resectable canine subcutaneous haemangiosarcoma. Vet. Comp. Oncol. 2010, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Lana, S.; U’Ren, L.; Plaza, S.; Elmslie, R.; Gustafson, D.; Morley, P.; Dow, S. Continuous low-dose oral chemotherapy for adjuvant therapy of splenic hemangiosarcoma in dogs. J. Vet. Intern. Med. 2007, 21, 764–769. [Google Scholar] [CrossRef] [PubMed]
- U’Ren, L.W.; Biller, B.J.; Elmslie, R.E.; Thamm, D.H.; Dow, S.W. Evaluation of a novel tumor vaccine in dogs with hemangiosarcoma. J. Vet. Intern. Med. 2007, 21, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Sahora, A.I.; Rusk, A.W.; Henkin, J.; McKeegan, E.M.; Shi, Y.; Khanna, C. Prospective study of thrombospondin-1 mimetic peptides, ABT-510 and ABT-898, in dogs with soft tissue sarcoma. J. Vet. Intern. Med. 2012, 26, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Barron, T.I.; Connolly, R.M.; Sharp, L.; Bennett, K.; Visvanathan, K. Beta blockers and breast cancer mortality: A population- based study. J. Clin. Oncol. 2011, 29, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, V.; Grazzini, M.; Gandini, S.; Benemei, S.; Asbury, C.D.; Marchionni, N.; Geppetti, P. Beta-adrenergic-blocking drugs and melanoma: Current state of the art. Expert. Rev. Anticancer. Ther. 2011, 12, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.S.; Karlan, B.Y.; Li, A.J. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol Oncol. 2012, 127, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Grytli, H.H.; Fagerland, M.W.; Fossa, S.D.; Tasken, K.A. Association between use of beta-blockers and prostate cancer-specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur. Urol. 2014, 65, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Grytli, H.H.; Fagerland, M.W.; Fossa, S.D.; Tasken, K.A.; Haheim, L.L. Use of beta-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate 2013, 73, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Lemeshow, S.; Sorensen, H.T.; Phillips, G.; Yang, E.V.; Antonsen, S.; Riis, A.H.; Lesinski, G.B.; Jackson, R.; Glaser, R. β-Blockers and survival among Danish patients with malignant melanoma: A population-based cohort study. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Melhem-Bertrandt, A.; Chavez-Macgregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Sood, A.K.; Conzen, S.D.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011, 29, 2645–2652. [Google Scholar] [CrossRef] [PubMed]
- Powe, D.G.; Voss, M.J.; Zanker, K.S.; Habashy, H.O.; Green, A.R.; Ellis, I.O.; Entschladen, F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 2010, 1, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.; Modiano, J.F. Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans-man and his best friend share more than companionship. Chromosome Res. 2008, 16, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.; Paoloni, M.; Mazcko, C.; Khanna, C. The comparative oncology trials consortium: Using spontaneously occurring cancers in dogs to inform the cancer drug development pathway. PLoS Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Gordon, I. Catching cancer by the tail: New perspectives on the use of kinase inhibitors. Clin. Cancer Res. 2009, 15, 3645–3647. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Lindblad-Toh, K.; Vail, D.; London, C.; Bergman, P.; Barber, L.; Breen, M.; Kitchell, B.; McNeil, E.; Modiano, J.F.; et al. The dog as a cancer model. Nat. Biotechnol. 2006, 24, 1065–1066. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.G.; Hill, S.J.; Summers, R.J. Evolution of beta-blockers: From anti-anginal drugs to ligand-directed signalling. Trends Pharmacol. Sci. 2011, 32, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.J.; McGrath, J.C. Previously unsuspected widespread cellular and tissue distribution of beta-adrenoceptors and its relevance to drug action. Trends Pharmacol. Sci. 2011, 32, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ma, Q.; Wang, Z.; Zhang, M.; Guo, K.; Wang, F.; Wu, E. Beta2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NFkappaB pathway. Mol. Cancer 2011. [Google Scholar] [CrossRef] [PubMed]
- Kondratenko, T.Y.; Zacharova, I.V.; Kuzina, N.V.; Katukov, V.; Severin, E.S.; Kornilova, Z.; Perelman, M.I. Alterations in human lung adrenergic receptors in cancer. Biochem. Mol. Biol. Int. 1993, 29, 123–130. [Google Scholar] [PubMed]
- Draoui, A.; Vandewalle, B.; Hornez, L.; Revillion, F.; Lefebvre, J. Beta-adrenergic receptors in human breast cancer: Identification, characterization and correlation with progesterone and estradiol receptors. Anticancer Res. 1991, 11, 677–680. [Google Scholar] [PubMed]
- Moretti, S.; Massi, D.; Farini, V.; Baroni, G.; Parri, M.; Innocenti, S.; Cecchi, R.; Chiarugi, P. Beta-adrenoceptors are upregulated in human melanoma and their activation releases pro-tumorigenic cytokines and metalloproteases in melanoma cell lines. Lab Invest. 2013, 93, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Nagmani, R.; Pasco, D.S.; Salas, R.D.; Feller, D.R. Evaluation of beta-adrenergic receptor subtypes in the human prostate cancer cell line-LNCaP. Biochem. Pharmacol. 2003, 65, 1489–1494. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Burdsall, J.; Workman, L.; Hollinger, B.; Neely, J. Beta-Adrenergic receptors in pediatric tumors: Uncoupled beta 1-adrenergic receptor in Ewing’s sarcoma. J. Natl. Cancer Inst. 1983, 71, 779–786. [Google Scholar] [PubMed]
- Chow, W.; Amaya, C.; Rains, S.; Chow, M.; Dickerson, E.B.; Bryan, B.A. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated beta blockade. JAMA Dermatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Stiles, J.M.; Amaya, C.; Rains, S.; Diaz, D.; Pham, R.; Battiste, J.; Modiano, J.F.; Kokta, V.; Boucheron, L.E.; Mitchell, D.C.; et al. Targeting of beta adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Barron, T.I.; Sharp, L.; Visvanathan, K. Beta-adrenergic blocking drugs in breast cancer: A perspective review. Ther. Adv. Med. Oncol. 2012, 4, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Tasken, K.A. Beta-adrenergic receptor signaling in prostate cancer. Front. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.W.; Kokolus, K.M.; Reed, C.B.; Hylander, B.L.; Ma, W.W.; Repasky, E.A. A nervous tumor microenvironment: The impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol. Immunother. 2014, 63, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Li, Z.; Lu, L.; Cho, C.H. Beta-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin. Cancer Biol. 2013, 23, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Gorden, B.H.; Kim, J.H.; Sarver, A.L.; Frantz, A.M.; Breen, M.; Lindblad-Toh, K.; O’Brien, T.D.; Sharkey, L.C.; Modiano, J.F.; Dickerson, E.B. Identification of three molecular and functional subtypes in canine hemangiosarcoma through gene expression profiling and progenitor cell characterization. Am. J. Pathol. 2014, 184, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, B.A.; Phang, T.L.; Fosmire, S.P.; Scott, M.C.; Trapp, S.C.; Duckett, M.M.; Robinson, S.R.; Slansky, J.E.; Sharkey, L.C.; Cutter, G.R.; et al. Gene expression profiling identifies inflammation and angiogenesis as distinguishing features of canine hemangiosarcoma. BMC Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by β-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, E.B.; Marley, K.; Edris, W.; Tyner, J.W.; Schalk, V.; Macdonald, V.; Loriaux, M.; Druker, B.J.; Helfand, S.C. Imatinib and dasatinib inhibit hemangiosarcoma and implicate PDGFR-beta and Src in tumor growth. Transl. Oncol. 2013, 6, 158–168. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [PubMed]
- Andersen, N.J.; Nickoloff, B.J.; Dykema, K.J.; Boguslawski, E.A.; Krivochenitser, R.I.; Froman, R.E.; Dawes, M.J.; Baker, L.H.; Thomas, D.G.; Kamstock, D.A.; et al. Pharmacologic inhibition of MEK signaling prevents growth of canine hemangiosarcoma. Mol. Cancer Ther. 2013, 12, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Leaute-Labreze, C.; Dumas de la Roque, E.; Hubiche, T.; Boralevi, F.; Thambo, J.B.; Taieb, A. Propranolol for severe hemangiomas of infancy. N. Engl. J. Med. 2008, 358, 2649–2651. [Google Scholar] [CrossRef] [PubMed]
- Amir, J.; Metzker, A.; Krikler, R.; Reisner, S.H. Strawberry hemangioma in preterm infants. Pediatr. Dermatol. 1986, 3, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Chiller, K.G.; Passaro, D.; Frieden, I.J. Hemangiomas of infancy: Clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch. Dermatol. 2002, 138, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.T.; Itinteang, T.; Leadbitter, P. Low-dose propranolol for infantile haemangioma. J. Plast. Reconstr. Aesthet. Surg. 2011, 64, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Hermans, D.J.; Bauland, C.G.; Zweegers, J.; van Beynum, I.M.; van der Vleuten, C.J. Propranolol in a case series of 174 patients with complicated infantile haemangioma: Indications, safety and future directions. Br. J. Dermatol. 2013, 168, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.E.; Itinteang, T.; Leadbitter, P.; Marsh, R.; Tan, S.T. Low-dose propranolol regimen for infantile haemangioma. J. Paediatr. Child Health 2014, 51, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.A.; Chen, B.S.; Saltzman, B.; Manning, S.C.; Parikh, S.R. Propranolol therapy for reducing the number of nasal infantile hemangioma invasive procedures. JAMA Otolaryngol. Head Neck Surg. 2014, 140, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Solman, L.; Murabit, A.; Gnarra, M.; Harper, J.I.; Syed, S.B.; Glover, M. Propranolol for infantile haemangiomas: Single centre experience of 250 cases and proposed therapeutic protocol. Arch. Dis. Child 2014, 99, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Banavali, S.; Pasquier, E.; Andre, N. Targeted therapy with propranolol and metronomic chemotherapy combination: Sustained complete response of a relapsing metastatic angiosarcoma. Ecancermedicalscience 2015. [Google Scholar] [CrossRef] [PubMed]
- Gorden, B.H.; Saha, J.; Khammanivong, A.; Schwartz, G.K.; Dickerson, E.B. Lysosomal drug sequestration as a mechanism of drug resistance in vascular sarcoma cells marked by high CSF-1R expression. Vasc. Cell 2014. [Google Scholar] [CrossRef] [PubMed]
- Lamerato-Kozicki, A.R.; Helm, K.M.; Jubala, C.M.; Cutter, G.C.; Modiano, J.F. Canine hemangiosarcoma originates from hematopoietic precursors with potential for endothelial differentiation. Exp. Hematol. 2006, 34, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Webb, I.J.; Bleul, C.; Springer, T.; Gutierrez-Ramos, J.C. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J. Exp. Med. 1997, 185, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.; Kollet, O.; Lapidot, T. Mutual, reciprocal SDF-1/CXCR4 interactions between hematopoietic and bone marrow stromal cells regulate human stem cell migration and development in NOD/SCID chimeric mice. Exp. Hematol. 2006, 34, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Sager, H.B.; Courties, G.; Dutta, P.; Iwamoto, Y.; Zaltsman, A.; von Zur Muhlen, C.; Bode, C.; Fricchione, G.L.; Denninger, J.; et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med. 2014, 20, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Im, K.S.; Graef, A.J.; Breen, M.; Lindblad-Toh, K.; Modiano, J.F.; Kim, J.H. Interactions between CXCR4 and CXCL12 promote cell migration and invasion in canine hemangiosarcoma. Vet. Comp. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, A.; Hsu, Y.M.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Graef, A.J.; LeVine, D.N.; Cohen, I.R.; Modiano, J.F.; Kim, J.H. Association of Sphingosine-1-phosphate (S1P)/S1P receptor-1 pathway with cell proliferation and survival in canine hemangiosarcoma. J. Vet. Intern. Med. 2015, 29, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Schappa, J.T.; Frantz, A.M.; Gorden, B.H.; Dickerson, E.B.; Vallera, D.A.; Modiano, J.F. Hemangiosarcoma and its cancer stem cell subpopulation are effectively killed by a toxin targeted through epidermal growth factor and urokinase receptors. Int. J. Cancer 2013, 133, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Chin, C.C.; Chen, C.N.; Kuo, Y.H.; Chen, T.C.; Yu, H.R.; Tung, S.Y.; Shen, C.H.; Hsieh, Y.Y.; Guo, S.E.; et al. Stromal cell-derived factor-1/CXC receptor 4 and beta1 integrin interaction regulates urokinase-type plasminogen activator expression in human colorectal cancer cells. J. Cell. Physiol. 2012, 227, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Mierzejewska, K.; Klyachkin, Y.M.; Ratajczak, J.; Abdel-Latif, A.; Kucia, M.; Ratajczak, M.Z. Sphingosine-1-phosphate-mediated mobilization of hematopoietic stem/progenitor cells during intravascular hemolysis requires attenuation of SDF-1-CXCR4 retention signaling in bone marrow. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Mottillo, E.P.; Zhao, J.; Gartung, A.; VanHecke, G.C.; Lee, J.F.; Maddipati, K.R.; Xu, H.; Ahn, Y.H.; Proia, R.L.; et al. Adipocyte lipolysis-stimulated interleukin-6 production requires sphingosine kinase 1 activity. J. Biol. Chem. 2014, 289, 32178–32185. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Boehmler, A.M.; Seitz, G.; Kuci, S.; Wiesner, T.; Brinkmann, V.; Kanz, L.; Mohle, R. The sphingosine 1-phosphate receptor agonist FTY720 supports CXCR4-dependent migration and bone marrow homing of human CD34+ progenitor cells. Blood 2004, 103, 4478–4486. [Google Scholar] [CrossRef] [PubMed]
- Golan, K.; Vagima, Y.; Ludin, A.; Itkin, T.; Cohen-Gur, S.; Kalinkovich, A.; Kollet, O.; Kim, C.; Schajnovitz, A.; Ovadya, Y.; et al. S1P promotes murine progenitor cell egress and mobilization via S1P1-mediated ROS signaling and SDF-1 release. Blood 2012, 119, 2478–2488. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Anada, Y.; Tani, M.; Ikeda, M.; Sano, T.; Kihara, A.; Igarashi, Y. Lack of sphingosine 1-phosphate-degrading enzymes in erythrocytes. Biochem. Biophys. Res. Commun. 2007, 357, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Kobayashi, N.; Yamaguchi, A.; Nishi, T. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T.; Sunden, Y.; Arai, Y.; Moriwaki, K.; Ishida, J.; et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Invest. 2012, 122, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, K.; Thangada, S.; Michaud, J.; Oo, M.L.; Ai, Y.; Lee, Y.M.; Wu, M.; Parikh, N.S.; Khan, F.; Proia, R.L.; et al. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem. J. 2006, 397, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Bryndza, E.; Abdel-Latif, A.; Ratajczak, J.; Maj, M.; Tarnowski, M.; Klyachkin, Y.M.; Houghton, P.; Morris, A.J.; Vater, A.; et al. Bioactive lipids S1P and C1P are prometastatic factors in human rhabdomyosarcoma, and their tissue levels increase in response to radio/chemotherapy. Mol. Cancer Res. 2013, 11, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Pearl, D.K.; Van Brocklyn, J.R. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Tjwa, M.; Sidenius, N.; Moura, R.; Jansen, S.; Theunissen, K.; Andolfo, A.; De Mol, M.; Dewerchin, M.; Moons, L.; Blasi, F.; et al. Membrane-anchored uPAR regulates the proliferation, marrow pool size, engraftment, and mobilization of mouse hematopoietic stem/progenitor cells. J. Clin. Invest. 2009, 119, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Selleri, C.; Montuori, N.; Ricci, P.; Visconte, V.; Baiano, A.; Carriero, M.V.; Rotoli, B.; Rossi, G.; Ragno, P. In vivo activity of the cleaved form of soluble urokinase receptor: A new hematopoietic stem/progenitor cell mobilizer. Cancer Res. 2006, 66, 10885–10890. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Selleri, C.; Montuori, N.; Ricci, P.; Visconte, V.; Carriero, M.V.; Sidenius, N.; Serio, B.; Blasi, F.; Rotoli, B.; Rossi, G.; et al. Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood 2005, 105, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Pliyev, B.K.; Antonova, O.A.; Menshikov, M. Participation of the urokinase-type plasminogen activator receptor (uPAR) in neutrophil transendothelial migration. Mol. Immunol. 2011, 48, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Obradovic, H.; Jaukovic, A.; Okic-Dordevic, I.; Trivanovic, D.; Kukolj, T.; Mojsilovic, S.; Ilic, V.; Santibanez, J.F.; Bugarski, D. Urokinase type plasminogen activator mediates Interleukin-17-induced peripheral blood mesenchymal stem cell motility and transendothelial migration. Biochim. Biophys. Acta. 2014, 1853, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Borgatti, A.; Winter, A.; Stuebner, K.; Koopmeiners, J.; Modiano, J.F.; Vallera, D.A. Preclinical Evaluation of a Novel Bispecific Targeted Toxin for The Treatment of Sarcomas. In Proceedings of the American Association for Cancer Research Annual Meeting, Philadelphia, PA, USA, 18–22 April 2015.
- Lee, J.W.; Shahzad, M.M.; Lin, Y.G.; Armaiz-Pena, G.; Mangala, L.S.; Han, H.D.; Kim, H.S.; Nam, E.J.; Jennings, N.B.; Halder, J.; et al. Surgical stress promotes tumor growth in ovarian carcinoma. Clin. Cancer Res. 2009, 15, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Vilardi, B.M.; Bravo-Calderon, D.M.; Bernabe, D.G.; Oliveira, S.H.; Oliveira, D.T. VEGF-C expression in oral cancer by neurotransmitter-induced activation of beta-adrenergic receptors. Tumour Biol. 2013, 34, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.V.; Kim, S.J.; Donovan, E.L.; Chen, M.; Gross, A.C.; Webster Marketon, J.I.; Barsky, S.H.; Glaser, R. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: Implications for stress-related enhancement of tumor progression. Brain Behav. Immun. 2009, 23, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.V.; Sood, A.K.; Chen, M.; Li, Y.; Eubank, T.D.; Marsh, C.B.; Jewell, S.; Flavahan, N.A.; Morrison, C.; Yeh, P.E.; et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006, 66, 10357–10364. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Armaiz-Pena, G.; Takahashi, R.; Lin, Y.G.; Trevino, J.; Li, Y.; Jennings, N.; Arevalo, J.; Lutgendorf, S.K.; Gallick, G.E.; et al. Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a Src-dependent mechanism. J. Biol. Chem. 2007, 282, 29919–29926. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, M.M.; Arevalo, J.M.; Armaiz-Pena, G.N.; Lu, C.; Stone, R.L.; Moreno-Smith, M.; Nishimura, M.; Lee, J.W.; Jennings, N.B.; Bottsford-Miller, J.; et al. Stress effects on FosB- and interleukin-8 (IL8)-driven ovarian cancer growth and metastasis. J. Biol. Chem. 2010, 285, 35462–35470. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, G.; Ciccarelli, M.; Sorriento, D.; Galasso, G.; Campanile, A.; Santulli, G.; Cipolletta, E.; Cerullo, V.; Cimini, V.; Altobelli, G.G.; et al. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: A novel role for the endothelial adrenergic system. Circ. Res. 2005, 97, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, M.; Sorriento, D.; Cipolletta, E.; Santulli, G.; Fusco, A.; Zhou, R.H.; Eckhart, A.D.; Peppel, K.; Koch, W.J.; Trimarco, B.; et al. Impaired neoangiogenesis in beta(2)-adrenoceptor gene-deficient mice: Restoration by intravascular human beta(2)-adrenoceptor gene transfer and role of NFkappaB and CREB transcription factors. Br. J. Pharmacol. 2012, 162, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013. [Google Scholar] [CrossRef] [PubMed]
- Calvani, M.; Pelon, F.; Comito, G.; Taddei, M.L.; Moretti, S.; Innocenti, S.; Nassini, R.; Gerlini, G.; Borgognoni, L.; Bambi, F.; Giannoni, E.; Filippi, L.; Chiarugi, P. Norepinephrine promotes tumor microenvironment reactivity through β3-adrenoreceptors during melanoma progression. Oncotarget 2015, 6, 4615–4632. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, B.A.; Trapp, S.; Phang, T.L.; Schappa, J.T.; Hunter, L.E.; Modiano, J.F. Gene expression profiles of sporadic canine hemangiosarcoma are uniquely associated with breed. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Padilla, M.L.; Dickerson, E.B.; Steinberg, H.; Breen, M.; Auerbach, R.; Helfand, S.C. Interleukin-12 inhibits tumor growth in a novel angiogenesis canine hemangiosarcoma xenograft model. Neoplasia 2004, 6, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Fosmire, S.P.; Dickerson, E.B.; Scott, A.M.; Bianco, S.R.; Pettengill, M.J.; Meylemans, H.; Padilla, M.; Frazer-Abel, A.A.; Akhtar, N.; Getzy, D.M.; et al. Canine malignant hemangiosarcoma as a model of primitive angiogenic endothelium. Lab. Invest. 2004, 84, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Thamm, D.H.; Dickerson, E.B.; Akhtar, N.; Lewis, R.; Auerbach, R.; Helfand, S.C.; MacEwen, E.G. Biological and molecular characterization of a canine hemangiosarcoma-derived cell line. Res. Vet. Sci. 2006, 81, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Frantz, A.M.; Anderson, K.L.; Graef, A.J.; Scott, M.C.; Robinson, S.; Sharkey, L.C.; O’Brien, T.D.; Dickerson, E.B.; Modiano, J.F. Interleukin-8 promotes canine hemangiosarcoma growth by regulating the tumor microenvironment. Exp. Cell Res. 2014, 323, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Langin, D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol. Res. 2006, 53, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Maziere, C.; Maziere, J.C.; Salmon, S.; Mora, L.; Auclair, M. The antihypertensive drug propranolol enhances LDL catabolism and alters cholesterol metabolism in human cultured fibroblasts. Atherosclerosis 1990, 81, 151–160. [Google Scholar] [CrossRef]
- Stiles, J.; Amaya, C.; Pham, R.; Rowntree, R.K.; Lacaze, M.; Mulne, A.; Bischoff, J.; Kokta, V.; Boucheron, L.E.; Mitchell, D.C.; Bryan, B.A. Propranolol treatment of infantile hemangioma endothelial cells: A molecular analysis. Exp. Ther. Med. 2012, 4, 594–604. [Google Scholar] [PubMed]
- Romero, I.L.; Mukherjee, A.; Kenny, H.A.; Litchfield, L.M.; Lengyel, E. Molecular pathways: trafficking of metabolic resources in the tumor microenvironment. Clin. Cancer Res. 2015, 21, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Kafara, P.; Steyaert, J.M.; Schwartz, L.; Lincet, H. The metabolic cooperation between cells in solid cancer tumors. Biochim. Biophys. Acta 2014, 1846, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dickerson, E.B.; Bryan, B.A. Beta Adrenergic Signaling: A Targetable Regulator of Angiosarcoma and Hemangiosarcoma. Vet. Sci. 2015, 2, 270-292. https://doi.org/10.3390/vetsci2030270
Dickerson EB, Bryan BA. Beta Adrenergic Signaling: A Targetable Regulator of Angiosarcoma and Hemangiosarcoma. Veterinary Sciences. 2015; 2(3):270-292. https://doi.org/10.3390/vetsci2030270
Chicago/Turabian StyleDickerson, Erin B., and Brad A. Bryan. 2015. "Beta Adrenergic Signaling: A Targetable Regulator of Angiosarcoma and Hemangiosarcoma" Veterinary Sciences 2, no. 3: 270-292. https://doi.org/10.3390/vetsci2030270
APA StyleDickerson, E. B., & Bryan, B. A. (2015). Beta Adrenergic Signaling: A Targetable Regulator of Angiosarcoma and Hemangiosarcoma. Veterinary Sciences, 2(3), 270-292. https://doi.org/10.3390/vetsci2030270