
Progress in Adaptive Immunotherapy for Cancer in Companion Animals: Success on the Path to a Cure


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
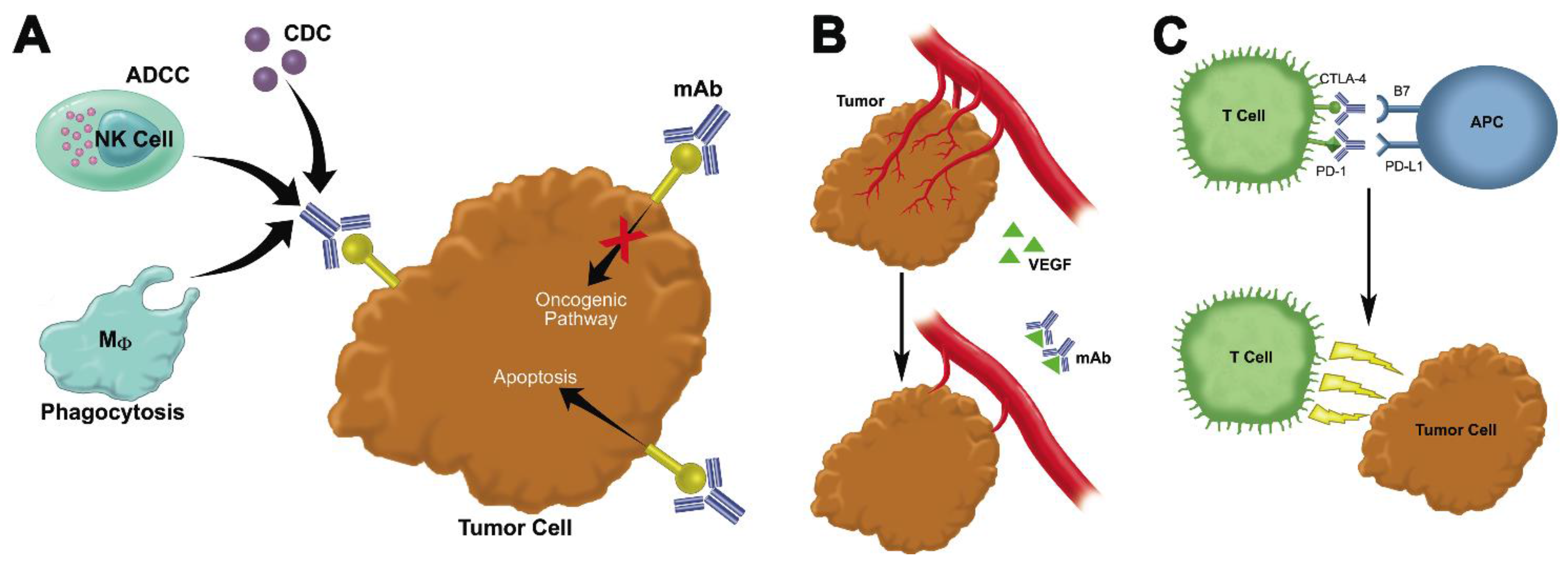
2. Passive Immunotherapy: Monoclonal Antibodies
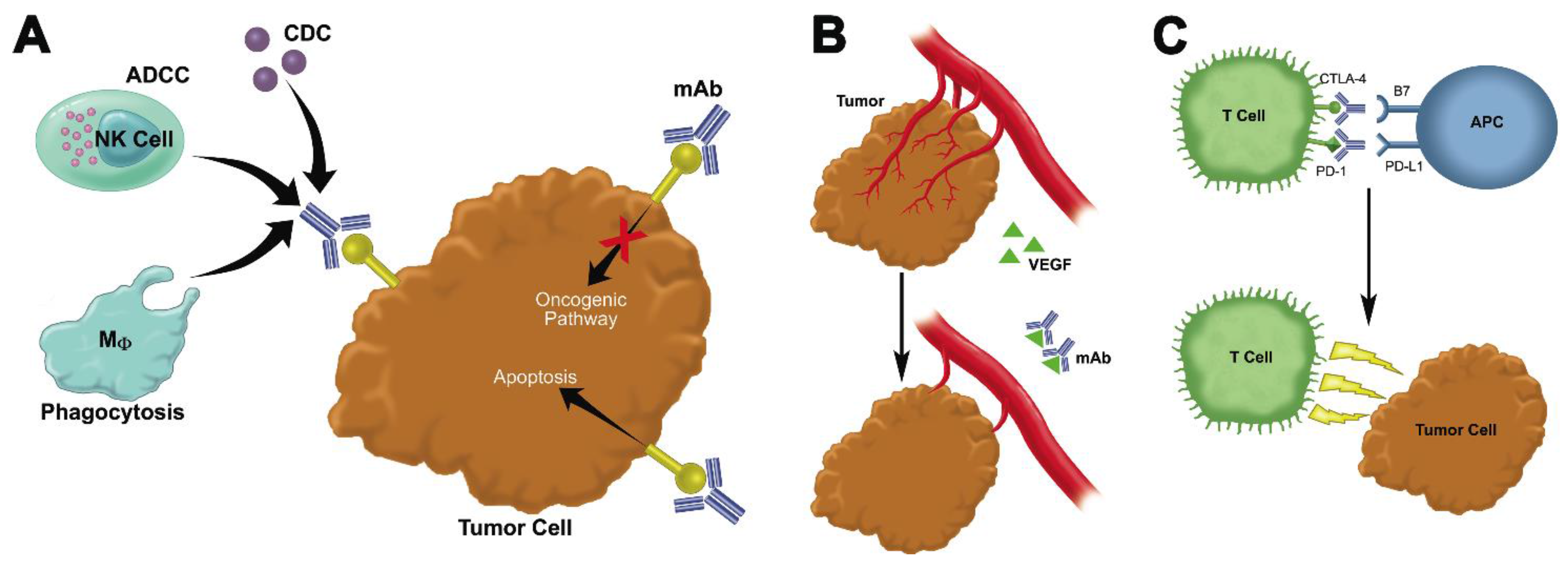
2.1. Monoclonal Antibodies That Bind to Malignant Cells and Antagonize Oncogenic Pathways
2.2. Monoclonal Antibodies That Block Growth-Promoting Pathways in the Tumor Stroma
2.3. Immune Checkpoint Inhibitors
2.4. Bispecifics, Trispecifics, Immunoconjugates, and Other Modified Antibodies That Enhance the Interaction between Immune Cells, Tumor Targets, and the Tumor Microenvironment
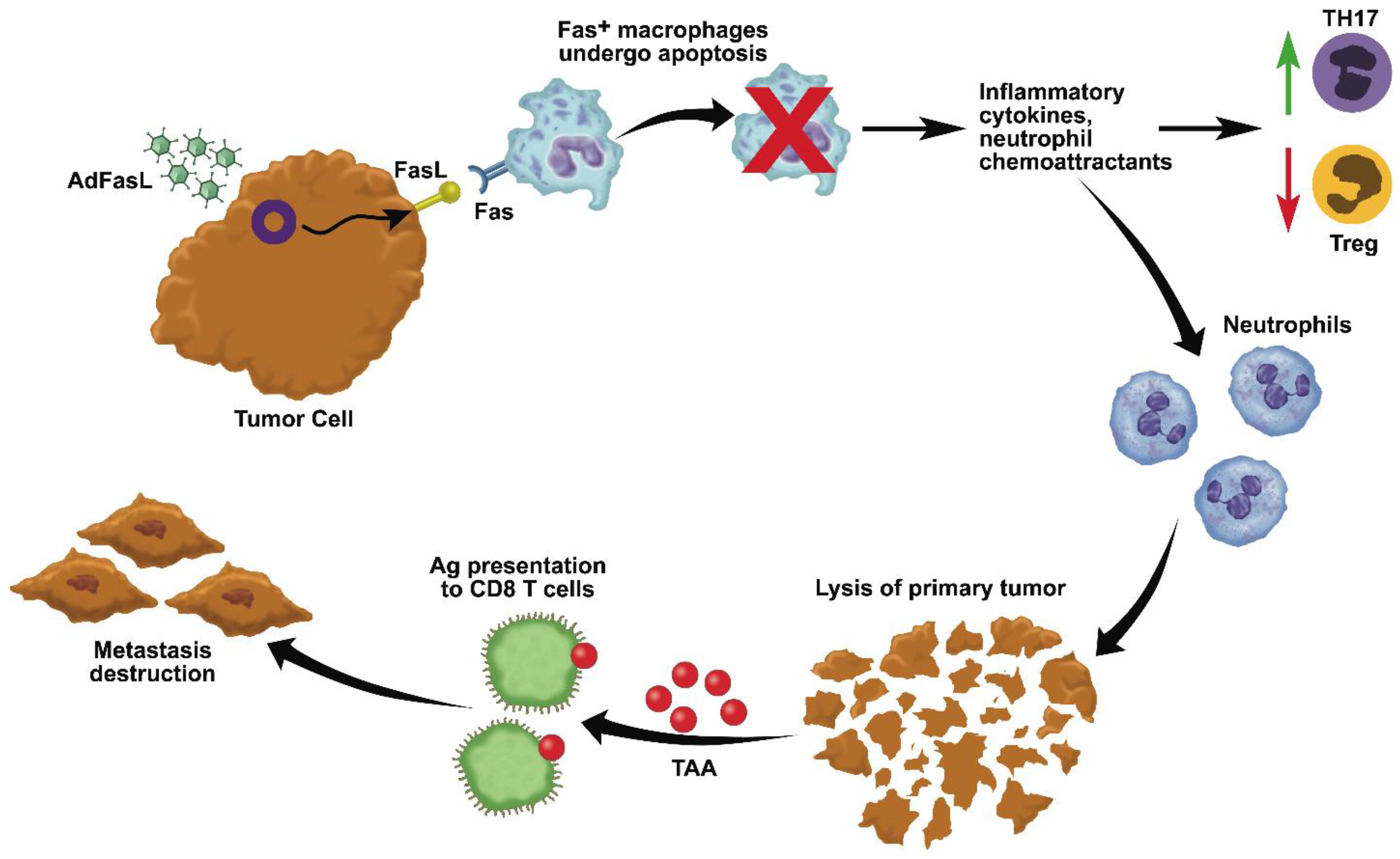
3. Active Immunotherapy: In Situ Immunization with Adenovirus-Fas Ligand
4. Administration of Attenuated Bacteria
5. Oncolytic Virotherapy
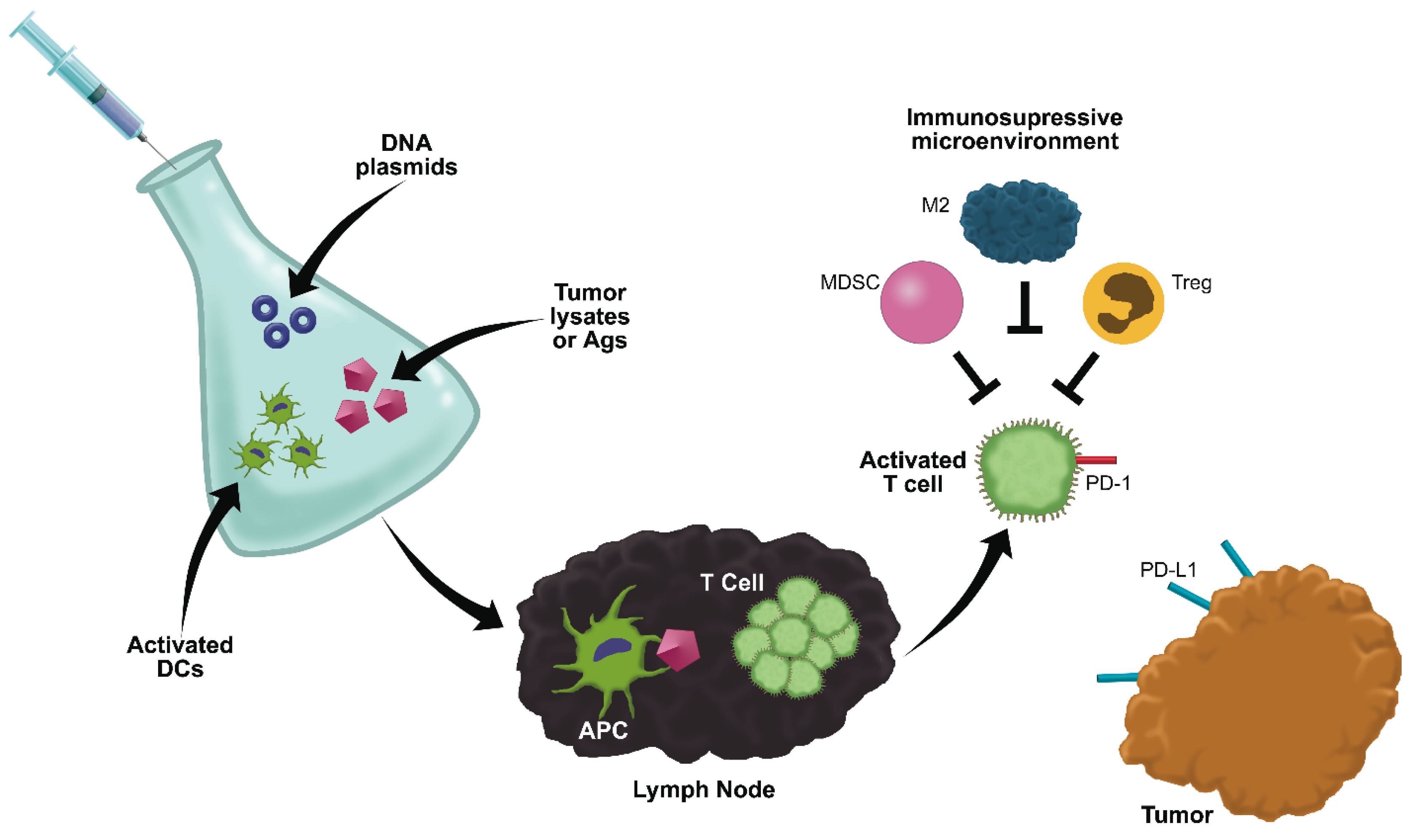
6. Anti-Cancer Vaccines
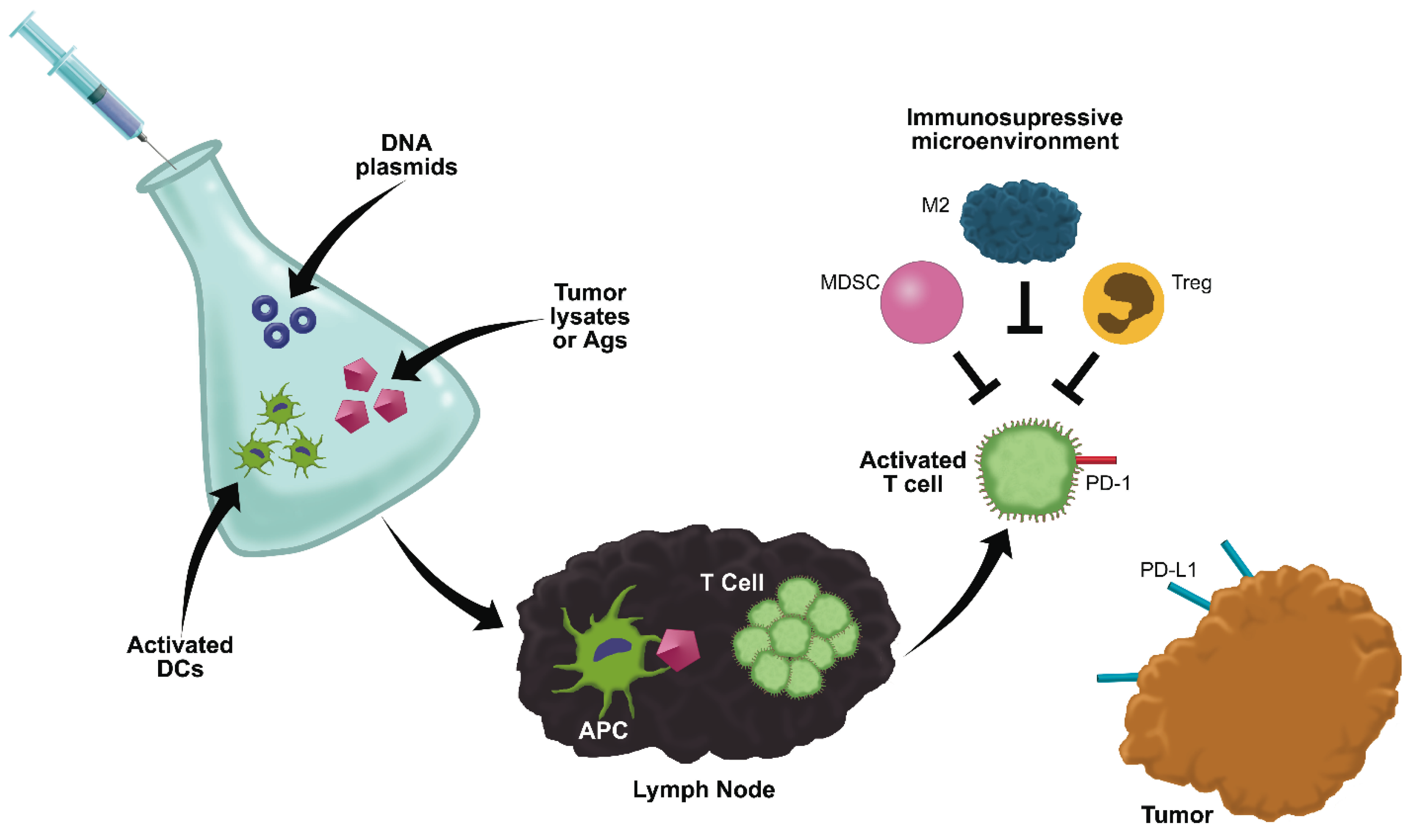
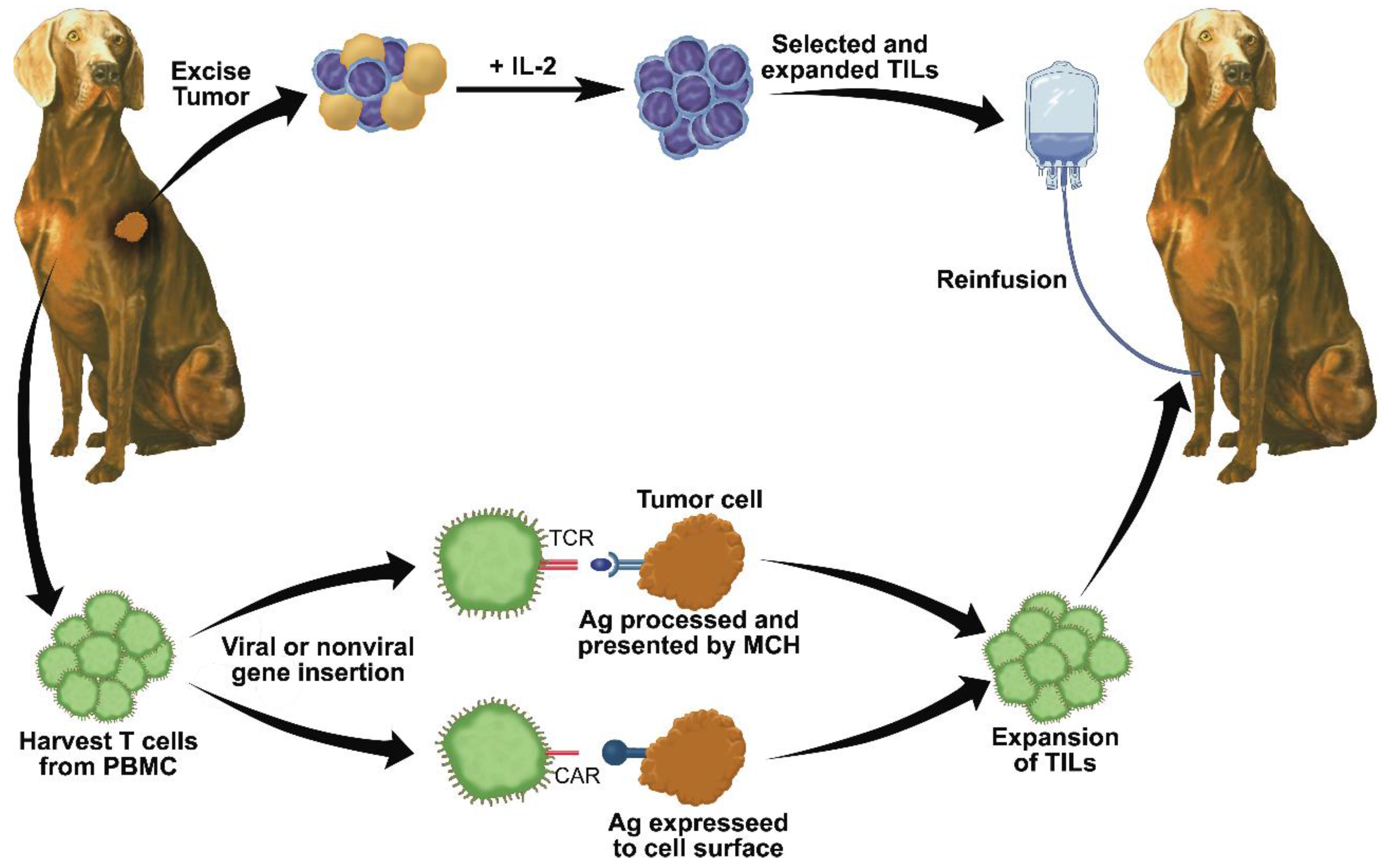
7. Adoptive T Cell Transfer
8. Adoptive Natural Killer Cell Transfer
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Couzin-Frankel, J. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Elert, E. Calling cells to arms. Nature 2013, 504, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Weiner, G.J. Cancer immunotherapy and breaking immune tolerance: New approaches to an old challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Wilson-Robles, H. Developing T cell cancer immunotherapy in the dog with lymphoma. ILAR J. 2014, 55, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Galon, J.; Sautes-Fridman, C.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Tumor-targeting monoclonal antibodies in cancer therapy. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Sliwkowski, M.X.; Mellman, I. Antibody therapeutics in cancer. Science 2013, 341, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.; Fazekas, J.; Wang, W.; Weichselbaumer, M.; Matz, M.; Mader, A.; Steinfellner, W.; Meitz, S.; Mechtcheriakova, D.; Sobanov, Y.; et al. Generation of a canine anti-EGFR (ErbB-1) antibody for passive immunotherapy in dog cancer patients. Mol. Cancer Ther. 2014, 13, 1777–1790. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Brewer, S.; Modiano, J.F.; Beall, M.J. Development of a novel anti-canine CD20 monoclonal antibody with diagnostic and therapeutic potential. Leuk Lymphoma 2015, 56, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Lui, G.; Molens, J.P.; Sotto, J.J.; Chaperot, L.; Plumas, J. Whole lymphoma B cells allow efficient cross-presentation of antigens by dendritic cells. Cytotherapy 2008, 10, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Deligne, C.; Metidji, A.; Fridman, W.H.; Teillaud, J.L. Anti-CD20 therapy induces a memory Th1 response through the IFN-gamma/IL-12 axis and prevents protumor regulatory T cell expansion in mice. Leukemia 2015, 29, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Efe, J.; Rue, S. A Chimeric (Canine) Anti-CD20 Monoclonal Antibody is Effective in A Xenograft Model of Canine Lymphoma; Melnick, A., Staudt, L.M., Weinstock, D., Eds.; American Society of Hematology: Colorado Springs, CO, USA, 2014; p. 102. [Google Scholar]
- Beall, M.J. Canine Anti-CD20 Antibodies. US20110091483 A1, 21 April 2011. Available online: http://www.google.com/patents/US20110091483 (accessed on 16 October 2015). [Google Scholar]
- Rue, S.; Eckleman, B.; Deveraux, Q.; Nasoff, N. Monoclonal Antibodies and Methods of Use. WO 2013063186 A1, 2013. Available online: https://data.epo.org/gpi/EP2771694A4-MONOCLONAL-ANTIBODIES-AND-METHODS-OF-USE (accessed on 16 October 2015). [Google Scholar]
- Hansen, G. Monoclonal Antibodies. US20110217298 A1, 2012. Available online: http://www.google.com/patents/US20110217298 (accessed on 16 October 2015). [Google Scholar]
- Ogilvie, G. Treatment of Canine B-Cell Lymphoma with Chemotherapy and A Canine Anti-CD20 Monoclonal Antibody: A Prospective Double-Blind, Randomized, Placebo-Controlled Study. In Proceedings of the Veterinary Cancer Society Annual Meeting, St. Louis, MO, USA, 10 October 2014.
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Aratana Therapeutics Announces Conditional Approval of Second Canine-Specific Antibody Therapy. In AT-005 Marks Aratana’s First Commercial Opportunity in T-Cell Canine Lymphoma; PR Newswire: Kansas City, MO, USA, 2014; Available online: http://www.aratana.com/ (accessed on 16 October 2015).
- Trivedi, S.; Jie, H.B.; Ferris, R.L. Tumor antigen-specific monoclonal antibodies and induction of T-cell immunity. Semin. Oncol. 2014, 41, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.; Craft, D.M.; Scase, T.J.; Bergman, P.J. Immunohistochemical detection of Her-2/neu expression in spontaneous feline mammary tumours. Vet. Comp. Oncol. 2005, 3, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C. Cats, cancer and comparative oncology. Vet. Sci. 2015, 2, 111–126. [Google Scholar] [CrossRef]
- Michishita, M.; Uto, T.; Nakazawa, R.; Yoshimura, H.; Ogihara, K.; Naya, Y.; Tajima, T.; Azakami, D.; Kishikawa, S.; Arai, T.; et al. Antitumor effect of bevacizumab in a xenograft model of canine hemangiopericytoma. J. Pharmacol. Sci. 2013, 121, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Scharf, V.F.; Farese, J.P.; Coomer, A.R.; Milner, R.J.; Taylor, D.P.; Salute, A.R.; Chang, M.N.; Neal, D.; Siemann, D.W. Effect of bevacizumab on angiogenesis and growth of canine osteosarcoma cells xenografted in athymic mice. Am. J. Vet. Res. 2013, 74, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. PNAS 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, K. Releasing the brakes. Nature 2013, 504, S6–S8. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Callahan, M.K.; Postow, M.A.; Wolchok, J.D. On being less tolerant: Enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Sci. Transl. Med. 2015, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Konnai, S.; Ikebuchi, R.; Okagawa, T.; Adachi, M.; Takagi, S.; Kagawa, Y.; Nakajima, C.; Suzuki, Y.; Murata, S.; et al. Expression of PD-L1 on canine tumor cells and enhancement of IFN-gamma production from tumor-infiltrating cells by PD-L1 blockade. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.S.; Choi, E.W.; Chung, J.Y.; Hwang, C.Y.; Lee, C.W.; Youn, H.Y. Cloning, expression and bioassay of canine CTLA4-Ig. Vet. Immunol. Immunopathol. 2007, 118, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Graves, S.S.; Stone, D.; Loretz, C.; Peterson, L.; McCune, J.S.; Mielcarek, M.; Storb, R. Establishment of long-term tolerance to srbc in dogs by recombinant canine CTLA4-Ig. Transplantation 2009, 88, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Huehls, A.; Coupet, T.; Sentman, C. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Knorr, D.A.; Bachanova, V.; Verneris, M.R.; Miller, J.S. Clinical utility of natural killer cells in cancer therapy and transplantation. Semin. Immunol. 2014, 26, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Mack, F.; Ritchie, M.; Sapra, P. The next generation of antibody drug conjugates. Semin. Oncol. 2014, 41, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Bethge, W.A.; Wilbur, D.S.; Storb, R.; Hamlin, D.K.; Santos, E.B.; Brechbiel, M.W.; Sandmaier, B.M. Radioimmunotherapy with bismuth-213 as conditioning for nonmyeloablative allogeneic hematopoietic cell transplantation in dogs: A dose deescalation study. Transplantation 2004, 78, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Bethge, W.A.; Wilbur, D.S.; Sandmaier, B.M. Radioimmunotherapy as non-myeloablative conditioning for allogeneic marrow transplantation. Leuk Lymphoma 2006, 47, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Burtner, C.; Chandrasekaran, D.; Santos, E.; Beard, B.; Adair, J.; Hamlin, D.; Wilbur, D.S.; Sandmaier, B.; Kiem, H.P. 211 astatine-conjugated monoclonal CD45 antibody-based nonmyeloablative conditioning for stem cell gene therapy. Hum. Gene Ther. 2015, 26, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Modiano, J.F.; Lamerato-Kozicki, A.R.; Jubala, C.M.; Coffey, D.; Borakove, M.; Schaack, J.; Bellgrau, D. Fas ligand gene transfer for cancer therapy. Cancer Ther. 2004, 2, 561–570. [Google Scholar]
- Modiano, J.F.; Bellgrau, D.; Cutter, G.R.; Lana, S.E.; Ehrhart, N.P.; Ehrhart, E.; Wilke, V.L.; Charles, J.B.; Munson, S.; Scott, M.C.; et al. Inflammation, apoptosis, and necrosis induced by neoadjuvant Fas ligand gene therapy improves survival of dogs with spontaneous bone cancer. Mol. Ther. 2012, 20, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Duke, R.C.; Newell, E.; Schleicher, M.; Meech, S.J.; Bellgrau, D. Transplantation of cells and tissues expressing Fas ligand. Transplant. Proc. 1999, 31, 1479–1481. [Google Scholar] [CrossRef]
- Seino, K.; Kayagaki, N.; Okumura, K.; Yagita, H. Antitumor effect of locally produced CD95 ligand. Nat. Med. 1997, 3, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Hohlbaum, A.M.; Gregory, M.S.; Ju, S.T.; Marshak-Rothstein, A. Fas ligand engagement of resident peritoneal macrophages in vivo induces apoptosis and the production of neutrophil chemotactic factors. J. Immunol. 2001, 167, 6217–6224. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Fontana, A.; Takeda, Y.; Yagita, H.; Yoshimoto, T.; Matsuzawa, A. Induction of antitumor immunity with Fas/APO-1 ligand (CD95l)-transfected neuroblastoma neuro-2a cells. J. Immunol. 1999, 162, 7350–7357. [Google Scholar] [PubMed]
- Arai, H.; Gordon, D.; Nabel, E.G.; Nabel, G.J. Gene transfer of Fas ligand induces tumor regression in vivo. Proc. Natl. Acad. Sci. 1997, 94, 13862–13867. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.K.; Gallimore, A.; Jones, E.; Sawitzki, B.; Cerundolo, V.; Screaton, G.R. Fas ligand breaks tolerance to self-antigens and induces tumor immunity mediated by antibodies. Cancer Cell 2002, 2, 315–322. [Google Scholar] [CrossRef]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liu, S.; Park, D.; Kang, Y.; Zheng, G. Depleting intratumoral CD4+CD25+ regulatory T cells via FasL protein transfer enhances the therapeutic efficacy of adoptive T cell transfer. Cancer Res. 2007, 67, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.R.; Sun, J.; Fosmire, S.P.; Hance, K.; Padilla, M.L.; Ritt, M.G.; Getzy, D.M.; Duke, R.C.; Withrow, S.J.; Lana, S.; et al. Enhancing antimelanoma immune responses through apoptosis. Cancer Gene Ther. 2003, 10, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, T.E.; Meech, S.J.; Srikanth, S.; Kraft, A.S.; Miller, G.J.; Schaack, J.; Duke, R. Adenovirus-mediated expression of Fas ligand induces apoptosis of human prostate cancer cells. Cell Death Differ. 1999, 6, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.M.; Paterson, Y. Attenuated Listeria monocytogenes: A powerful and versatile vector for the future of tumor immunotherapy. Front. Cell. Infect. Microbiol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Thamm, D.H.; Kurzman, I.D.; King, I.; Li, Z.; Sznol, M.; Dubielzig, R.R.; Vail, D.M.; MacEwen, E.G. Systemic administration of an attenuated, tumor-targeting Salmonella typhimurium to dogs with spontaneous neoplasia: Phase I evaluation. Clin. Cancer Res. 2005, 11, 4827–4834. [Google Scholar] [CrossRef] [PubMed]
- Saltzman, D.A. Cancer immunotherapy based on the killing of Salmonella typhimurium-infected tumour cells. Expert Opin. Biol. Ther. 2005, 5, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, B.S.; Banton, K.L.; Frykman, N.L.; Leonard, A.S.; Saltzman, D.A. Attenuated Salmonella typhimurium with IL-2 gene reduces pulmonary metastases in murine osteosarcoma. Clin. Orthop. Relat. Res. 2008, 466, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Fritz, S.E.; Henson, M.; Greengard, E.; Winter, A.L.; Stuebner, K.M.; Yoon, U.; Wilke, V.L.; Borgatti, A.; Augustin, L.B.; Modiano, J.F.; et al. A phase I clinical study to evaluate safety of orally administered, genetically engineered Salmonella enterica serovar typhimurium for canine osteosarcoma. J. Vet. Med. Sci. 2015. submitted. [Google Scholar]
- Gnanandarajah, J.S.; Ndikuyeze, G.; Engiles, J.B.; Wallecha, A.; Mason, N. A recombinant Her2/neu expressing Listeria monocytogenes (lm-llo) immunotherapy delays metastatic disease and prolongs overall survival in a spontaneous canine model of osteosarcoma—A phase I clinical trial. J. ImmunoTher. Cancer 2014, 2. [Google Scholar] [CrossRef]
- Pol, J.; Bloy, N.; Obrist, F.; Eggermont, A.; Galon, J.; Cremer, I.; Erbs, P.; Limacher, J.M.; Preville, X.; Zitvogel, L.; et al. Trial watch: Oncolytic viruses for cancer therapy. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Gentschev, I.; Patil, S.S.; Petrov, I.; Cappello, J.; Adelfinger, M.; Szalay, A.A. Oncolytic virotherapy of canine and feline cancer. Viruses 2014, 6, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Paterson, J.M.; Lemay, C.G.; Falls, T.J.; McGuire, A.; Parato, K.A.; Stojdl, D.F.; Daneshmand, M.; Speth, K.; Kirn, D.; et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007, 15, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Westberg, S.; Sadeghi, A.; Svensson, E.; Segall, T.; Dimopoulou, M.; Korsgren, O.; Hemminki, A.; Loskog, A.S.; Tötterman, T.H.; von Euler, H. Treatment efficacy and immune stimulation by AdCD40L gene therapy of spontaneous canine malignant melanoma. J. Immunother. 2013, 36, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.; Li, C.Y.; Larue, S.M.; Poulson, J.M.; Avery, P.R.; Pruitt, A.F.; Zhang, X.; Ullrich, R.L.; Thrall, D.E.; Dewhirst, M.W.; et al. A phase I trial of hyperthermia-induced interleukin-12 gene therapy in spontaneously arising feline soft tissue sarcomas. Mol. Cancer Ther. 2007, 6, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Jourdier, T.M.; Moste, C.; Bonnet, M.C.; Delisle, F.; Tafani, J.P.; Devauchelle, P.; Tartaglia, J.; Moingeon, P. Local immunotherapy of spontaneous feline fibrosarcomas using recombinant poxviruses expressing interleukin 2 (IL-2). Gene Ther. 2003, 10, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.K.; Naik, S.; Galyon, G.D.; Jenks, N.; Steele, M.; Peng, K.W.; Federspiel, M.J.; Donnell, R.; Russell, S.J. Safety studies on intravenous administration of oncolytic recombinant vesicular stomatitis virus in purpose-bred beagle dogs. Hum. Gene Ther. Clin. Dev. 2013, 24, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.J. Anticancer vaccines. Vet. Clin. North Am. Small Anim. Pract. 2007, 37, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Kissick, H.T.; Sanda, M.G. The role of active vaccination in cancer immunotherapy: Lessons from clinical trials. Curr. Opin. Immunol. 2015, 35, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Mulders, P.F.; de Santis, M.; Powles, T.; Fizazi, K. Targeted treatment of metastatic castration-resistant prostate cancer with Sipuleucel-T immunotherapy. Cancer Immunol. Immunother. 2015, 64, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.J.; McKnight, J.; Novosad, A.; Charney, S.; Farrelly, J.; Craft, D.M.; Wulderk, M.; Jeffers, Y.; Sadelain, M.; Hohenhaus, A.E.; et al. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: A phase I trial. Clin. Cancer Res. 2003, 9, 1284–1290. [Google Scholar] [PubMed]
- Bergman, P.J.; Camps-Palau, M.A.; McKnight, J.A.; Leibman, N.F.; Craft, D.M.; Leung, C.; Liao, J.; Riviere, I.; Sadelain, M.; Hohenhaus, A.E.; et al. Development of a xenogeneic DNA vaccine program for canine malignant melanoma at the animal medical center. Vaccine 2006, 24, 4582–4585. [Google Scholar] [CrossRef] [PubMed]
- Grosenbaugh, D.A.; Leard, A.T.; Bergman, P.J.; Klein, M.K.; Meleo, K.; Susaneck, S.; Hess, P.R.; Jankowski, M.K.; Jones, P.D.; Leibman, N.F.; et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am. J. Vet. Res. 2011, 72, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Ottnod, J.M.; Smedley, R.C.; Walshaw, R.; Hauptman, J.G.; Kiupel, M.; Obradovich, J.E. A retrospective analysis of the efficacy of Oncept vaccine for the adjunct treatment of canine oral malignant melanoma. Vet. Comp. Oncol. 2013, 11, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Gregor, P.; Wolchok, J.D.; Orlandi, F.; Craft, D.M.; Leung, C.; Houghton, A.N.; Bergman, P.J. Vaccination with human tyrosinase DNA induces antibody responses in dogs with advanced melanoma. Cancer Immunol. 2006, 6, 1–17. [Google Scholar]
- Vail, D.M. Levels of evidence in canine oncology trials—A case in point. Vet. Comp. Oncol. 2013, 11, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, D.; Gavazza, A.; Mesiti, G.; Lubas, G.; Scarselli, E.; Conforti, A.; Bendtsen, C.; Ciliberto, G.; La Monica, N.; Aurisicchio, L. A vaccine targeting telomerase enhances survival of dogs affected by B-cell lymphoma. Mol. Ther. 2010, 18, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, D.; Mesiti, G.; Ciliberto, G.; La Monica, N.; Aurisicchio, L. Telomerase and Her-2/neu as targets of genetic cancer vaccines in dogs. Vaccine 2010, 28, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Gavazza, A.; Lubas, G.; Fridman, A.; Peruzzi, D.; Impellizeri, J.A.; Luberto, L.; Marra, E.; Roscilli, G.; Ciliberto, G.; Aurisicchio, L. Safety and efficacy of a genetic vaccine targeting telomerase plus chemotherapy for the therapy of canine B-cell lymphoma. Hum. Gene Ther. 2013, 24, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.M.; Pluhar, G.E.; Seiler, C.E.; Goulart, M.R.; SantaCruz, K.S.; Schutten, M.M.; Meints, J.P.; O’Sullivan, M.G.; Bentley, R.T.; Packer, R.A.; et al. Vaccination for invasive canine meningioma induces in situ production of antibodies capable of antibody-dependent cell-mediated cytotoxicity. Cancer Res. 2013, 73, 2987–2997. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, S.; Rodriguez-Lecompte, J.C.; Woods, P.; Foley, R.; Kruth, S.; Liaw, P.C.Y.; Gauldie, J. Bone marrow-derived dendritic cell vaccination of dogs with naturally occurring melanoma by using human gp100 antigen. Vet. Intern. Med. 2005, 19, 56–63. [Google Scholar]
- Sorenmo, K.U.; Krick, E.; Coughlin, C.M.; Overley, B.; Gregor, T.P.; Vonderheide, R.H.; Mason, N.J. CD40-activated B cell cancer vaccine improves second clinical remission and survival in privately owned dogs with Non-Hodgkin’s lymphoma. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.; Venanzi, F.M.; Bagashova, E.; Rud, O.; Mariotti, F.; Vullo, C.; Catone, G.; Sherman, M.Y.; Concetti, A.; Chursov, A.; et al. Pilot study of p62 DNA vaccine in dogs with mammary tumors. Oncotarget 2015, 5, 12803–12810. [Google Scholar] [CrossRef]
- Heinzerling, L.M.; Feige, K.; Rieder, S.; Akens, M.K.; Dummer, R.; Stranzinger, G.; Moelling, K. Tumor regression induced by intratumoral injection of DNA coding for human interleukin 12 into melanoma metastases in gray horses. J. Mol. Med. 2001, 78, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.-M.V.; Feige, K.; Wunderlin, P.; Hodl, A.; Meli, M.L.; Seltenhammer, M.; Grest, P.; Nicolson, L.; Schelling, C.; Heinzerling, L.M. Double-blind placebo-controlled study with interleukin-18 and interleukin-12-encoding plasmid DNA shows antitumor effect in metastatic melanoma in gray horses. J. Immunother. 2011, 34, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Glikin, G.C.; Finocchiaro, L.M. Clinical trials of immunogene therapy for spontaneous tumors in companion animals. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Riddell, S.R.; Schumacher, T.N. Adoptive cellular therapy: A race to the fnish line. Sci. Transl. Med. 2015, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Sheppard, S.; Hartline, C.A.; Huls, H.; Johnson, M.; Palla, S.L.; Maiti, S.; Ma, W.; Davis, R.E.; Craig, S.; et al. Adoptive T-cell therapy improves treatment of canine Non-Hodgkin lymphoma post chemotherapy. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Vera, J.F.; Gerken, C.; Rooney, C.M.; Miller, T.; Pfent, C.; Wang, L.L.; Wilson-Robles, H.M.; Gottschalk, S. Toward immunotherapy with redirected T cells in a large animal model: Ex vivo activation, expansion, and genetic modification of canine T cells. J. Immunother. 2014, 37, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Michael, H.T.; Ito, D.; McCullar, V.; Zhang, B.; Miller, J.S.; Modiano, J.F. Isolation and characterization of canine natural killer cells. Vet. Immunol. Immunopathol. 2013, 155, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.J.; Lee, S.H.; Park, J.Y.; Kim, J.S.; Lee, J.J.; Suh, G.H.; Lee, Y.K.; Cho, D.; Kim, S.K. Interleukin-21 induces proliferation and modulates receptor expression and effector function in canine natural killer cells. Vet. Immunol. Immunopathol. 2015, 165, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Shin, D.J.; Kim, S.K. Generation of recombinant canine interleukin-15 and evaluation of its effects on the proliferation and function of canine NK cells. Vet. Immunol. Immunopathol. 2015, 165, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Trad, M.; Hanke, N.T.; Larmonier, C.B.; Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T cell transfer in breast cancer. Cancer Res. 2014, 74, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Hannesdottir, L.; Tymoszuk, P.; Parajuli, N.; Wasmer, M.H.; Philipp, S.; Daschil, N.; Datta, S.; Koller, J.B.; Tripp, C.H.; Stoitzner, P.; et al. Lapatinib and doxorubicin enhance the STAT1-dependent antitumor immune response. Eur. J. Immunol. 2013, 43, 2718–2729. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Mirsoian, A.; Bouchlaka, M.N.; Sckisel, G.D.; Chen, M.; Pai, C.C.; Maverakis, E.; Spencer, R.G.; Fishbein, K.W.; Siddiqui, S.; Monjazeb, A.M.; et al. Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. J. Exp. Med. 2014, 211, 2373–2383. [Google Scholar] [CrossRef] [PubMed]
- Rosinski, S.L.; Storb, R.; Strong, R.K.; Sale, G.E.; Stone, D.M.; Gewe, M.M.; Friend, D.J.; Abrams, V.K.; Randolph-Habecker, J.; Graves, S.S. Anti-CD28 antibody-initiated cytokine storm in canines. Transplant. Direct 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbe, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, K.L.; Modiano, J.F. Progress in Adaptive Immunotherapy for Cancer in Companion Animals: Success on the Path to a Cure. Vet. Sci. 2015, 2, 363-387. https://doi.org/10.3390/vetsci2040363
Anderson KL, Modiano JF. Progress in Adaptive Immunotherapy for Cancer in Companion Animals: Success on the Path to a Cure. Veterinary Sciences. 2015; 2(4):363-387. https://doi.org/10.3390/vetsci2040363
Chicago/Turabian StyleAnderson, Katie L., and Jaime F. Modiano. 2015. "Progress in Adaptive Immunotherapy for Cancer in Companion Animals: Success on the Path to a Cure" Veterinary Sciences 2, no. 4: 363-387. https://doi.org/10.3390/vetsci2040363
APA StyleAnderson, K. L., & Modiano, J. F. (2015). Progress in Adaptive Immunotherapy for Cancer in Companion Animals: Success on the Path to a Cure. Veterinary Sciences, 2(4), 363-387. https://doi.org/10.3390/vetsci2040363