

Genomic Sequencing and Analysis of Enzootic Nasal Tumor Virus Type 2 Provides Evidence for Recombination within the Prevalent Chinese Strains

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. RNA Extraction
2.3. The Complete Genome Amplification of ENTV-2 CQ2
2.4. Sequence Assembly and Phylogenetic Analysis
2.5. Sequence Annotation and Comparative Analysis of ENTV-2 Whole Genome Sequences
2.6. Recombination Analysis
3. Results
3.1. Molecular Detection and Genomic Sequencing of ENTV-2
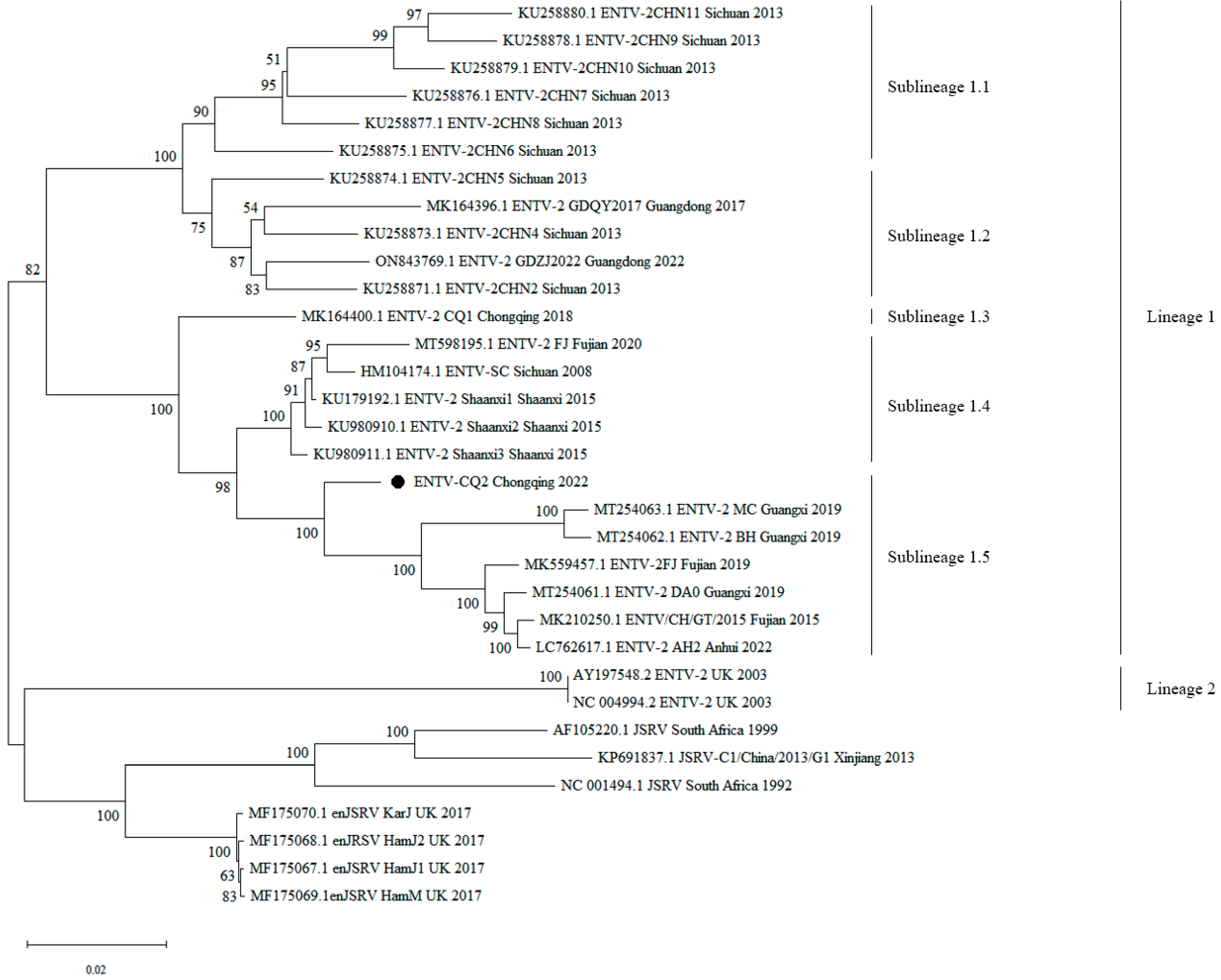
3.2. Phylogenetic Analysis of ENTV-2 CQ2
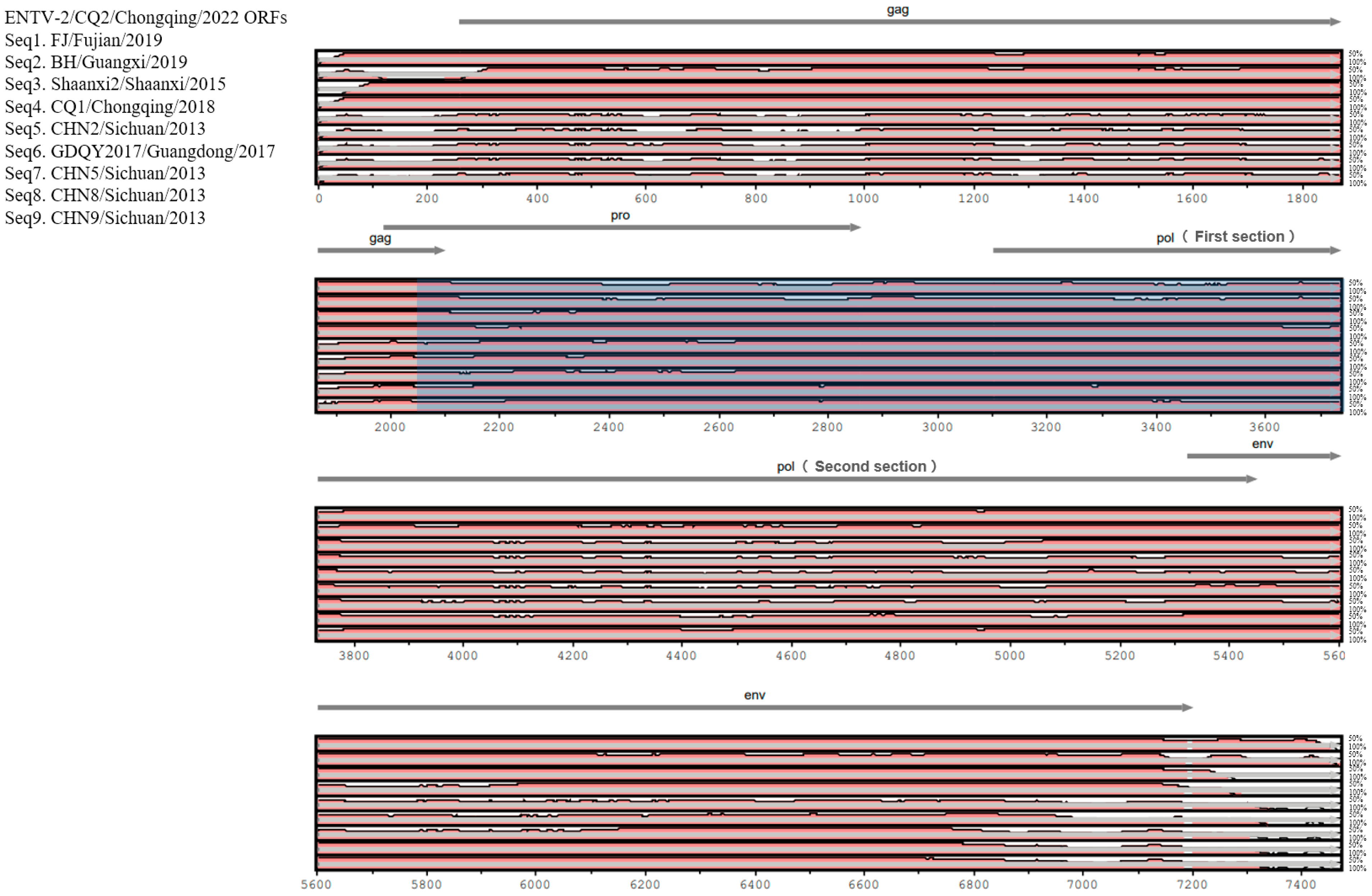
3.3. Comparative Genomic Analysis of ENTV-2 Strains
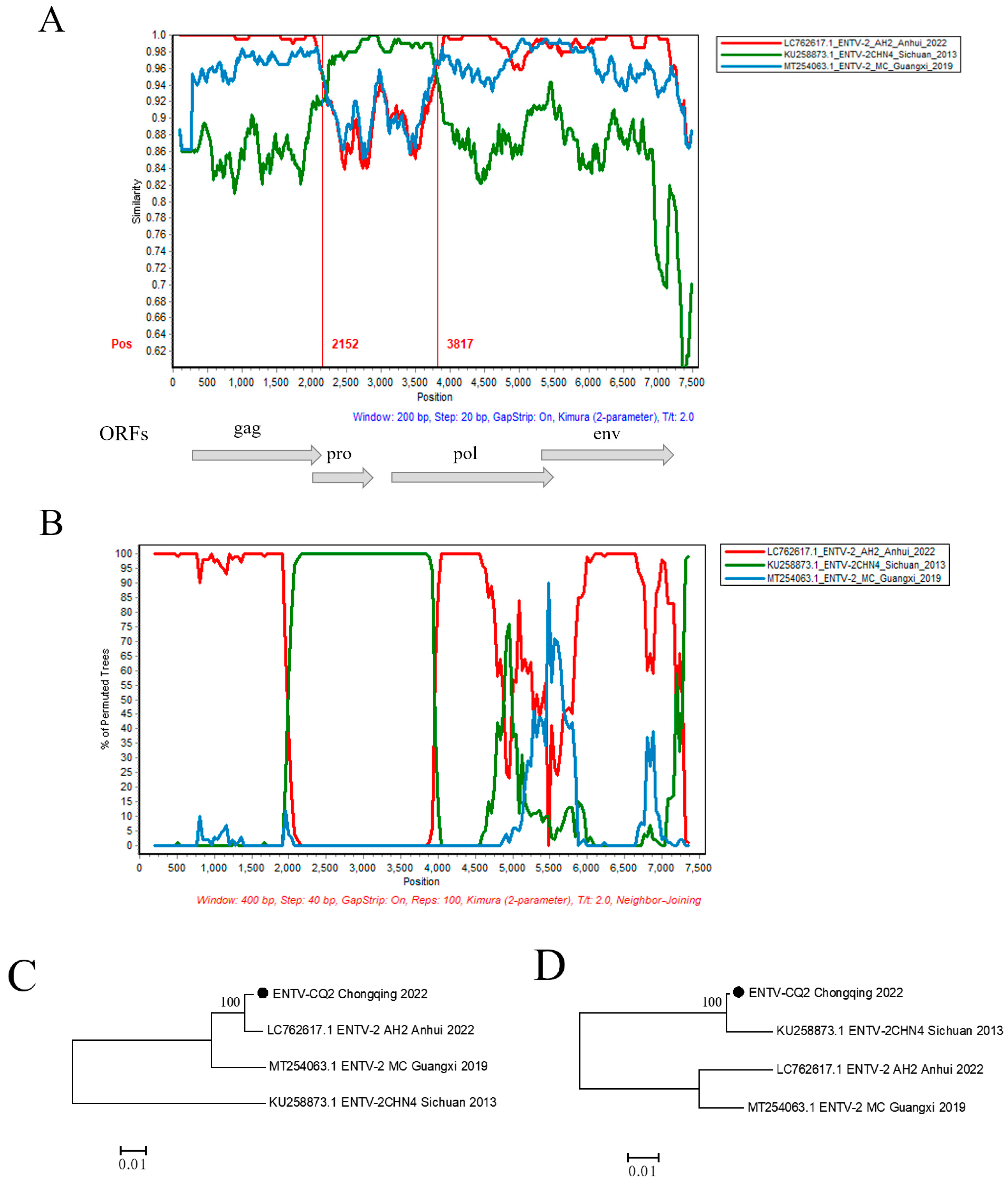
3.4. Recombination Analysis among ENTV-2 Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Las, H.M.; Ortin, A.; Cousens, C.; Minguijon, E.; Sharp, J.M. Enzootic nasal adenocarcinoma of sheep and goats. Curr. Top. Microbiol. 2003, 275, 201–223. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.R.; Linnerth-Petrik, N.M.; Laporte, A.N.; Menzies, P.I.; Foster, R.A.; Wootton, S.K. Full-length genome sequence analysis of enzootic nasal tumor virus reveals an unusually high degree of genetic stability. Virus Res. 2010, 151, 74–87. [Google Scholar] [CrossRef]
- He, Y.; Zhang, Q.; Wang, J.; Zhou, M.; Fu, M.; Xu, X. Full-length genome sequence analysis of enzootic nasal tumor virus isolated from goats in China. Virol. J. 2017, 14, 141. [Google Scholar] [CrossRef]
- York, D.F.; Vigne, R.; Verwoerd, D.W.; Querat, G. Nucleotide sequence of the jaagsiekte retrovirus, an exogenous and endogenous type D and B retrovirus of sheep and goats. J. Virol. 1992, 66, 4930–4939. [Google Scholar] [CrossRef] [PubMed]
- Ortin, A.; Cousens, C.; Minguijon, E.; Pascual, Z.; Villarreal, M.P.; Sharp, J.M.; Heras, M.L. Characterization of enzootic nasal tumour virus of goats: Complete sequence and tissue distribution. J. Gen. Virol. 2003, 84, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Vitellozzi, G.; Mughetti, L.; Palmarini, M.; Mandara, M.T.; Mechelli, L.; Sharp, J.M.; Manocchio, I. Enzootic intranasal tumour of goats in Italy. Zentralbl. Veterinarmed. B 1993, 40, 459–468. [Google Scholar] [CrossRef]
- de Cecco, B.S.; Lorenzett, M.P.; Henker, L.C.; Weber, M.N.; Mosena, A.; Baumbach, L.; Canal, C.W.; Driemeier, D.; Pavarini, S.P.; Sonne, L. Detection of enzootic nasal tumor virus (ENTV) in a sheep flock in southern Brazil. Trop. Anim. Health PRO 2019, 51, 2095–2098. [Google Scholar] [CrossRef] [PubMed]
- De Las, H.M.; Borobia, M.; Ortin, A. Neoplasia-Associated Wasting Diseases with Economic Relevance in the Sheep Industry. Animals 2021, 11, 381. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Q.; Liu, W.; Chen, Y.; Jiang, J.; Wu, D.; Lin, Y.; Fang, Y.; Zheng, X.; Huang, S.; et al. Epidemiological Investigation on Goat Endemic Intranasal Tumor Virus Type 2 in Fuqing City of Fujian Province. China Anim. Health Insp. 2023, 40, 25–30. (In Chinese) [Google Scholar] [CrossRef]
- Ye, C.; Huang, Q.; Chen, T.; Jiang, J.; Hou, F.; Xu, D.; Peng, Y.; Fang, R.; Chen, J. First detection and genotypic analysis of goat enzootic nasal tumor virus 2 in Chongqing, China. Arch. Virol. 2019, 164, 1647–1650. [Google Scholar] [CrossRef]
- Wang, B.; Ye, N.; Cao, S.J.; Wen, X.T.; Huang, Y.; Yan, Q.G. Identification of novel and differentially expressed MicroRNAs in goat enzootic nasal adenocarcinoma. BMC Genom. 2016, 17, 896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Meng, J.; Li, Z.; He, Y.; Wang, D.; Li, N.; Sun, J.; Bai, F.; Wang, J. Detection and Sequence Analysis of Goat Enzootic Nasal Tumor Virus in Jiangcheng County, Yunnan Province. China Anim. Health Insp. 2022, 39, 48–52. (In Chinese) [Google Scholar] [CrossRef]
- Lei, H.; Su, M.; Ning, L.; Kang, Y.; Chen, K.; Zeng, Y. Investigation and diagnosis of Enzootic Nasal Tumor in Goats. Prog. Vet. Med. 2006, 2, 112–114. (In Chinese) [Google Scholar] [CrossRef]
- He, R.; Du, Y.; Gan, L.; Mohsin, M.A.; He, B.X. Development of a SYBR Green-based real-time quantitative polymerase chain reaction assay to detect enzootic nasal tumor virus in goats. Can. J. Vet. Res. 2021, 85, 145–150. [Google Scholar] [PubMed]
- Hou, H.; Zhu, D.; Zhang, D.; Hu, X.; Zhao, R.; Dai, Y. Cloning and Analysis of gag Gene of Enzootic Nasal Tumor Virus in Goats. China Herbiv. Sci. 2018, 38, 53–55. (In Chinese) [Google Scholar] [CrossRef]
- Zhai, S.L.; Lv, D.H.; Xu, Z.H.; Yu, J.S.; Wen, X.H.; Zhang, H.; Chen, Q.L.; Jia, C.L.; Zhou, X.R.; Zhai, Q.; et al. A Novel Enzootic Nasal Tumor Virus Circulating in Goats from Southern China. Viruses 2019, 11, 956. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, S.; Bonhoeffer, S. Contribution of recombination to the evolutionary history of HIV. Curr. Opin. Hiv. Aids 2015, 10, 84–89. [Google Scholar] [CrossRef]
- Burke, D.S. Recombination in HIV: An important viral evolutionary strategy. Emerg. Infect. Dis. 1997, 3, 253–259. [Google Scholar] [CrossRef]
- Zhang, M.; Foley, B.; Schultz, A.K.; Macke, J.P.; Bulla, I.; Stanke, M.; Morgenstern, B.; Korber, B.; Leitner, T. The role of recombination in the emergence of a complex and dynamic HIV epidemic. Retrovirology 2010, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; van der Meer, F.; Checkley, S.; Joseph, T.; King, R.; Ravi, M.; Peters, D.; Fonseca, K.; Gagnon, C.A.; Provost, C.; et al. Analysis of Whole-Genome Sequences of Infectious laryngotracheitis Virus Isolates from Poultry Flocks in Canada: Evidence of Recombination. Viruses 2020, 12, 1302. [Google Scholar] [CrossRef]
- Yan, T.; Guo, L.; Jiang, X.; Wang, H.; Yao, Z.; Zhu, S.; Diao, Y.; Tang, Y. Discovery of a novel recombinant avian orthoreovirus in China. Vet. Microbiol. 2021, 260, 109094. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Ye, C.; Chen, T.; Jiang, J.; Peng, Y.; Chen, J.; Fang, R. EvaGreen-based real-time PCR assay for sensitive detection of enzootic nasal tumor virus 2. Mol. Cell. Probes 2019, 44, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.; Rheinstein, P.H. Mouse mammary tumor viral env sequences are not present in the human genome but are present in breast tumors and normal breast tissues. Virus Res. 2019, 266, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Swanstrom, R.; Wills, J.W. Synthesis, Assembly, and Processing of Viral Proteins-Retroviruses-NCBI Bookshelf; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Gifford, R.; Tristem, M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 2003, 26, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.D. Identification of Hyal2 as the cell-surface receptor for jaagsiekte sheep retrovirus and ovine nasal adenocarcinoma virus. Curr. Top. Microbiol. 2003, 275, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Monot, M.; Archer, F.; Gomes, M.; Mornex, J.F.; Leroux, C. Advances in the study of transmissible respiratory tumours in small ruminants. Vet. Microbiol. 2015, 181, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.K.; Duh, F.M.; Vigdorovich, V.; Danilkovitch-Miagkova, A.; Lerman, M.I.; Miller, A.D. Candidate tumor suppressor HYAL2 is a glycosylphosphatidylinositol (GPI)-anchored cell-surface receptor for jaagsiekte sheep retrovirus, the envelope protein of which mediates oncogenic transformation. Proc. Natl. Acad. Sci. USA 2001, 98, 4443–4448. [Google Scholar] [CrossRef]
- Dirks, C.; Duh, F.M.; Rai, S.K.; Lerman, M.I.; Miller, A.D. Mechanism of cell entry and transformation by enzootic nasal tumor virus. J. Virol. 2002, 76, 2141–2149. [Google Scholar] [CrossRef]
- Rosenberg, N.; Jolicoeur, P. Retroviral Pathogenesis-Retroviruses-NCBI Bookshelf; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Maeda, N.; Inoshima, Y.; De Las, H.M.; Maenaka, K. Enzootic nasal tumor virus type 2 envelope of goats acts as a retroviral oncogene in cell transformation. Virus Genes 2021, 57, 50–59. [Google Scholar] [CrossRef]
- White, K.A.; Enjuanes, L.; Berkhout, B. RNA virus replication, transcription and recombination. RNA Biol. 2011, 8, 182–183. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) | Position in CQ2 (OR682176) (bp) | Product Length (bp) |
|---|---|---|---|
| ENTV-2-1-F | ACAAGGCATCAGCCATTTTGGT | ||
| ENTV-2-1-R | AACCTCACCAAGTCGCTGC | 1–1002 | 1002 |
| ENTV-2-2-F | GGGGAACAAATTCGAACTCAT TATACT | ||
| ENTV-2-2-R | CCCATCTCAGGTGCTAGTATT GTAT | 433–2110 | 1678 |
| ENTV-2-3-F | GCAACCCTGCAGGTTCCAA | ||
| ENTV-2-3-R | TGTGGGTGCTCGAGGGGTA | 1999–3865 | 1867 |
| ENTV-2-4-F | ATTGCTGATGAAAAAATT | ||
| ENTV-2-4-R | TATTGAGGGAGAACAAA | 3497–4740 | 1244 |
| ENTV-2-5-F | GCAAATGATTGAAACTGT | ||
| ENTV-2-5-R | ACTATTGCCATGACCAAA | 4393–6152 | 1760 |
| ENTV-2-6-F | CTCCTTGGACTTTATGTCGAGC | ||
| ENTV-2-6-R | TGTTTTATTGTGTCATAGTATA | 5944–7468 | 1525 |
| Gene | Loci in the Genome of CQ2 | CQ2 | CHN4 | AH2 | MC |
|---|---|---|---|---|---|
| pro | 2273 | C a (K) b | C (K) | T (R) | T (R) |
| 2369 | A (E) | A (E) | T (D) | T (D) | |
| 2503–2504 | AA (E) | AA (E) | GT (G) | GT (G) | |
| 2608–2609 | GG (R) | GG (R) | CA (T) | CA (T) | |
| 2738 | A (E) | A (E) | T (D) | T (D) | |
| 2799 | A (I) | A (I) | G (V) | G (V) | |
| 2829–2831 | AGT (S) | AGT (S) | GAG (E) | GAG (E) | |
| pol | 3176 | A (I) | A (I) | G (V) | G (V) |
| 3294 | G (R) | G (R) | A (K) | A (K) | |
| 3356 | T (L) | T (L) | A (I) | A (I) | |
| 3461 | A (I) | A (I) | G (V) | G (V) | |
| 3494 | G (V) | G (V) | A (I) | A (I) | |
| 3555 | A (Y) | A (Y) | T (F) | T (F) | |
| 3564–3565 | TT (V) | TT (V) | CC (A) | CC (A) | |
| 3746 | T (S) | T (S) | C (P) | C (P) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Niu, J.; Liu, Y.; Dai, Y.; Ni, H.; Wang, J.; Fang, R.; Ye, C. Genomic Sequencing and Analysis of Enzootic Nasal Tumor Virus Type 2 Provides Evidence for Recombination within the Prevalent Chinese Strains. Vet. Sci. 2024, 11, 248. https://doi.org/10.3390/vetsci11060248
Li Y, Niu J, Liu Y, Dai Y, Ni H, Wang J, Fang R, Ye C. Genomic Sequencing and Analysis of Enzootic Nasal Tumor Virus Type 2 Provides Evidence for Recombination within the Prevalent Chinese Strains. Veterinary Sciences. 2024; 11(6):248. https://doi.org/10.3390/vetsci11060248
Chicago/Turabian StyleLi, Yixuan, Jingyi Niu, Yiyu Liu, Yu Dai, Hongbo Ni, Jinliang Wang, Rendong Fang, and Chao Ye. 2024. "Genomic Sequencing and Analysis of Enzootic Nasal Tumor Virus Type 2 Provides Evidence for Recombination within the Prevalent Chinese Strains" Veterinary Sciences 11, no. 6: 248. https://doi.org/10.3390/vetsci11060248
APA StyleLi, Y., Niu, J., Liu, Y., Dai, Y., Ni, H., Wang, J., Fang, R., & Ye, C. (2024). Genomic Sequencing and Analysis of Enzootic Nasal Tumor Virus Type 2 Provides Evidence for Recombination within the Prevalent Chinese Strains. Veterinary Sciences, 11(6), 248. https://doi.org/10.3390/vetsci11060248