
Comparative Analysis of the Ovary Transcriptome among Wanyue Black and Yorkshire Gilts Using RNA-Seq

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Serum Hormone Detection
2.3. RNA Extraction, Library Construction, and Sequencing
2.4. Analysis of RNA-Seq Data
2.5. Bioinformatics Analysis
2.6. Quantitative Real-Time RT-PCR (qRT-PCR)
2.7. Statistical Analysis
2.8. Statement of Use of AI or AI-Assisted Technologies
3. Results
3.1. Levels of Reproductive Hormones
3.2. The Summary of RNA Sequencing Data
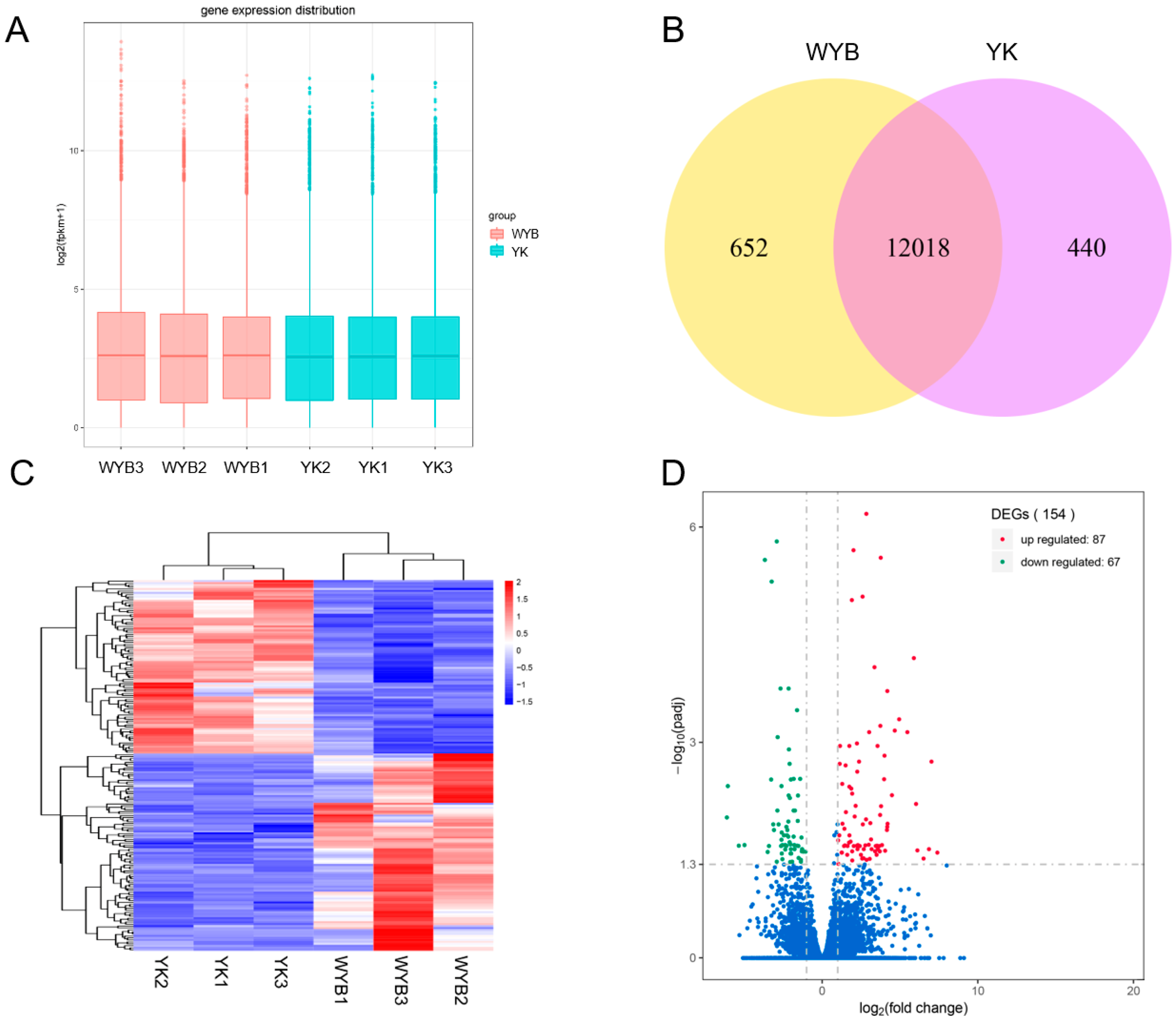
3.3. Identification and Analysis of DEGs
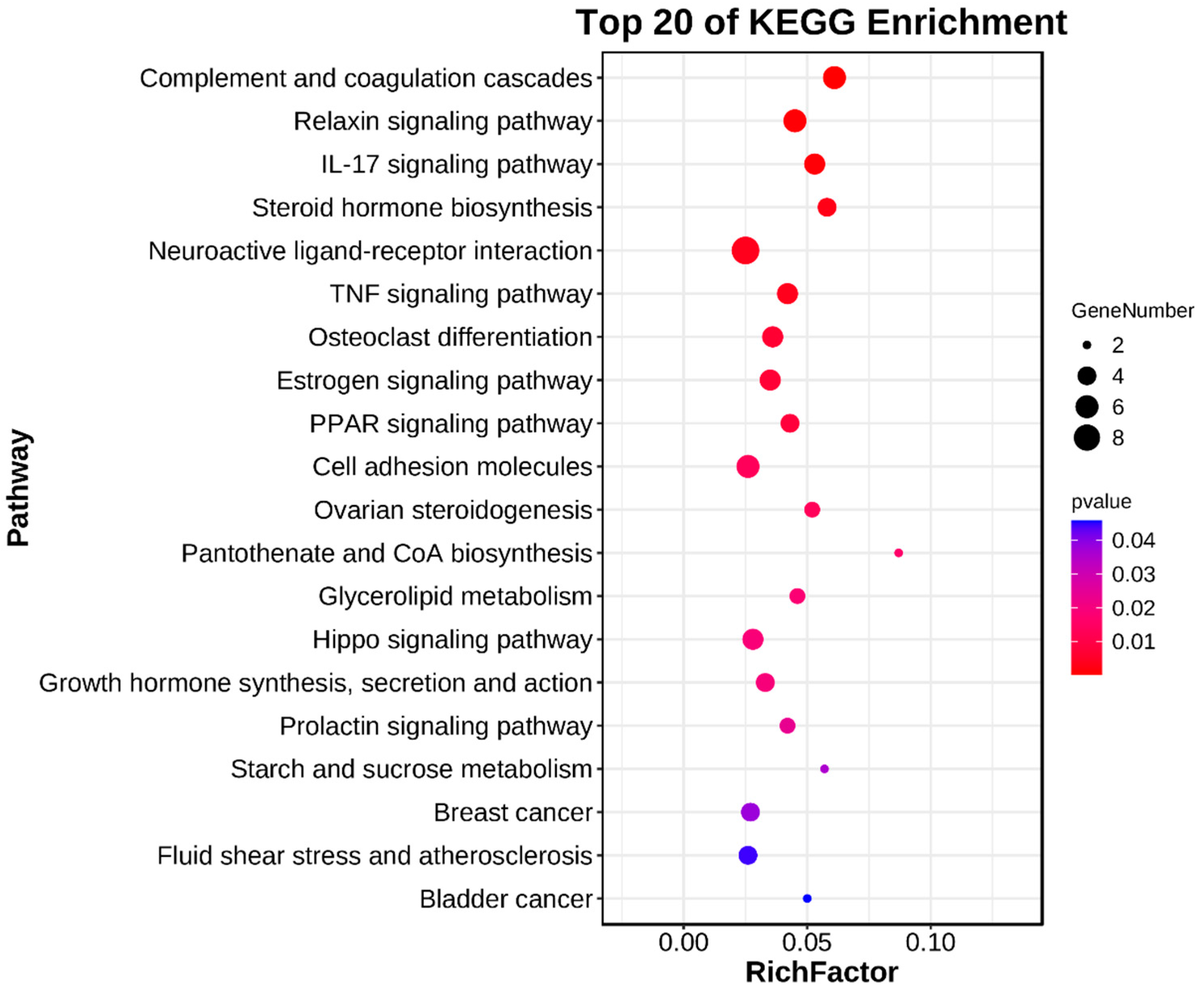
3.4. Enrichment Analyses of DEGs
3.5. PPI Network Analysis
3.6. RNA-Seq Data Validation by RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nonneman, D.J.; Schneider, J.F.; Lents, C.A.; Wiedmann, R.T.; Vallet, J.L.; Rohrer, G.A. Genome-wide association and identification of candidate genes for age at puberty in swine. BMC Genet. 2016, 17, 50. [Google Scholar] [CrossRef]
- Li, Q.; Yuan, X.; Chen, Z.; Zhang, A.; Zhang, Z.; Zhang, H.; Li, J. Heritability estimates and effect on lifetime reproductive performance of age at puberty in sows. Anim. Reprod. Sci. 2018, 195, 207–215. [Google Scholar] [CrossRef]
- Perry, G.A. Factors affecting puberty in replacement beef heifers. Theriogenology 2016, 86, 373–378. [Google Scholar] [CrossRef]
- Blakemore, S.J.; Burnett, S.; Dahl, R.E. The role of puberty in the developing adolescent brain. Hum. Brain Mapp. 2010, 31, 926–933. [Google Scholar] [CrossRef]
- Hu, K.; Sun, W.; Li, Y.; Zhang, B.; Zhang, M.; Guo, C.; Wang, X. Study on the Mechanism of Sarsasapogenin in Treating Precocious Puberty by Regulating the HPG Axis. Evid.-Based Complement. Altern. Med. eCAM 2020, 2020, 1978043. [Google Scholar] [CrossRef]
- Młodawska, W.; Grzesiak, M.; Kochan, J.; Nowak, A. Intrafollicular level of steroid hormones and the expression of androgen receptor in the equine ovary at puberty. Theriogenology 2018, 121, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ernst, E.; Kjærsgaard, M.; Birkebæk, N.H.; Clausen, N.; Andersen, C.Y. Case report: Stimulation of puberty in a girl with chemo- and radiation therapy induced ovarian failure by transplantation of a small part of her frozen/thawed ovarian tissue. Eur. J. Cancer 2013, 49, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Baudry, M.; Bi, X.; Aguirre, C. Progesterone-estrogen interactions in synaptic plasticity and neuroprotection. Neuroscience 2013, 239, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Pan, X.; Li, N.; Gong, W.; Chen, Y.; Yuan, X. Identification of Circular RNAs in Hypothalamus of Gilts during the Onset of Puberty. Genes 2021, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Mark-Kappeler, C.J.; Hoyer, P.B.; Pepling, M.E. The steroid hormone environment during primordial follicle formation in perinatal mouse ovaries. Biol. Reprod. 2014, 91, 68. [Google Scholar] [CrossRef] [PubMed]
- Prunier, A.; Chopineau, M.; Mounier, A.M.; Mormède, P. Patterns of plasma LH, FSH, oestradiol and corticosteroids from birth to the first oestrous cycle in Meishan gilts. J. Reprod. Fertil. 1993, 98, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Sirotkin, A.V.; Pivko, J. Hormonal indexes as predictors of porcine reproductive traits. Anim. Sci. J. 2023, 94, e13879. [Google Scholar] [CrossRef] [PubMed]
- Seminara, S.B.; Messager, S.; Chatzidaki, E.E.; Thresher, R.R.; Acierno, J.S., Jr.; Shagoury, J.K.; Colledge, W.H. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 2003, 349, 1614–1627. [Google Scholar] [CrossRef]
- Kaiser, U.B.; Kuohung, W. KiSS-1 and GPR54 as new players in gonadotropin regulation and puberty. Endocrine 2005, 26, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Long, J.R.; Xu, H.; Zhao, L.J.; Liu, P.Y.; Shen, H.; Liu, Y.J.; Deng, H.W. The oestrogen receptor alpha gene is linked and/or associated with age of menarche in different ethnic groups. J. Med. Genet. 2005, 42, 796–800. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xita, N.; Tsatsoulis, A.; Stavrou, I.; Georgiou, I. Association of SHBG gene polymorphism with menarche. Mol. Hum. Reprod. 2005, 11, 459–462. [Google Scholar] [CrossRef]
- Yang, Y.; Adeola, A.C.; Xie, H.B.; Zhang, Y.P. Genomic and transcriptomic analyses reveal selection of genes for puberty in Bama Xiang pigs. Zool. Res. 2018, 39, 424–430. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, X.; Pei, Y.; Wang, H.; He, K.; Zhao, A. Identification of Differentially Expressed Genes in Porcine Ovaries at Proestrus and Estrus Stages Using RNA-Seq Technique. BioMed Res. Int. 2018, 2018, 9150723. [Google Scholar] [CrossRef]
- Chu, Q.; Zhou, B.; Xu, F.; Chen, R.; Shen, C.; Liang, T.; Schinckel, A.P. Genome-wide differential mRNA expression profiles in follicles of two breeds and at two stages of estrus cycle of gilts. Sci. Rep. 2017, 7, 5052. [Google Scholar] [CrossRef]
- Fan, B.; Wang, Z.G.; Li, Y.J.; Zhao, X.L.; Liu, B.; Zhao, S.H.; Li, K. Genetic variation analysis within and among Chinese indigenous swine populations using microsatellite markers. Anim. Genet. 2002, 33, 422–427. [Google Scholar] [CrossRef]
- Hunter, M.G.; Biggs, C.; Foxcroft, G.R.; McNeilly, A.S.; Tilton, J.E. Comparisons of endocrinology and behavioural events during the periovulatory period in Meishan and large-white hybrid gilts. J. Reprod. Fertil. 1993, 97, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Bazer, F.W.; Thatcher, W.W.; Martinat-Botte, F.; Terqui, M. Sexual maturation and morphological development of the reproductive tract in large white and prolific Chinese Meishan pigs. J. Reprod. Fertil. 1988, 83, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Pan, X.; Kong, Y.; Jiang, Y.; Zhong, Y.; Zhang, H.; Zhang, Z.; Yuan, X.; Li, J. Pituitary-Derived Circular RNAs Expression and Regulatory Network Prediction during the Onset of Puberty in Landrace × Yorkshire Crossbred Pigs. Front. Genet. 2020, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ye, J.; Qin, P.; Yan, X.; Gong, X.; Li, X.; Liu, Y.; Li, Y.; Yu, T.; Zhang, Y.; et al. Analysis of serum reproductive hormones and ovarian genes in pubertal female goats. J. Ovarian Res. 2023, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, E.K.; Belsham, D.D. Hypothalamic reproductive neurons communicate through signal transduction to control reproduction. Mol. Cell. Endocrinol. 2020, 518, 110971. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.L., Jr.; Garza, F., Jr.; St George, R.L.; Rabb, M.H.; Barry, B.E.; French, D.D. Relationships among LH, FSH and prolactin secretion, storage and response to secretagogue and hypothalamic GnRH content in ovariectomized pony mares administered testosterone, dihydrotestosterone, estradiol, progesterone, dexamethasone or follicular fluid. Domest. Anim. Endocrinol. 1991, 8, 189–199. [Google Scholar] [CrossRef]
- Li, W.; Zeng, W.; Jin, X.; Xu, H.; Fang, X.; Ma, Z.; Ma, L. High-Altitude Stress Orchestrates mRNA Expression and Alternative Splicing of Ovarian Follicle Development Genes in Tibetan Sheep. Animals 2022, 12, 2812. [Google Scholar] [CrossRef]
- Choi, Y.; Jeon, H.; Akin, J.W.; Curry, T.E.; Jo, M. The FOS/AP-1 Regulates Metabolic Changes and Cholesterol Synthesis in Human Periovulatory Granulosa Cells. Endocrinology 2021, 162, bqab127. [Google Scholar] [CrossRef]
- Choi, Y.; Rosewell, K.L.; Brännström, M.; Akin, J.W.; Curry, T.E., Jr.; Jo, M. FOS, a Critical Downstream Mediator of PGR and EGF Signaling Necessary for Ovulatory Prostaglandins in the Human Ovary. J. Clin. Endocrinol. Metab. 2018, 103, 4241–4252. [Google Scholar] [CrossRef]
- Lu, W.; Zhou, Y.; Liu, Y.; Zhang, H.; Yuan, Z.; Han, Y.; Weng, Q. Seasonal changes of vitamin D(3) and ovarian steroidogenesis in the wild ground squirrels (Citellus dauricus Brandt). J. Steroid Biochem. Mol. Biol. 2023, 234, 106385. [Google Scholar] [CrossRef]
- Liu, W.; Du, C.; Nan, L.; Li, C.; Wang, H.; Fan, Y.; Zhang, S. Influence of Estrus on Dairy Cow Milk Exosomal miRNAs and Their Role in Hormone Secretion by Granulosa Cells. Int. J. Mol. Sci. 2023, 24, 9608. [Google Scholar] [CrossRef]
- Yu, D.; Jiang, X.; Ge, W.; Qiao, B.; Zhang, D.; Liu, H.; Kuang, H. Gestational exposure to acrylamide suppresses luteal endocrine function through dysregulation of ovarian angiogenesis, oxidative stress and apoptosis in mice. Food Chem. Toxicol. 2022, 159, 112766. [Google Scholar] [CrossRef]
- Song, Y.; Hu, R.; Li, F.; Huang, Y.; Liu, Z.; Geng, Y.; Zhang, M. In view of ovarian steroidogenesis and luteal construction to explore the effects of Bushen Huoxue recipe in mice of ovarian hyperstimulation. J. Ethnopharmacol. 2024, 318, 116913. [Google Scholar] [CrossRef]
- Trocka, D.; Satie, A.P.; Ravel, C. The human zona pellucida: New genetic findings. Med. Sci. 2023, 39, 522–529. [Google Scholar] [CrossRef]
- Sun, L.; Tong, K.; Liu, W.; Tian, Y.; Liu, D.; Huang, G.; Li, J. Novel variants in ZP1, ZP2 and ZP3 associated with empty follicle syndrome and abnormal zona pellucida. Reprod. Biomed. Online 2023, 46, 847–855. [Google Scholar] [CrossRef]
- Hao, X.; Yuan, F.; Cui, Y.; Zhang, M. Oocyte-secreted factor TGFB2 enables mouse cumulus cell expansion in vitro. Mol. Reprod. Dev. 2022, 89, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Zhang, X.; Zhao, C.; Geng, R.; Wu, K.; Yuan, M.; Ai, N.; Ge, W. Rescue of bmp15 deficiency in zebrafish by mutation of inha reveals mechanisms of BMP15 regulation of folliculogenesis. PLoS Genet. 2023, 19, e1010954. [Google Scholar] [CrossRef]
- Kristensen, S.G.; Kumar, A.; Mamsen, L.S.; Kalra, B.; Pors, S.E.; Bøtkjær, J.A.; Macklon, K.T.; Fedder, J.; Ernst, E.; Hardy, K.; et al. Intrafollicular Concentrations of the Oocyte-secreted Factors GDF9 and BMP15 Vary Inversely in Polycystic Ovaries. J. Clin. Endocrinol. Metab. 2022, 107, e3374–e3383. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Nakamura, T.; Bayasula Nakanishi, N.; Kasahara, Y.; Nagai, T.; Murase, T.; Osuka, S.; Goto, M.; Iwase, A.; Kikkawa, F. Molecular mechanism of FSHR expression induced by BMP15 in human granulosa cells. J. Assist. Reprod. Genet. 2019, 36, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yi, H. BMP15 regulates FSHR through TGF-β receptor II and SMAD4 signaling in prepubertal ovary of Rongchang pigs. Res. Vet. Sci. 2022, 143, 66–73. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| β-actin | GGACTTCGAGCAGGAGATGG | AGGAAGGAGGGCTGGAAGAG |
| RLN2 | CTGAGCCAACTTCCCAGAGA | CCACCCACAGACGGACTAAT |
| ZP2 | CACTGGAGTGACTCGCTACT | GTCACGTGTGTGGCATTACA |
| TIMP1 | GTTTCCCTGCACATCCATCC | TCTGGAAGCCCTTGTCAGAG |
| CLU | AAATCACTGCTCAGCTCCCT | ATCGTCTCATTGCACAAGCC |
| KITLG | CCTCGTGGAATGCATGGAAG | TGCCACCATCTCCAAATCCT |
| UGT1A6 | CCCAACCCACTGTCCTATGT | CAGAGGAAGCCCTCTGACAA |
| AKR1D1 | CCTCTCTTGCTTGGTTGCTC | GTTAGGCTGAGGGACTTGGT |
| HSD3B1 | TTCAATCGCCACTTCGTGAC | CCAGGTCAGTGAGTCTTGGT |
| LOC100515166 | AGATCGAGAGCCTGAACGAG | GCCTCATACTGCTCCCTCAT |
| SHC2 | CATCCTGGGCAAGAGCAATC | GGCCATCAATGGAGATGCTG |
| FOS | AGCTGACTGACACACTCCAA | ATCAAGGGAAGCCACAGACA |
| MMP9 | TTCTTCTCTGGACGCCAAGT | TTCACGTCGAACCTCCAGAA |
| BMP15 | TTCACTTGGACTCTGGGCAT | ATTTGCAACACAGCCCAGTT |
| CYP19A1 | TATCCTTGCACCCGGATGAG | AGCTAGCAAAGATGGGTGGT |
| GO Term | P.adj | Count | Genes |
|---|---|---|---|
| Reproduction | 0.0081 | 22 | SERPINA5; TDRD9; SALL1; MMP9; ZP3; FKBP6; ZP2; AREG; RXFP2; ZP4; CLDN11; TIMP1; BMP15; ACSL4; FOSL1; JUNB; DND1; WNT2; CEBPB; KITLG; SOCS3; VGF |
| Reproductive system development | 0.0139 | 11 | SALL1; ZP3; RXFP2; BMP15; FOSL1; JUNB; WNT2; CEBPB; KITLG; SOCS3; VGF |
| Ovarian follicle development | 0.0397 | 4 | ZP3; BMP15; CEBPB; VGF |
| KEGG Pathway | p | Count | Genes |
|---|---|---|---|
| Steroid hormone biosynthesis | 0.0028 | 4 | HSD3B1, AKR1D1, CYP11A1, UGT1A6 |
| Estrogen signaling pathway | 0.0071 | 5 | FOS, KRT14, MMP9, SHC2, LOC100515166 |
| Ovarian steroidogenesis | 0.0133 | 3 | HSD3B1, CYP11A1, BMP15 |
| Growth hormone synthesis, Secretion, and action | 0.0194 | 4 | FOS, JUNB, SOCS3, SHC2 |
| Prolactin signaling pathway | 0.0236 | 3 | FOS, SOCS3, SHC2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Chen, S.; Liu, Y.; Xie, F.; Wen, H.; Zhao, S.; Zheng, X.; Ding, Y.; Yin, Z.; Zhang, X. Comparative Analysis of the Ovary Transcriptome among Wanyue Black and Yorkshire Gilts Using RNA-Seq. Vet. Sci. 2024, 11, 115. https://doi.org/10.3390/vetsci11030115
Zhang H, Chen S, Liu Y, Xie F, Wen H, Zhao S, Zheng X, Ding Y, Yin Z, Zhang X. Comparative Analysis of the Ovary Transcriptome among Wanyue Black and Yorkshire Gilts Using RNA-Seq. Veterinary Sciences. 2024; 11(3):115. https://doi.org/10.3390/vetsci11030115
Chicago/Turabian StyleZhang, Huibin, Shuo Chen, Yangguang Liu, Fan Xie, Haoyu Wen, Shiming Zhao, Xianrui Zheng, Yueyun Ding, Zongjun Yin, and Xiaodong Zhang. 2024. "Comparative Analysis of the Ovary Transcriptome among Wanyue Black and Yorkshire Gilts Using RNA-Seq" Veterinary Sciences 11, no. 3: 115. https://doi.org/10.3390/vetsci11030115
APA StyleZhang, H., Chen, S., Liu, Y., Xie, F., Wen, H., Zhao, S., Zheng, X., Ding, Y., Yin, Z., & Zhang, X. (2024). Comparative Analysis of the Ovary Transcriptome among Wanyue Black and Yorkshire Gilts Using RNA-Seq. Veterinary Sciences, 11(3), 115. https://doi.org/10.3390/vetsci11030115