
Metagenome-Based Analysis of the Microbial Community Structure and Drug-Resistance Characteristics of Livestock Feces in Anhui Province, China


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Library Construction, and Metagenome Sequencing
2.3. Assembly and Annotation of Metagenome Sequencing Data
2.4. Construction of Non-Redundant Gene Set
2.5. Individualized Analysis
3. Results
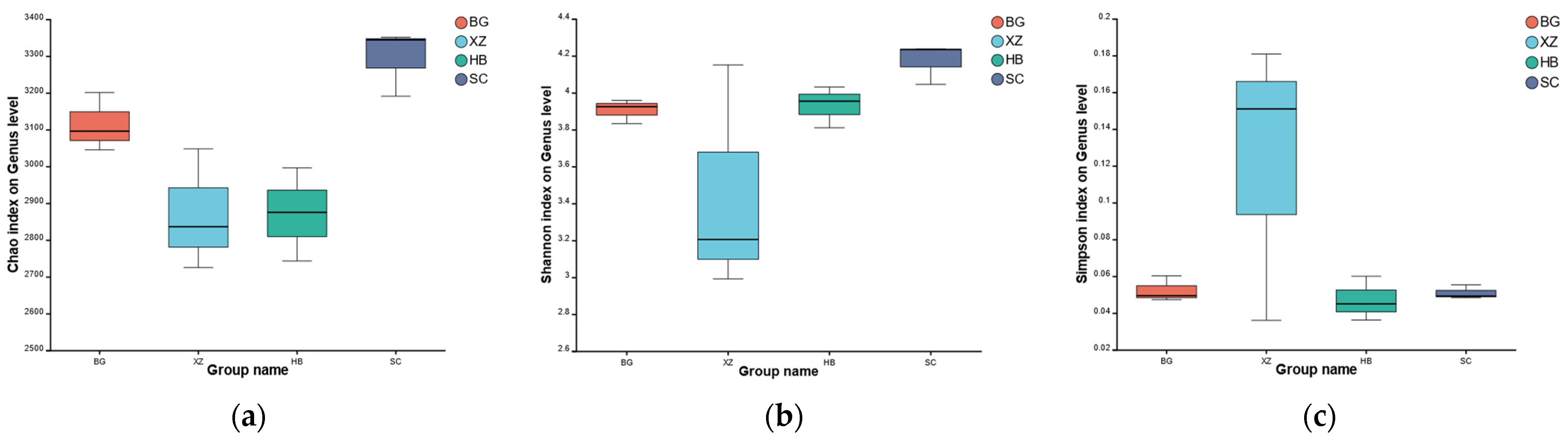
3.1. Metagenome Sequencing Results and Alpha Diversity
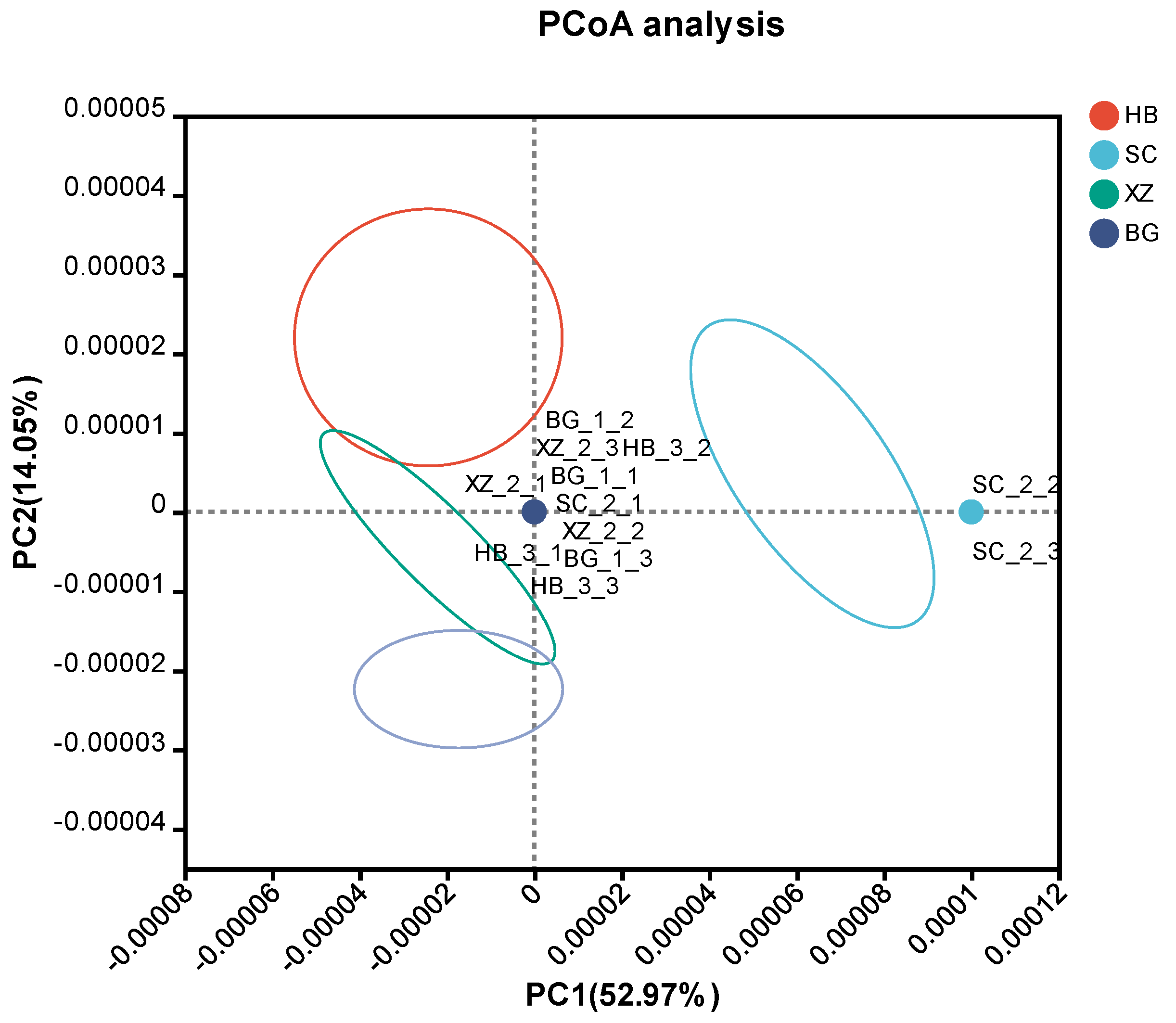
3.2. Beta Diversity
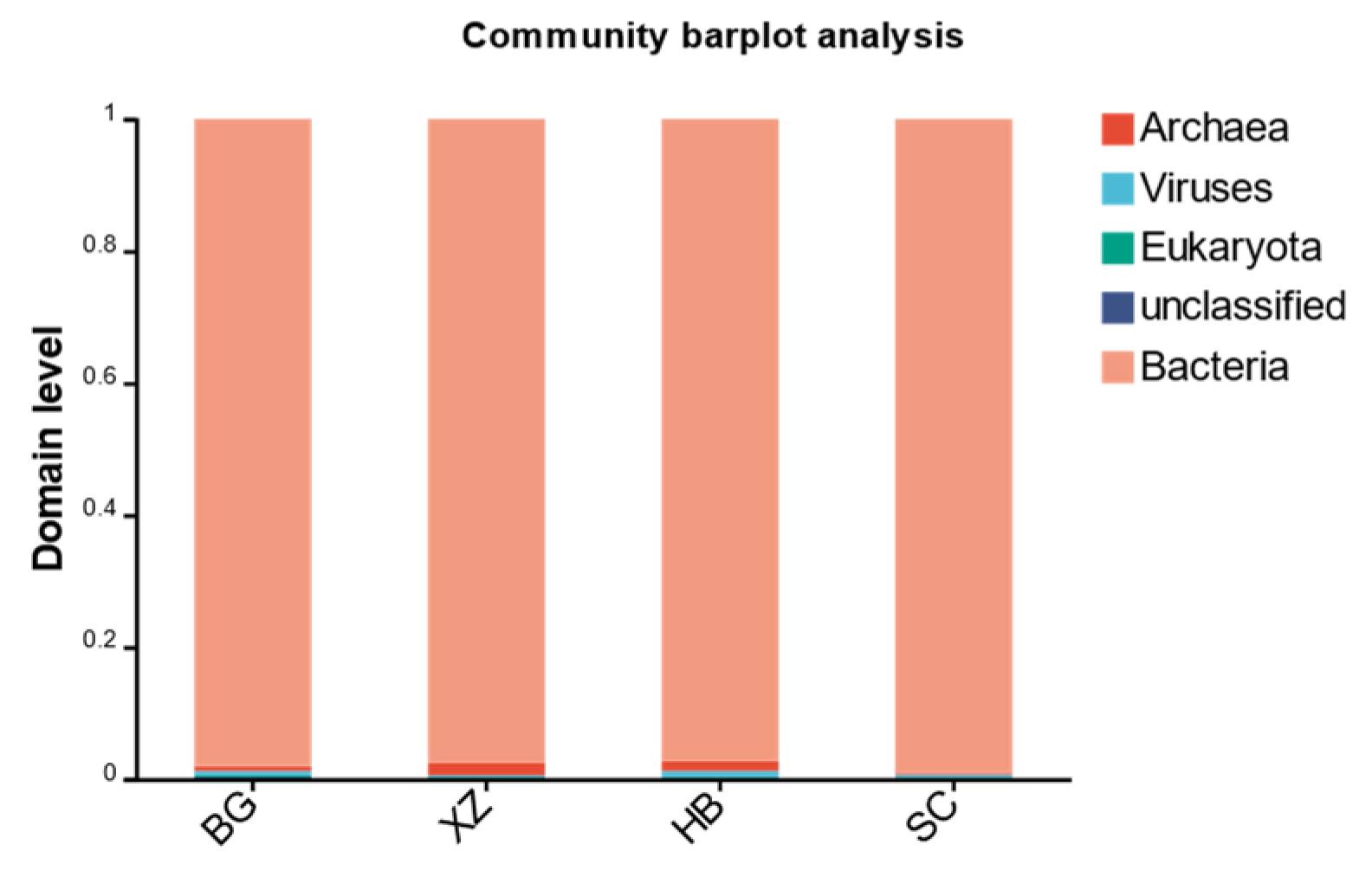
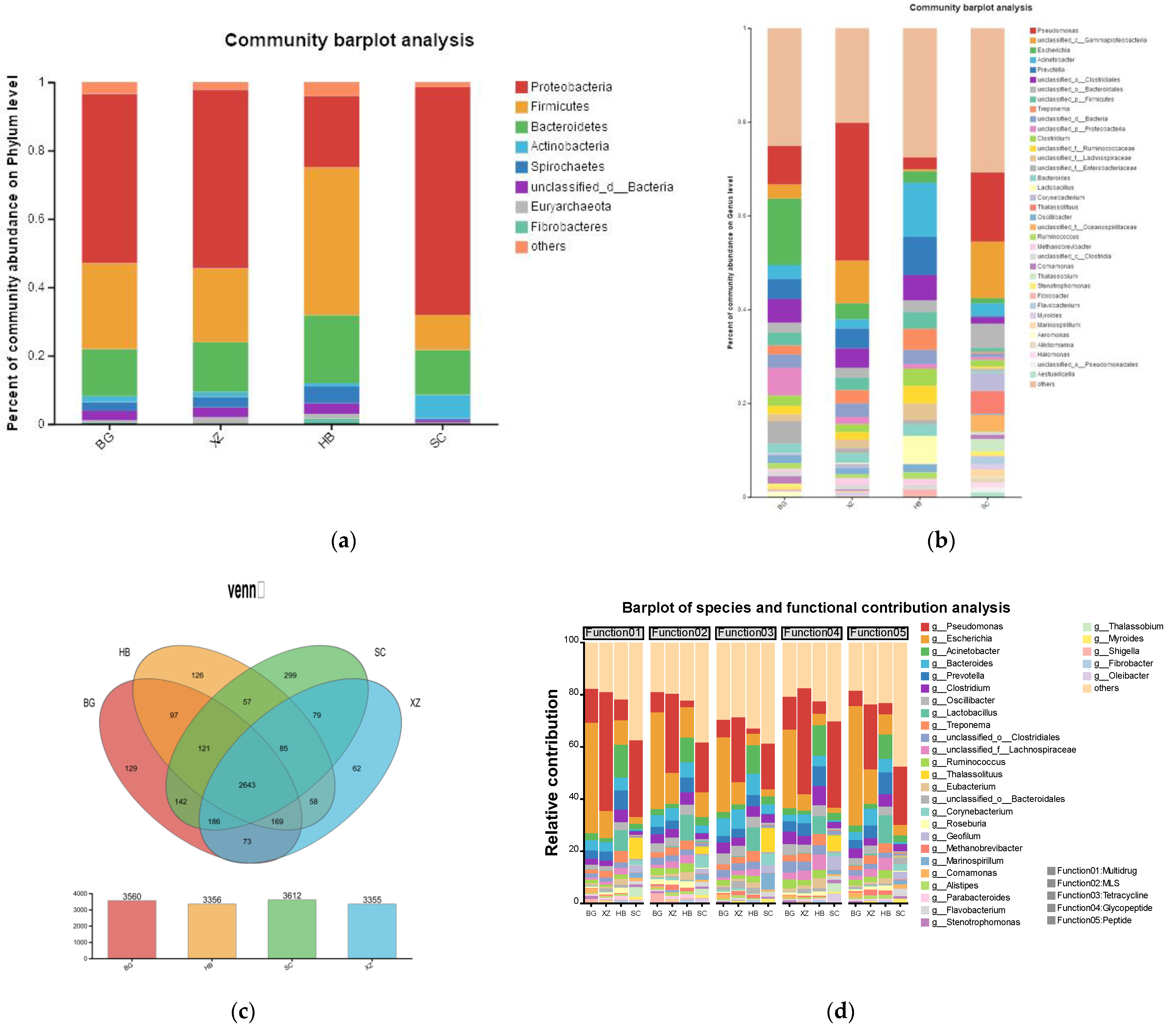
3.3. Microbial Community Structure
3.4. Bacterial Community Composition
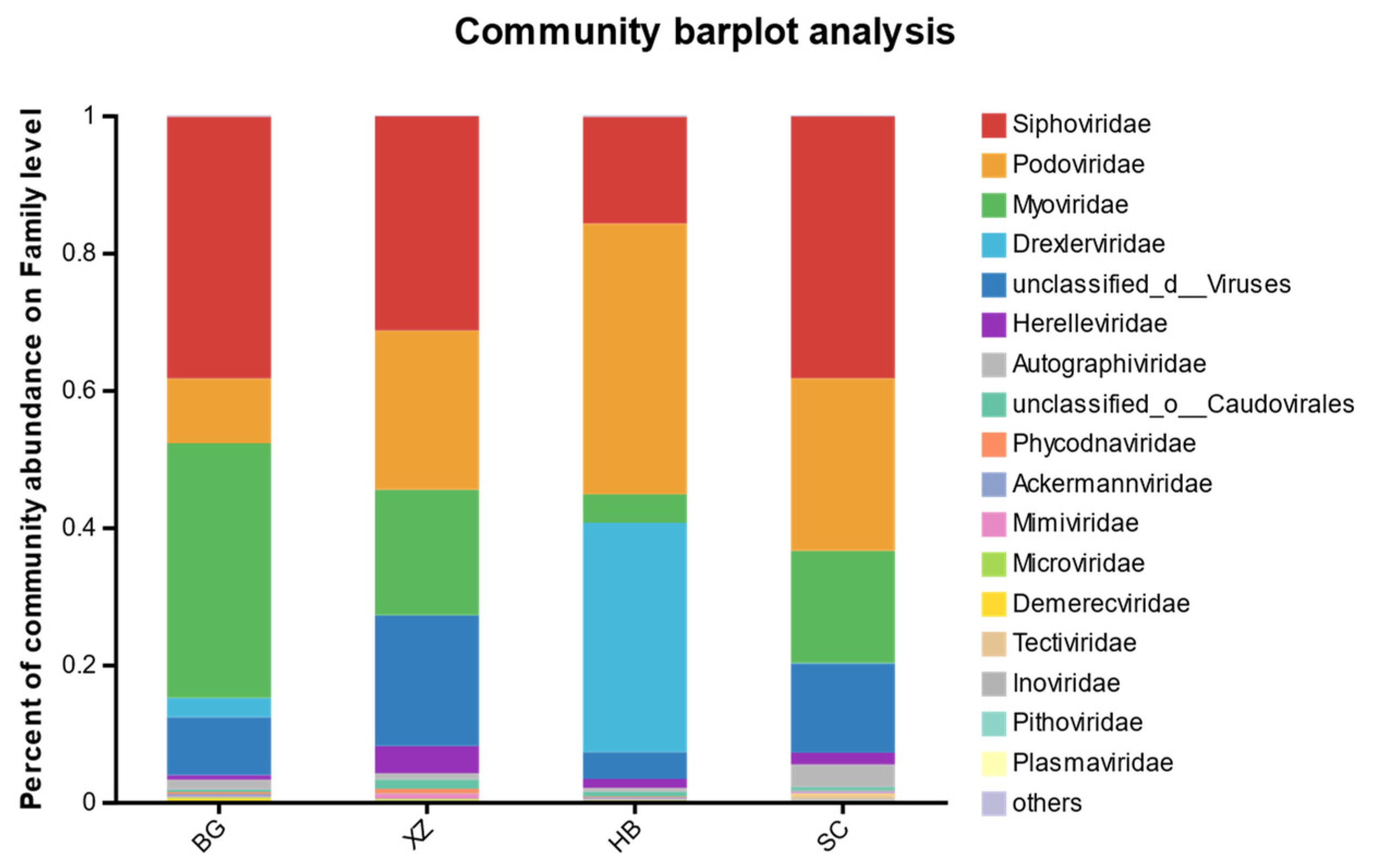
3.5. Viral Community Composition
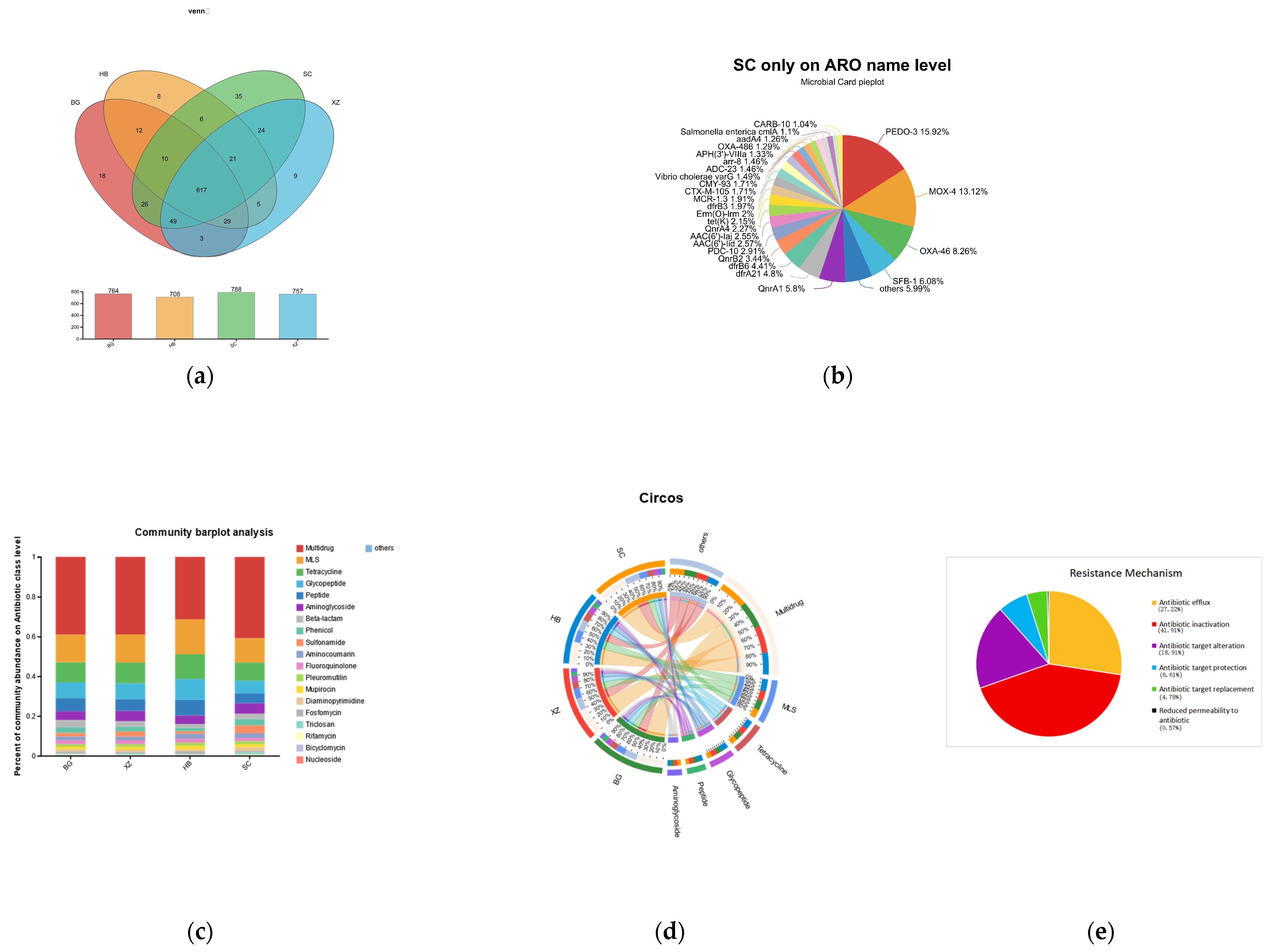
3.6. ARGs
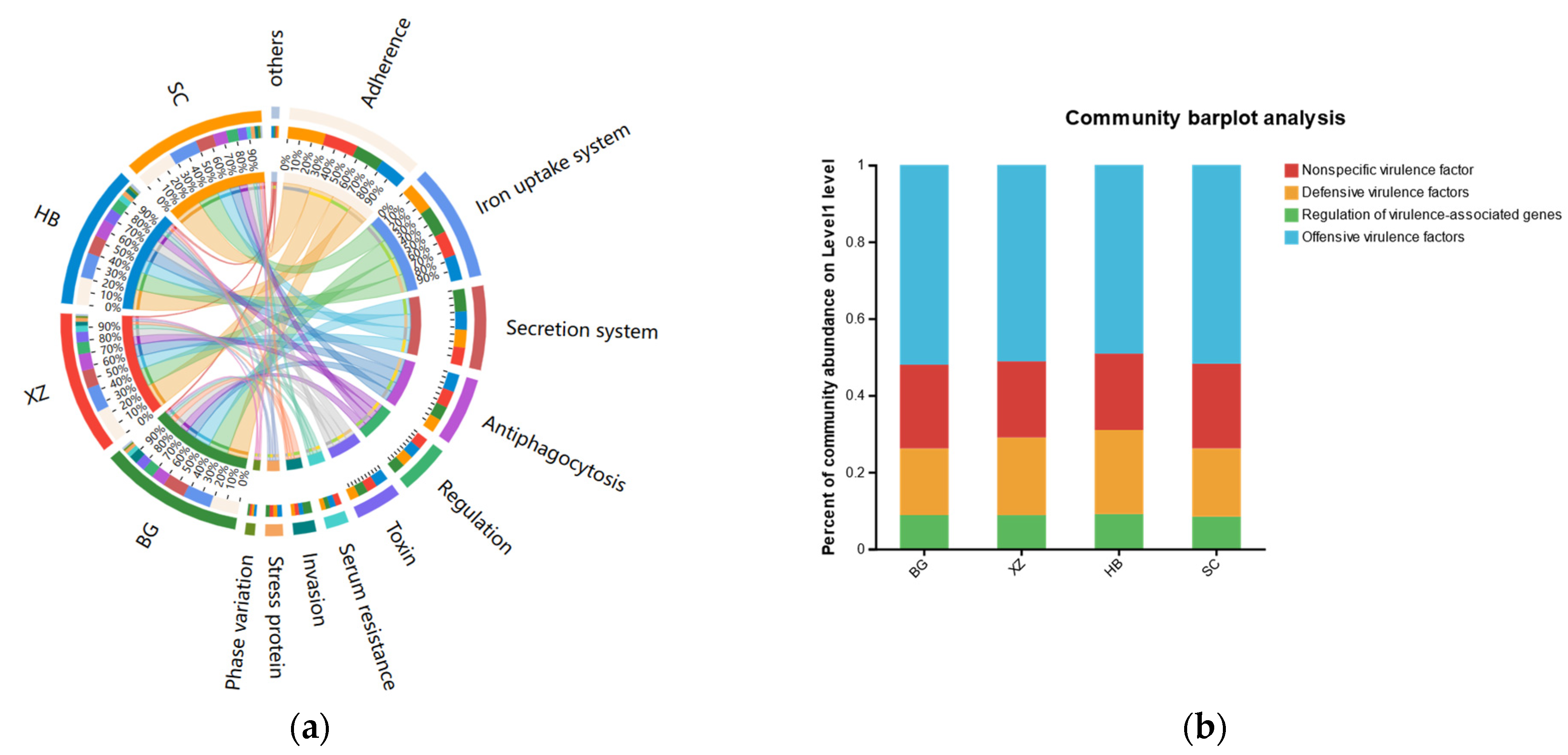
3.7. Microbial Virulence Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pan, M.; Xiao, L.; Lin, Y.; Cao, Y.; Li, X.; Ye, J.; Zhang, M.; Runmin, K. Study on the characteristics of drug resistance gene contamination in the feces of large-scale piofarms in Sichuan Province using metagenomic methoo. Heilongjiang Anim. Sci. Vet. Med. 2020, 18, 58–64. [Google Scholar]
- Xie, X.; Xu, S.; Wang, W.; Yang, J.; Zhao, Z.; Wang, M.; Huabao, Z. Research progress on microbial degradation of antibiotics in livestock and poultry breeding wastes. Acta Agric. Zhejiangensis 2023, 35, 1975–1992. [Google Scholar]
- Amos, G.; Zhang, L.; Hawkey, P.; Gaze, W.; Wellington, E. Functional metagenomic analysis reveals rivers are a reservoir for diverse antibiotic resistance genes. Vet. Microbiol. 2014, 171, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Shi, Y.; Li, W.; Liu, J.; Yaqi, C. Environmental behavior and impacts of antibiotics. Environ. Chem. 2013, 32, 1619–1633. [Google Scholar]
- Pan, X.; Qiang, Z.; Ben, W.; Chen, M. Residual veterinary antibiotics in swine manure from concentrated animal feeding operations in Shandong Province, China. Chemosphere 2011, 84, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.P.; Adeli, A.; McLaughlin, M.R. Microbial ecology, bacterial pathogens, and antibiotic resistant genes in swine manure wastewater as influenced by three swine management systems. Water Res. 2014, 57, 96–103. [Google Scholar] [CrossRef] [PubMed]
- He, L.Y.; Ying, G.G.; Liu, Y.S.; Su, H.C.; Chen, J.; Liu, S.S.; Zhao, J.L. Discharge of swine wastes risks water quality and food safety: Antibiotics and antibiotic resistance genes from swine sources to the receiving environments. Environ. Int. 2016, 92–93, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Gao, S.; Fan, S.; Ma, L.; Dengpan, B. Environmental Fate of Antibiotics and Antibiotic Resistance Genes in Animal Manure. Curr. Biotechnol. 2018, 9, 146–151. [Google Scholar]
- Kumar, R.R.; Park, B.J.; Cho, J.Y. Application and Environmental Risks of Livestock Manure. J. Korean Soc. Appl. Biol. Chem. 2013, 56, 497–503. [Google Scholar] [CrossRef]
- Biswas, D.; Hannon, S.J.; Townsend, H.G.; Potter, A.; Allan, B.J. Genes coding for virulence determinants of Campylobacter jejuni in human clinical and cattle isolates from Alberta, Canada, and their potential role in colonization of poultry. Int. Microbiol. 2011, 14, 25–32. [Google Scholar]
- Khoshbakht, R.; Tabatabaei, M.; Hosseinzadeh, S.; Shekarforoush, S.S.; Aski, H.S. Distribution of nine virulence-associated genes in Campylobacter jejuni and C. coli isolated from broiler feces in Shiraz, Southern Iran. Foodborne Pathog. Dis. 2013, 10, 764–770. [Google Scholar] [CrossRef]
- Li, X.; Zhang, G.; Zhu, Y.; Bi, J.; Hao, H.; Hou, H. Effect of the luxI/R gene on AHL-signaling molecules and QS regulatory mechanism in Hafnia alvei H4. AMB Express 2019, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Calanche, J.B.; Beltran, J.A.; Hernandez Arias, A.J. Aquaculture and sensometrics: The need to evaluate sensory attributes and the consumers’ Preferences. Rev. Aquac. 2019, 12, 805–821. [Google Scholar] [CrossRef]
- Albertsen, M.; Hugenholtz, P.; Skarshewski, A.; Nielsen, K.L.; Tyson, G.W.; Nielsen, P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013, 31, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [CrossRef]
- Huang, Z.; Qiu, J.; Li, J.; Xu, D.; Liu, Q. Exploration of microbial diversity based on 16S rRNA gene sequence analysis. Acta Microbiol. Sin. 2020, 61, 1044–1063. [Google Scholar]
- He, R.; Yuan, K.; Lin, L.; Yang, Y.; Zou, S.; Luan, T.; Baowei, C. Functional metagenomics: One of the most robust tools for discovering new antibioticsresistance genes. Environ. Chem. 2018, 38, 1548–1556. [Google Scholar]
- Qiu, W.; Zhong, M.-T.; Lun, Y.-Z. Analysis of application and existing problems of clinical metagenomics. Chin. J. Microecol. 2020, 33, 594–598. [Google Scholar]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Chen, L.; Wang, D.; Garmaeva, S.; Kurilshikov, A.; Vich Vila, A.; Gacesa, R.; Sinha, T.; Lifelines Cohort, S.; Segal, E.; Weersma, R.K.; et al. The long-term genetic stability and individual specificity of the human gut microbiome. Cell 2021, 184, 2302.e12–2315.e12. [Google Scholar] [CrossRef]
- Yang, D.; Van Gompel, L.; Luiken, R.E.C.; Sanders, P.; Joosten, P.; van Heijnsbergen, E.; Wouters, I.M.; Scherpenisse, P.; Chauvin, C.; Wadepohl, K.; et al. Association of antimicrobial usage with faecal abundance of aph(3′)-III, ermB, sul2 and tetW resistance genes in veal calves in three European countries. Int. J. Antimicrob. Agents 2020, 56, 106131. [Google Scholar] [CrossRef]
- Fishbein, S.R.S.; Mahmud, B.; Dantas, G. Antibiotic perturbations to the gut microbiome. Nat. Rev. Microbiol. 2023, 21, 772–788. [Google Scholar] [CrossRef]
- Van Gompel, L.; Luiken, R.E.C.; Sarrazin, S.; Munk, P.; Knudsen, B.E.; Hansen, R.B.; Bossers, A.; Aarestrup, F.M.; Dewulf, J.; Wagenaar, J.A.; et al. The antimicrobial resistome in relation to antimicrobial use and biosecurity in pig farming, a metagenome-wide association study in nine European countries. J. Antimicrob. Chemother. 2019, 74, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, N.; Nekouei, O.; Gow, S.; Agunos, A.; Checkley, S. Risk factors associated with the A2C resistance pattern among E. coli isolates from broiler flocks in Canada. Prev. Vet. Med. 2017, 148, 115–120. [Google Scholar] [CrossRef] [PubMed]
- AbuOun, M.; O’Connor, H.M.; Stubberfield, E.J.; Nunez-Garcia, J.; Sayers, E.; Crook, D.W.; Smith, R.P.; Anjum, M.F. Characterizing Antimicrobial Resistant Escherichia coli and Associated Risk Factors in a Cross-Sectional Study of Pig Farms in Great Britain. Front. Microbiol. 2020, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Ying, G.G.; Singer, A.C.; Zhu, Y.G. Review of antibiotic resistance in China and its environment. Environ. Int. 2018, 110, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Postma, M.; Backhans, A.; Collineau, L.; Loesken, S.; Sjolund, M.; Belloc, C.; Emanuelson, U.; Grosse Beilage, E.; Nielsen, E.O.; Stark, K.D.C.; et al. Evaluation of the relationship between the biosecurity status, production parameters, herd characteristics and antimicrobial usage in farrow-to-finish pig production in four EU countries. Porc. Health Manag. 2016, 2, 9. [Google Scholar] [CrossRef]
- Tóth, A.G.; Csabai, I.; Judge, M.F.; Maróti, G.; Becsei, A.; Spisák, S.; Solymosi, N. Mobile Antimicrobial Resistance Genes in Probiotics. Antibiotics 2021, 10, 1287. [Google Scholar] [CrossRef] [PubMed]
- Lawther, K.; Santos, F.G.; Oyama, L.B.; Rubino, F.; Morrison, S.; Creevey, C.J.; McGrath, J.W.; Huws, S.A. Resistome Analysis of Global Livestock and Soil Microbiomes. Front. Microbiol. 2022, 13, 897905. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Base (bp) | Contigs | Contigs Bases (bp) | N50 (bp) | N90 (bp) | Max (bp) | Min (bp) | ORFs |
|---|---|---|---|---|---|---|---|---|---|
| BG | 44,576,741 | 6,731,087,941 | 339,630 | 236,303,250 | 737 | 357 | 74,979 | 300 | 448,655 |
| HB | 42,737,859 | 6,453,416,659 | 358,414 | 286,392,037 | 928 | 376 | 177,876 | 300 | 478,813 |
| SC | 47,829,571 | 7,222,265,271 | 336,596 | 278,158,946 | 988 | 374 | 121,958 | 300 | 449,776 |
| Bacteria | Archaea | Viruses | Eukaryota | |
|---|---|---|---|---|
| BG | 0.980559 | 0.007228 | 0.006983 | 0.002385 |
| XZ | 0.975898 | 0.018320 | 0.002967 | 0.001471 |
| HB | 0.973169 | 0.014486 | 0.009305 | 0.002093 |
| SC | 0.990551 | 0.002702 | 0.004980 | 0.000697 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, Y.; Qi, Z.; Sang, J.; Yu, Z.; Li, M.; Wang, Z.; Tu, J.; Song, X.; Qi, K. Metagenome-Based Analysis of the Microbial Community Structure and Drug-Resistance Characteristics of Livestock Feces in Anhui Province, China. Vet. Sci. 2024, 11, 87. https://doi.org/10.3390/vetsci11020087
Shao Y, Qi Z, Sang J, Yu Z, Li M, Wang Z, Tu J, Song X, Qi K. Metagenome-Based Analysis of the Microbial Community Structure and Drug-Resistance Characteristics of Livestock Feces in Anhui Province, China. Veterinary Sciences. 2024; 11(2):87. https://doi.org/10.3390/vetsci11020087
Chicago/Turabian StyleShao, Ying, Zhao Qi, Jinhui Sang, Zhaorong Yu, Min Li, Zhenyu Wang, Jian Tu, Xiangjun Song, and Kezong Qi. 2024. "Metagenome-Based Analysis of the Microbial Community Structure and Drug-Resistance Characteristics of Livestock Feces in Anhui Province, China" Veterinary Sciences 11, no. 2: 87. https://doi.org/10.3390/vetsci11020087
APA StyleShao, Y., Qi, Z., Sang, J., Yu, Z., Li, M., Wang, Z., Tu, J., Song, X., & Qi, K. (2024). Metagenome-Based Analysis of the Microbial Community Structure and Drug-Resistance Characteristics of Livestock Feces in Anhui Province, China. Veterinary Sciences, 11(2), 87. https://doi.org/10.3390/vetsci11020087