
Microbiota Characterization of the Cow Mammary Gland Microenvironment and Its Association with Somatic Cell Count

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. SCC in the Samples
2.3. Bacterial DNA Isolation from the Milk Samples
2.4. DNA Extraction and PCR Amplification
2.5. Illumina MiSeq Sequencing
2.6. Statistical Analysis
3. Results
3.1. SCC Measurement
3.2. Analysis of Bacterial Community Diversity
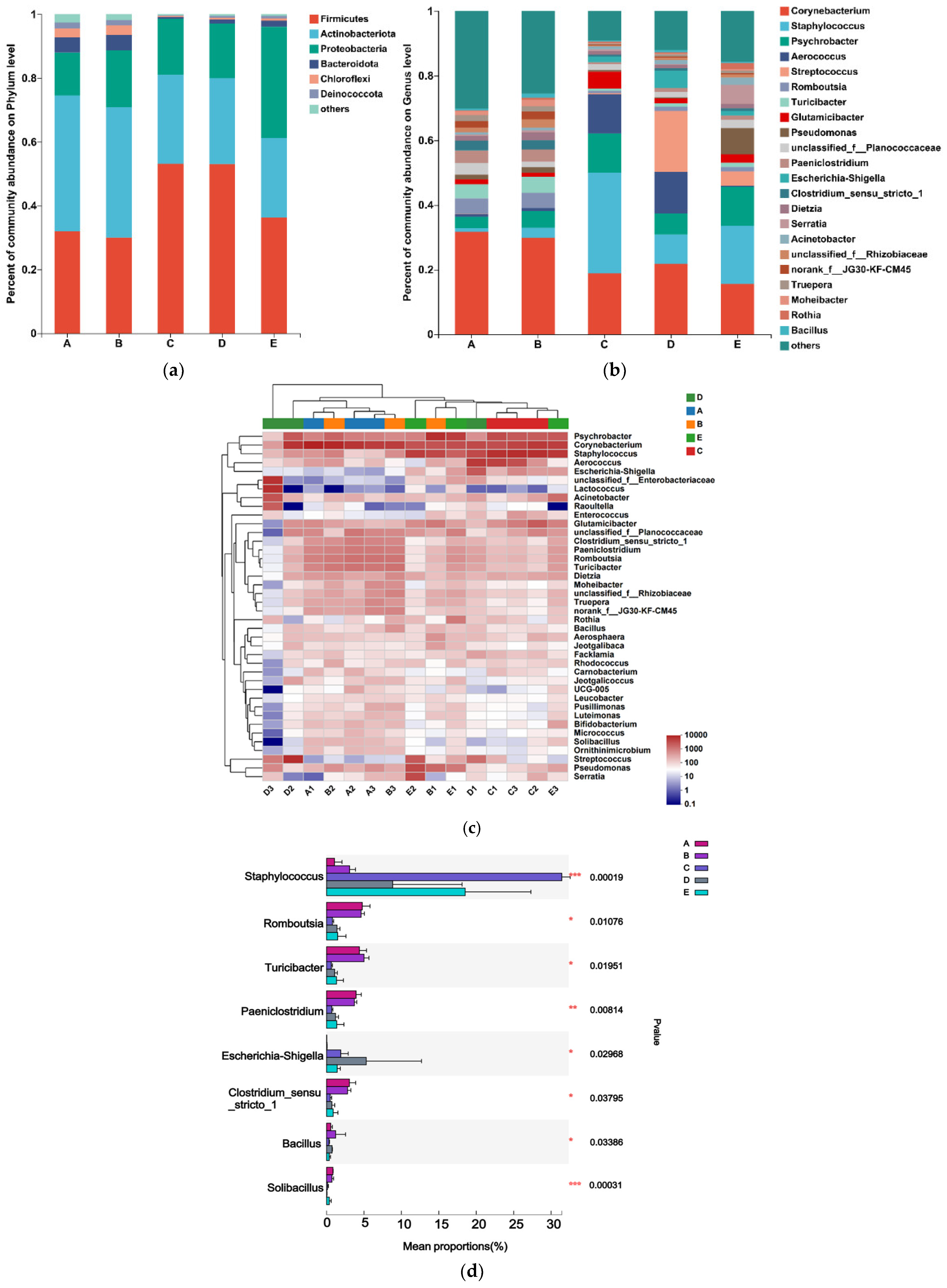
3.3. Analysis of Microbial Community Composition
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sinha, M.K.; Thombare, N.N.; Mondal, B. Subclinical mastitis in dairy animals: Incidence, economics, and predisposing factors. Sci. World J. 2014, 2014, 523984. [Google Scholar] [CrossRef] [PubMed]
- Sharun, K.; Dhama, K.; Tiwari, R.; Gugjoo, M.B.; Yatoo, M.I.; Patel, S.K.; Pathak, M.; Karthik, K.; Khurana, S.K.; Singh, R.; et al. Advances in therapeutic and managemental approaches of bovine mastitis: A comprehensive review. Vet. Q. 2021, 41, 107–136. [Google Scholar] [CrossRef] [PubMed]
- Neculai-Valeanu, A.S.; Ariton, A.M. Udder Health Monitoring for Prevention of Bovine Mastitis and Improvement of Milk Quality. Bioengineering 2022, 9, 608. [Google Scholar] [CrossRef] [PubMed]
- Hadrich, J.C.; Wolf, C.A.; Lombard, J.; Dolak, T.M. Estimating milk yield and value losses from increased somatic cell count on US dairy farms. J. Dairy Sci. 2018, 101, 3588–3596. [Google Scholar] [CrossRef] [PubMed]
- Boutinaud, M.; Jammes, H. Potential uses of milk epithelial cells: A review. Reprod. Nutr. Dev. 2002, 42, 133–147. [Google Scholar] [CrossRef]
- Hoque, M.N.; Istiaq, A.; Clement, R.A.; Sultana, M.; Crandall, K.A.; Siddiki, A.Z.; Hossain, M.A. Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci. Rep. 2019, 9, 13536. [Google Scholar] [CrossRef] [PubMed]
- Green, M.J.; Green, L.E.; Schukken, Y.H.; Bradley, A.J.; Peeler, E.J.; Barkema, H.W.; de Haas, Y.; Collis, V.; Medley, G. Somatic cell count distributions during lactation predict clinical mastitis. J. Dairy Sci. 2004, 87, 1256–1264. [Google Scholar] [CrossRef]
- Kandeel, S.A.; Megahed, A.A.; Arnaout, F.K.; Constable, P.D. Evaluation and Comparison of 2 On-Farm Tests for Estimating Somatic Cell Count in Quarter Milk Samples from Lactating Dairy Cattle. J. Vet. Intern. Med. 2018, 32, 506–515. [Google Scholar] [CrossRef]
- Hagnestam-Nielsen, C.; Emanuelson, U.; Berglund, B.; Strandberg, E. Relationship between somatic cell count and milk yield in different stages of lactation. J. Dairy Sci. 2009, 92, 3124–3133. [Google Scholar] [CrossRef]
- Schukken, Y.H.; Wilson, D.J.; Welcome, F.; Garrison-Tikofsky, L.; Gonzalez, R.N. Monitoring udder health and milk quality using somatic cell counts. Vet. Res. 2003, 34, 579–596. [Google Scholar] [CrossRef]
- Gonzalo, C.; Ariznabarreta, A.; Carriedo, J.A.; San, P.F. Mammary pathogens and their relationship to somatic cell count and milk yield losses in dairy ewes. J. Dairy Sci. 2002, 85, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Günther, J.; Petzl, W.; Bauer, I.; Ponsuksili, S.; Zerbe, H.; Schuberth, H.; Brunner, R.M.; Seyfert, H.-M. Differentiating Staphylococcus aureus from Escherichia coli mastitis: S. aureus triggers unbalanced immune-dampening and host cell invasion immediately after udder infection. Sci. Rep. 2017, 7, 4811. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.S.; Silva, D.M.; Silva, M.P.; Vidigal, P.; Silva, J.; Guerra, S.T.; Ribeiro, M.G.; Mendes, T.A.d.O.; Ribon, A.d.O.B. Comparative genomics of Staphylococcus aureus associated with subclinical and clinical bovine mastitis. PLoS ONE 2019, 14, e220804. [Google Scholar] [CrossRef] [PubMed]
- Burvenich, C.; Van Merris, V.; Mehrzad, J.; Diez-Fraile, A.; Duchateau, L. Severity of E. coli mastitis is mainly determined by cow factors. Vet. Res. 2003, 34, 521–564. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, T.; Aubry, E.; van Vorst, K.; Amon, T.; Fulde, M. The Role of Streptococcus spp. in Bovine Mastitis. Microorganisms 2021, 9, 1497. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Barkema, H.W.; Zhang, L.; Liu, G.; Deng, Z.; Cai, L.; Shan, R.; Zhang, S.; Zou, J.; Kastelic, J.P.; et al. Incidence of clinical mastitis and distribution of pathogens on large Chinese dairy farms. J. Dairy Sci. 2017, 100, 4797–4806. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, G.; Machado, V.S.; Santisteban, C.; Schukken, Y.H.; Bicalho, R.C. Microbial diversity of bovine mastitic milk as described by pyrosequencing of metagenomic 16s rDNA. PLoS ONE 2012, 7, e47671. [Google Scholar] [CrossRef] [PubMed]
- Church, D.L.; Cerutti, L.; Gurtler, A.; Griener, T.; Zelazny, A.; Emler, S. Performance and Application of 16S rRNA Gene Cycle Sequencing for Routine Identification of Bacteria in the Clinical Microbiology Laboratory. Clin. Microbiol. Rev. 2020, 33, e00053-19. [Google Scholar] [CrossRef]
- Catozzi, C.; Sanchez, B.A.; Francino, O.; Lecchi, C.; De Carlo, E.; Vecchio, D.; Martucciello, A.; Fraulo, P.; Bronzo, V.; Cuscó, A.; et al. The microbiota of water buffalo milk during mastitis. PLoS ONE 2017, 12, e184710. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.; Tuddenham, S.; Sears, C.L.; Zhao, N. Meta-analysis methods for multiple related markers: Applications to microbiome studies with the results on multiple alpha-diversity indices. Stat. Med. 2021, 40, 2859–2876. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Fuentes, S.; Grievink, W.; van Niftrik, L.; Tindall, B.J.; Timmerman, H.M.; Rijkers, G.T.; Smidt, H. Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the genus Clostridium into the genera Romboutsia gen. nov., Intestinibacter gen. nov., Terrisporobacter gen. nov. and Asaccharospora gen. nov. Int. J. Syst. Evol. Microbiol. 2014, 64 Pt 5, 1600–1616. [Google Scholar] [CrossRef]
- Guan, Z.; Chen, L.; Gerritsen, J.; Smidt, H.; Goldfine, H. The cellular lipids of Romboutsia. Biochim. Biophys. Acta 2016, 1861 Pt A, 1076–1082. [Google Scholar] [CrossRef]
- Wiersema, M.L.; Koester, L.R.; Schmitz-Esser, S.; Koltes, D.A. Comparison of intestinal permeability, morphology, and ileal microbial communities of commercial hens housed in conventional cages and cage-free housing systems. Poult. Sci. 2021, 100, 1178–1191. [Google Scholar] [CrossRef]
- Tanaka, K.; Harata, G.; Miyazawa, K.; He, F.; Tanigaki, S.; Kobayashi, Y. The gut microbiota of non-obese Japanese pregnant women with gestational diabetes mellitus. Biosci. Microbiota Food Health 2022, 41, 4–11. [Google Scholar] [CrossRef]
- Kemis, J.H.; Linke, V.; Barrett, K.L.; Boehm, F.J.; Traeger, L.L.; Keller, M.P.; Rabaglia, M.E.; Schueler, K.L.; Stapleton, D.S.; Gatti, D.M.; et al. Genetic determinants of gut microbiota composition and bile acid profiles in mice. PLoS Genet. 2019, 15, e1008073. [Google Scholar] [CrossRef]
- Maki, J.J.; Lippolis, J.D.; Looft, T. Proteomic response of Turicibacter bilis MMM721 to chicken bile and its bile acids. BMC Res. Notes 2022, 15, 236. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.Y.; Zhang, R.Y.; Wang, D.S.; Zhu, W.Y. Impact of subacute ruminal acidosis (SARA) adaptation on rumenmicrobiota indairy cattle using pyrosequencing. Anaerobe 2013, 24, 12–19. [Google Scholar] [CrossRef]
- Gagnon, N.; Talbot, G.; Ward, P.; Roy, D.; Dupuis, M.; Farnworth, E.; Tompkins, T.A.; Lessard, M. Evaluation ofbacterial diversityin the gut of piglets supplemented with probiotics using ribosomal intergenic spacer analysis. Can. J. Anim. Sci. 2007, 87, 207–219. [Google Scholar] [CrossRef]
- Bosshard, P.P.; Zbinden, R.; Altwegg, M. Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int. J. Syst. Evol. Microbiol. 2002, 52, 1263–1266. [Google Scholar] [PubMed]
- Piepers, S.; Peeters, K.; Opsomer, G.; Barkema, H.W.; Frankena, K.; De Vliegher, S. Pathogen group specific risk factors at herd, heifer and quarter levels for intramammary infections in early lactating dairy heifers. Prev. Vet. Med. 2011, 99, 91–101. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Bao, H.; Wei, R.; Zhou, Y.; Zhang, H.; Wang, R. Population structure and antimicrobial profile of Staphylococcus aureus strains associated with bovine mastitis in China. Microb. Pathog. 2016, 97, 103–109. [Google Scholar] [CrossRef]
- Welter, D.K.; Ruaud, A.; Henseler, Z.M.; De Jong, H.N.; van Coeverden de Groot, P.; Michaux, J.; Gormezano, L.; Waters, J.L.; Youngblut, N.D.; Ley, R.E. Free-Living, Psychrotrophic Bacteria of the Genus Psychrobacter Are Descendants of Pathobionts. Msystems 2021, 6, e00258-21. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Indicators | Group A | Group B | Group C | Group D | Group E |
|---|---|---|---|---|---|
| Somatic cells | <1 × 105 | 1 × 105–2 × 105 | 2 × 105–5 × 105 | 5 × 105–1 × 106 | >1 × 106 |
| Quarters | 75 | 73 | 26 | 15 | 11 |
| proportion | 37.5% | 36.5% | 13.0% | 7.5% | 5.5% |
| Mean Abundance | |||||
|---|---|---|---|---|---|
| Group | Firmicutes | Actinobacteriota | Proteobacteria | Bacteroidota | Others |
| A | 0.3192 | 0.4257 | 0.1354 | 0.0475 | 0.0722 |
| B | 0.3001 | 0.4078 | 0.1797 | 0.0476 | 0.0648 |
| C | 0.5309 | 0.2790 | 0.1752 | 0.0066 | 0.0083 |
| D | 0.5306 | 0.2685 | 0.1719 | 0.0133 | 0.0157 |
| E | 0.3622 | 0.2500 | 0.3484 | 0.0201 | 0.0193 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Liu, H.; Cao, G.; Cui, Y.; Wang, H.; Chen, X.; Xu, F.; Li, X. Microbiota Characterization of the Cow Mammary Gland Microenvironment and Its Association with Somatic Cell Count. Vet. Sci. 2023, 10, 699. https://doi.org/10.3390/vetsci10120699
Liu J, Liu H, Cao G, Cui Y, Wang H, Chen X, Xu F, Li X. Microbiota Characterization of the Cow Mammary Gland Microenvironment and Its Association with Somatic Cell Count. Veterinary Sciences. 2023; 10(12):699. https://doi.org/10.3390/vetsci10120699
Chicago/Turabian StyleLiu, Jing, Huan Liu, Guangjie Cao, Yifang Cui, Huanhuan Wang, Xiaojie Chen, Fei Xu, and Xiubo Li. 2023. "Microbiota Characterization of the Cow Mammary Gland Microenvironment and Its Association with Somatic Cell Count" Veterinary Sciences 10, no. 12: 699. https://doi.org/10.3390/vetsci10120699
APA StyleLiu, J., Liu, H., Cao, G., Cui, Y., Wang, H., Chen, X., Xu, F., & Li, X. (2023). Microbiota Characterization of the Cow Mammary Gland Microenvironment and Its Association with Somatic Cell Count. Veterinary Sciences, 10(12), 699. https://doi.org/10.3390/vetsci10120699