Collagen Structure-Function Mapping Informs Applications for Regenerative Medicine




Abstract
1. Introduction
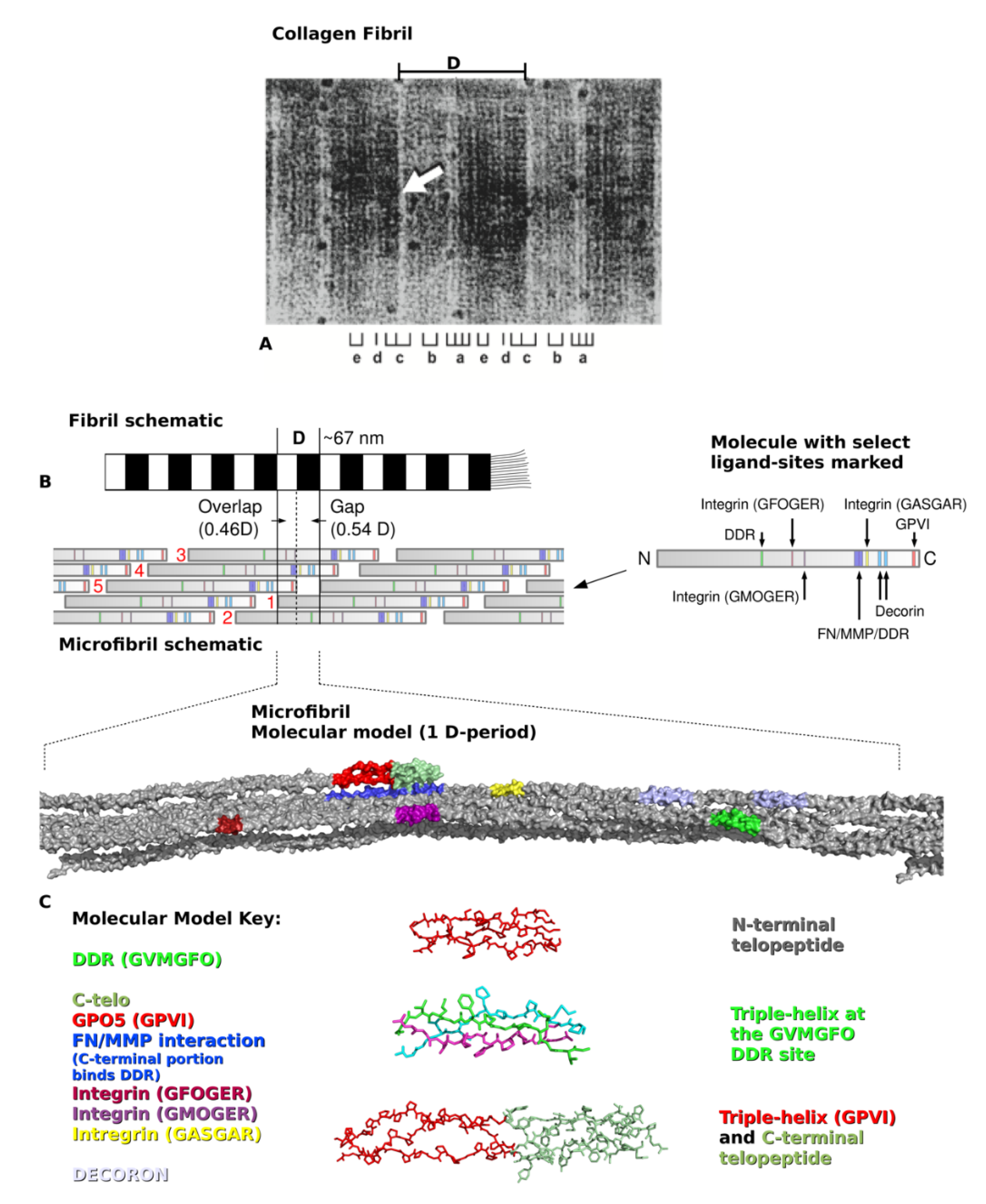
2. Type I Collagen Structure and Assembly
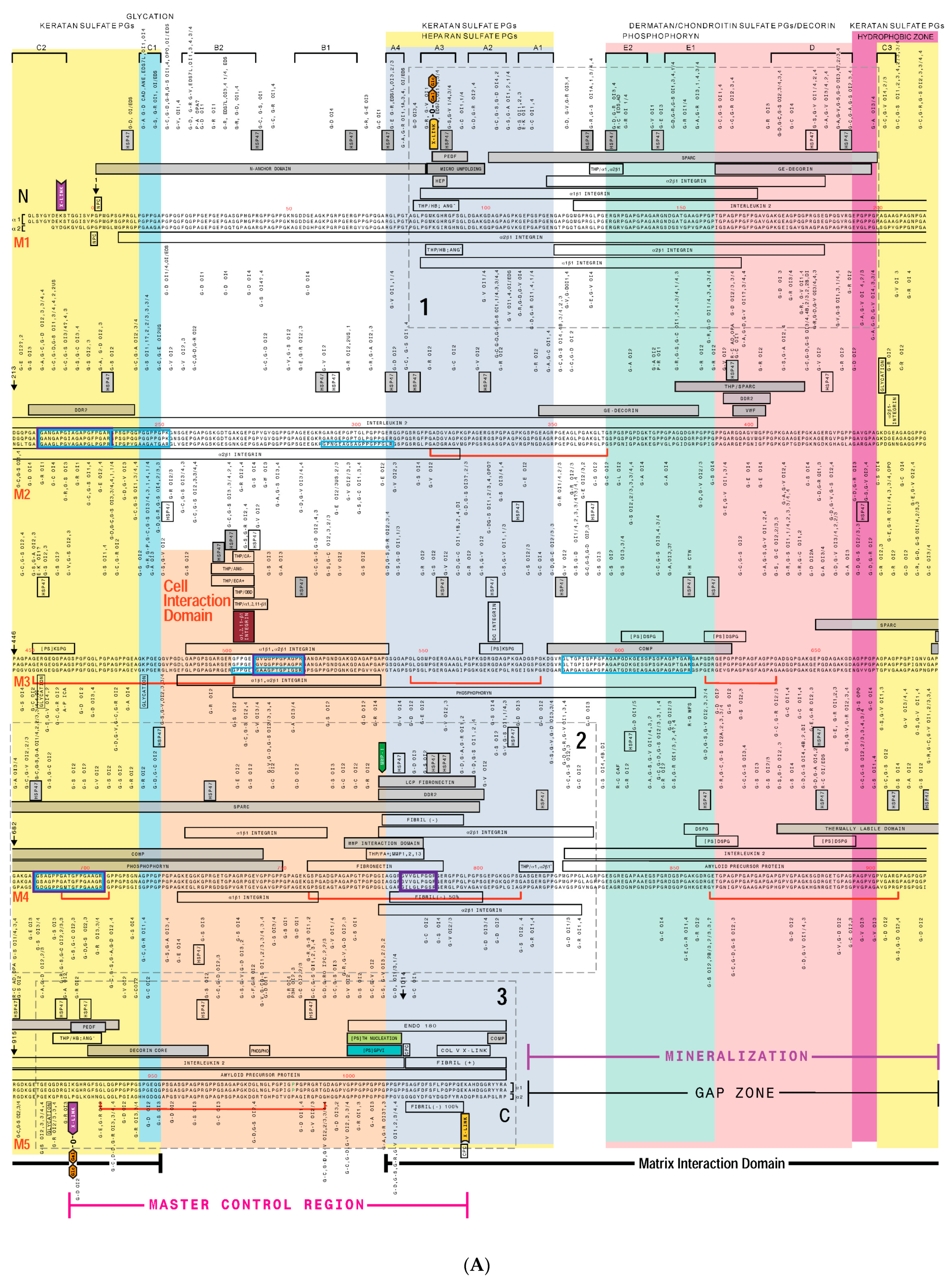
3. Creating a “Road Map” or Interactome of Type I Collagen
Human Mutation Patterns
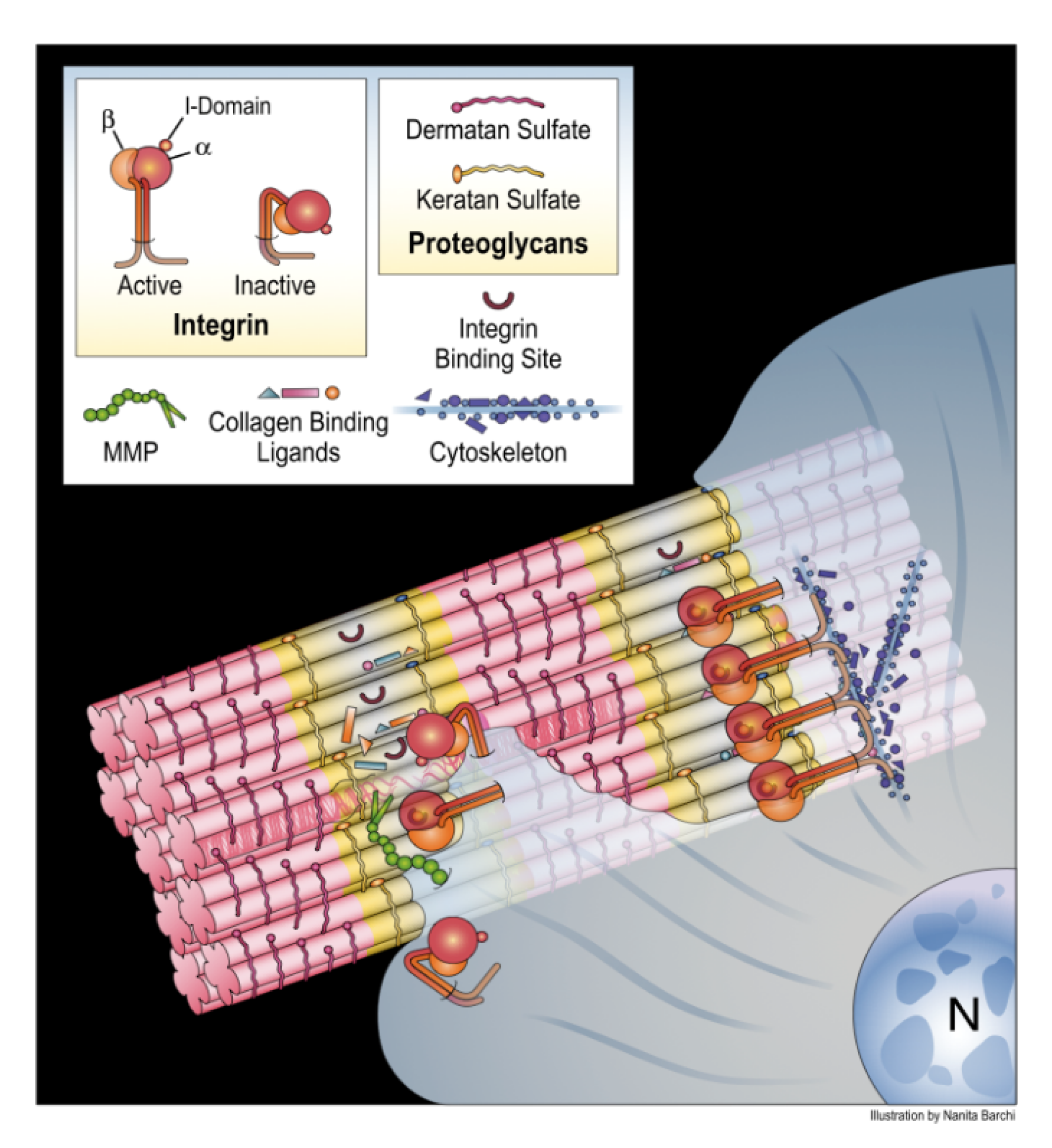
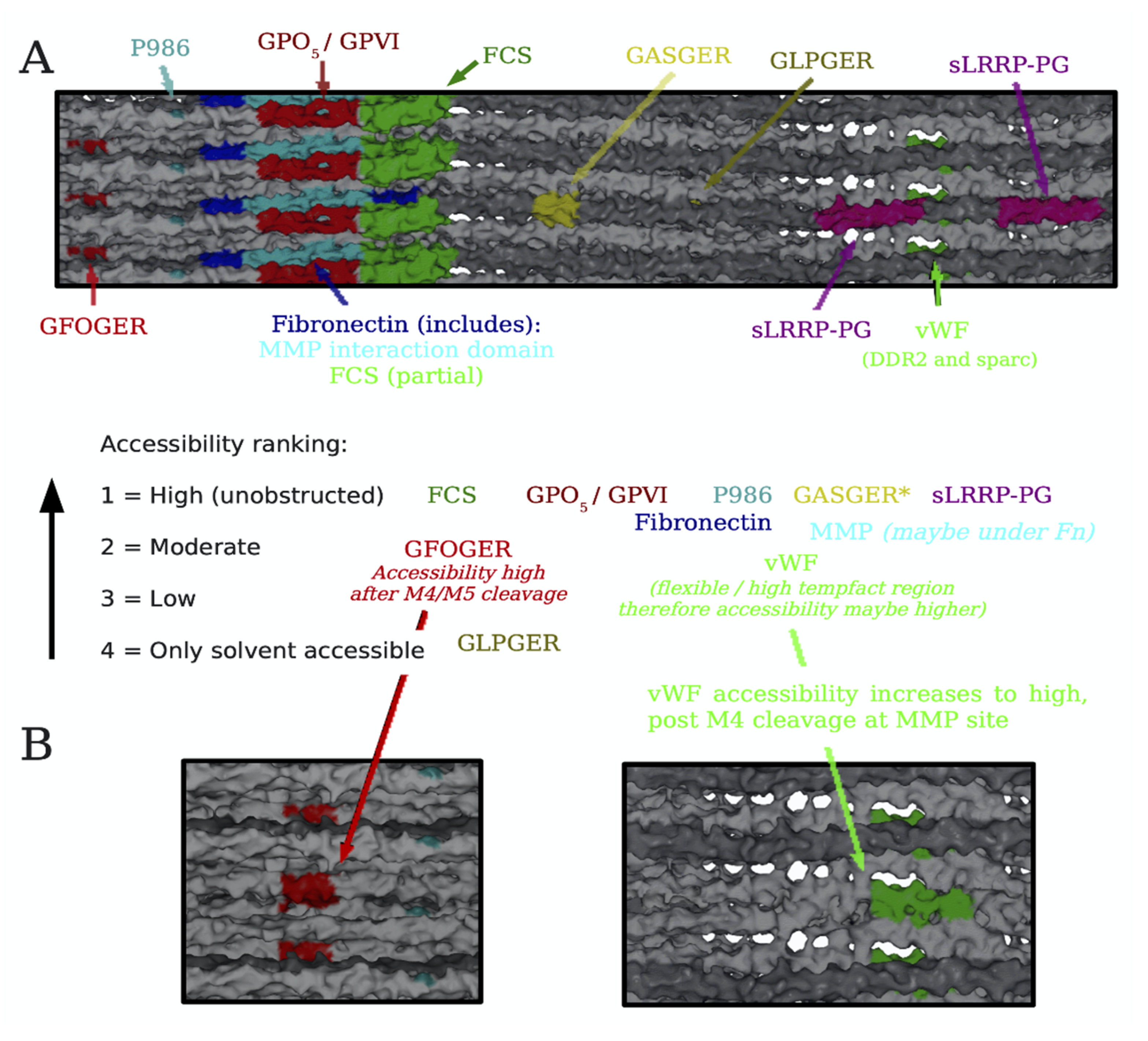
4. Translating the Collagen Interactome into the Three-Dimensional (3D) “Living” Fibril
Conditional Cell and Matrix Interaction Domains of the Type I Collagen Fibril
5. Type III Collagen Interactome
6. Collagen Structure-Function Studies Inform Bioengineering Applications
6.1. Anti-Collagen Antibodies
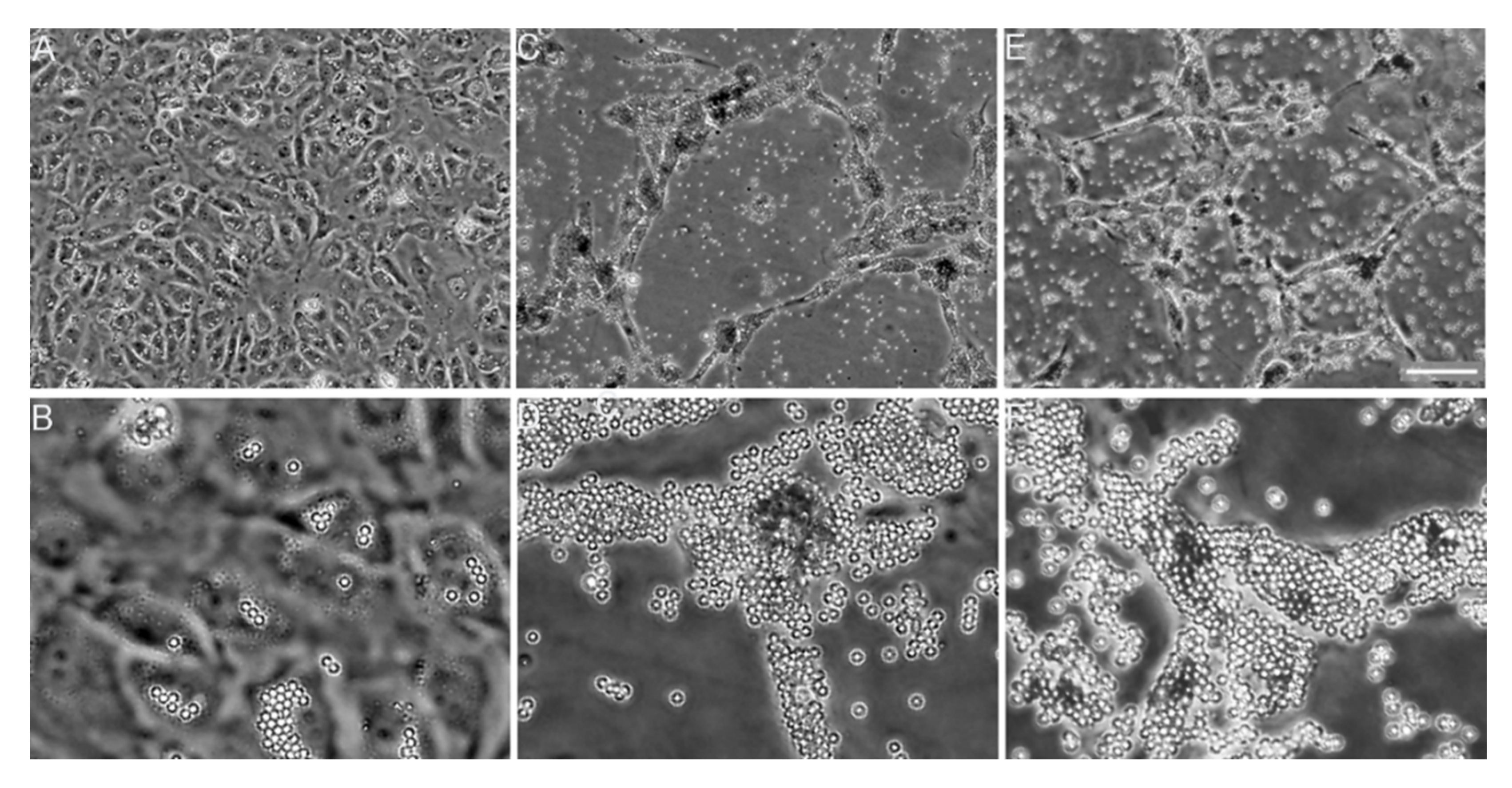
6.2. Collagen Fibrillogenesis as a Potential Anti-Fibrotic Target
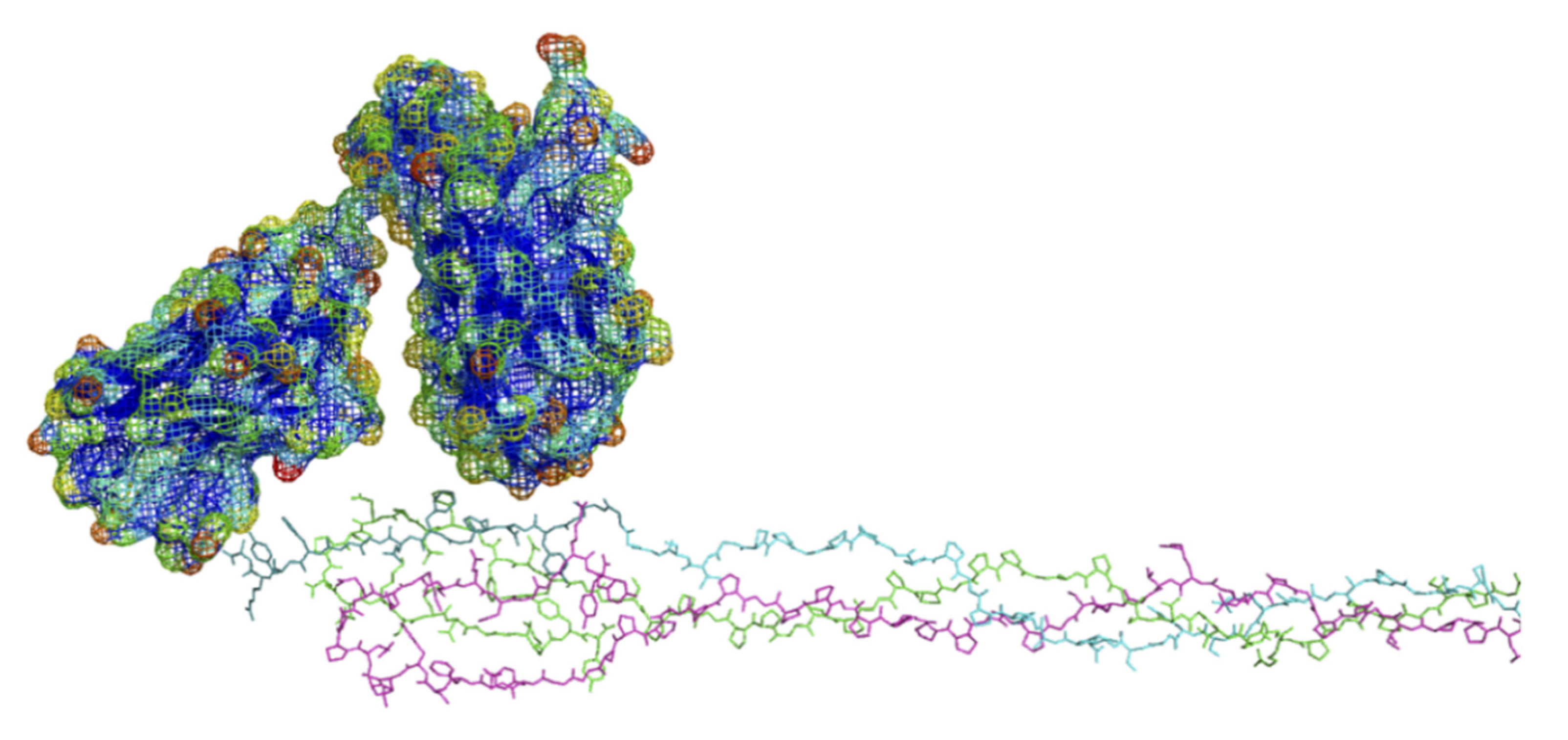
6.3. Modeling of Anti-α2Ct Antibody-Collagen Fibril Interactions
6.3.1. Collagen C-Terminus
6.3.2. Antibody Variant Used for Modeling
6.4. A Broad View on Blocking Matrix Assembly as an Anti-Fibrotic Therapy
7. Collagen-Induced Angiogenesis
Integrin Receptor Ligation and Clustering in Collagen-Induced Angiogenesis
8. Bioengineering Applications: Angiogenic Polymers
8.1. Recombinant Super-Angiogenic Collagens
8.2. Collagen Mimetics as Angiogenesis Substrates
9. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kadler, K.E.; Baldock, C.; Bella, J.; Boot-Handford, R.P. Collagens at a glance. J. Cell Sci. 2007, 120 (Pt 12), 1955–1958. [Google Scholar] [CrossRef]
- Marini, J.C.; Forlino, A.; Cabral, W.A.; Barnes, A.M.; San Antonio, J.D.; Milgrom, S.; Hyland, J.C.; Korkko, J.; Prockop, D.J.; De Paepe, A.; et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007, 28, 209–221. [Google Scholar] [CrossRef]
- Piez, K.A.; Reddi, A.H. Extracellular Matrix Biochemistry; Elsevier: New York, NY, USA, 1984; p. xvi. 473p. [Google Scholar]
- Jacenko, O.; Olsen, B.R.; LuValle, P. Organization and regulation of collagen genes. Crit. Rev. Eukaryot. Gene Expr. 1991, 1, 327–353. [Google Scholar]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, G.A.; Sweeney, S.M.; Korkko, J.; Ala-Kokko, L.; San Antonio, J.D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 2002, 277, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Twardowski, T.; Fertala, A.; Orgel, J.P.; San Antonio, J.D. Type I collagen and collagen mimetics as angiogenesis promoting superpolymers. Curr. Pharm. Des. 2007, 13, 3608–3621. [Google Scholar] [CrossRef] [PubMed]
- Ayad, S. The Extracellular Matrix Factsbook, 2nd ed.; Academic Press: San Diego, CA, USA, 1998; p. x. 301p. [Google Scholar]
- Chen, J.M.; Burger, C.; Krishnan, C.; Chu, B.; Hsiao, B.; Glimcher, M. In vitro mineralization of collagen in demineralized fish bone. Macromol. Chem. Phys. 2005, 206, 43–51. [Google Scholar] [CrossRef]
- Silver, F.H.; Landis, W.J. Deposition of apatite in mineralizing vertebrate extracellular matrices: A model of possible nucleation sites on type I collagen. Connect. Tissue Res. 2011, 52, 242–254. [Google Scholar] [CrossRef]
- Li, Y.; Aparicio, C. Discerning the subfibrillar structure of mineralized collagen fibrils: A model for the ultrastructure of bone. PLoS ONE 2013, 8, e76782. [Google Scholar] [CrossRef]
- Boskey, A.L.; Villarreal-Ramirez, E. Intrinsically disordered proteins and biomineralization. Matrix Biol. 2016, 52–54, 43–59. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound Healing: A Cellular Perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef] [PubMed]
- Abou Neel, E.A.; Bozec, L.; Knowles, J.C.; Syed, O.; Mudera, V.; Day, R.; Hyun, J.K. Collagen—Emerging collagen based therapies hit the patient. Adv. Drug Deliv. Rev. 2013, 65, 429–456. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Traub, W. Organization of hydroxyapatite crystals within collagen fibrils. FEBS Lett. 1986, 206, 262–266. [Google Scholar] [CrossRef]
- Beniash, E. Biominerals—Hierarchical nanocomposites: The example of bone. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 47–69. [Google Scholar] [CrossRef]
- Orgel, J.P.; Irving, T.C.; Miller, A.; Wess, T.J. Microfibrillar structure of type I collagen in situ. Proc. Natl. Acad. Sci. USA 2006, 103, 9001–9005. [Google Scholar] [CrossRef]
- Scott, J.E. Proteoglycan-fibrillar collagen interactions. Biochem. J. 1988, 252, 313–323. [Google Scholar] [CrossRef]
- Petruska, J.A.; Hodge, A.J. A Subunit Model for the Tropocollagen Macromolecule. Proc. Natl. Acad. Sci. USA 1964, 51, 871–876. [Google Scholar] [CrossRef]
- Orgel, J.; Madhurapantula, R.S. A structural prospective for collagen receptors such as DDR and their binding of the collagen fibril. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118478. [Google Scholar] [CrossRef]
- Sweeney, S.M.; Orgel, J.P.; Fertala, A.; McAuliffe, J.D.; Turner, K.R.; Di Lullo, G.A.; Chen, S.; Antipova, O.; Perumal, S.; Ala-Kokko, L.; et al. Candidate cell and matrix interaction domains on the collagen fibril, the predominant protein of vertebrates. J. Biol. Chem. 2008, 283, 21187–21197. [Google Scholar] [CrossRef]
- Chapman, J.A. The staining pattern of collagen fibrils. I. An analysis of electron micrographs. Connect. Tissue Res. 1974, 2, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fertala, A.; Ratner, B.D.; Sage, E.H.; Jiang, S. Identifying the SPARC binding sites on collagen I and procollagen I by atomic force microscopy. Anal. Chem. 2005, 77, 6765–6771. [Google Scholar] [CrossRef] [PubMed]
- Konitsiotis, A.D.; Raynal, N.; Bihan, D.; Hohenester, E.; Farndale, R.W.; Leitinger, B. Characterization of high affinity binding motifs for the discoidin domain receptor DDR2 in collagen. J. Biol. Chem. 2008, 283, 6861–6868. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, G.; Kovac, L.; Radziejewski, C.; Samuelsson, S.J. Binding of discoidin domain receptor 2 to collagen I: An atomic force microscopy investigation. Biochemistry 2002, 41, 11091–11098. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gurusiddappa, S.; Rich, R.L.; Owens, R.T.; Keene, D.R.; Mayne, R.; Hook, A.; Hook, M. Multiple binding sites in collagen type I for the integrins alpha1beta1 and alpha2beta1. J. Biol. Chem. 2000, 275, 38981–38989. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Morton, L.F.; Onley, D.J.; Peachey, A.R.; Messent, A.J.; Smethurst, P.A.; Tuckwell, D.S.; Farndale, R.W.; Barnes, M.J. Identification in collagen type I of an integrin alpha2 beta1-binding site containing an essential GER sequence. J. Biol. Chem. 1998, 273, 33287–33294. [Google Scholar] [CrossRef]
- Danielson, K.G.; Baribault, H.; Holmes, D.F.; Graham, H.; Kadler, K.E.; Iozzo, R.V. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 1997, 136, 729–743. [Google Scholar] [CrossRef]
- Glimcher, M.J.; Hodge, A.J.; Schmitt, F.O. Macromolecular Aggregation States in Relation to Mineralization: The Collagen-Hydroxyapatite System as Studied in Vitro. Proc. Natl. Acad. Sci. USA 1957, 43, 860–867. [Google Scholar] [CrossRef]
- Glimcher, M. Molecular biology of mineralized tissues with particular reference to bone. Rev. Mod. Phys. 1959, 31, 359–393. [Google Scholar] [CrossRef]
- Cohen-Solal, L.; Maroteaux, P.; Glimcher, M. Presence of gamm-glutamyl-phosphate in the collagens of bone and other calcified tissues and its absence in the collagens of unmineralized tissues. In The Chemistry and Biology of Mineralized Connective Tissue; Veis, A., Ed.; Elsevier North Holland, Inc.: Amsterdam, The Netherlands, 1981; pp. 7–11. [Google Scholar]
- Cohen-Solal, L.; Cohen-Solal, M.; Glimcher, M.J. Identification of gamma-glutamyl phosphate in the alpha 2 chains of chicken bone collagen. Proc. Natl. Acad. Sci. USA 1979, 76, 4327–4330. [Google Scholar] [CrossRef]
- Landis, W.J.; Moradian-Oldak, J.; Weiner, S. Topographic imaging of mineral and collagen in the calcifying turkey tendon. Connect. Tissue Res. 1991, 25, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Landis, W.J.; Silver, F.H. Mineral deposition in the extracellular matrices of vertebrate tissues: Identification of possible apatite nucleation sites on type I collagen. Cells Tissues Organs 2009, 189, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Yalak, G.; Olsen, B.R. Proteomic database mining opens up avenues utilizing extracellular protein phosphorylation for novel therapeutic applications. J. Transl. Med. 2015, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Bouleftour, W.; Juignet, L.; Bouet, G.; Granito, R.N.; Vanden-Bossche, A.; Laroche, N.; Aubin, J.E.; Lafage-Proust, M.H.; Vico, L.; Malaval, L. The role of the SIBLING, Bone Sialoprotein in skeletal biology—Contribution of mouse experimental genetics. Matrix Biol. 2016, 52–54, 60–77. [Google Scholar] [CrossRef]
- Reznikov, N.; Bilton, M.; Lari, L.; Stevens, M.M.; Kroger, R. Fractal-like hierarchical organization of bone begins at the nanoscale. Science 2018, 360, eaao2189. [Google Scholar] [CrossRef]
- Xu, Y.; Nudelman, F.; Eren, E.D.; Wirix, M.J.M.; Cantaert, B.; Nijhuis, W.H.; Hermida-Merino, D.; Portale, G.; Bomans, P.H.H.; Ottmann, C.; et al. Intermolecular channels direct crystal orientation in mineralized collagen. Nat. Commun. 2020, 11, 5068. [Google Scholar] [CrossRef]
- Olszta, M.C.; Jee, S.S.; Kumar, R.; Kim, Y.Y.; Kaufman, M.; Douglas, E.; Glower, L. Bone structure and formation: A new perspective. Mater. Sci. Eng. Rep. 2007, 58, 77–116. [Google Scholar] [CrossRef]
- Li, Y.; Thula, T.T.; Jee, S.; Perkins, S.L.; Aparicio, C.; Douglas, E.P.; Gower, L.B. Biomimetic mineralization of woven bone-like nanocomposites: Role of collagen cross-links. Biomacromolecules 2012, 13, 49–59. [Google Scholar] [CrossRef]
- Scott, J.E.; Tenni, R. Osteogenesis imperfecta mutations may probe vital functional domains (e.g., proteoglycan binding sites) of type 1 collagen fibrils. Cell Biochem. Funct. 1997, 15, 283–286. [Google Scholar] [CrossRef]
- Byers, P.H.; Wallis, G.A.; Willing, M.C. Osteogenesis imperfecta: Translation of mutation to phenotype. J. Med. Genet. 1991, 28, 433–442. [Google Scholar] [CrossRef]
- Marini, J.C.; Lewis, M.B.; Wang, Q.; Chen, K.J.; Orrison, B.M. Serine for glycine substitutions in type I collagen in two cases of type IV osteogenesis imperfecta (OI). Additional evidence for a regional model of OI pathophysiology. J. Biol. Chem. 1993, 268, 2667–2673. [Google Scholar] [PubMed]
- Orgel, J.P.; Wess, T.J.; Miller, A. The in situ conformation and axial location of the intermolecular cross-linked non-helical telopeptides of type I collagen. Structure 2000, 8, 137–142. [Google Scholar] [CrossRef]
- Barrea, R.A.; Gore, D.; Kondrashkina, E.; Weng, T.; Heurich, R.; Vukonich, M.; Orgel, J.; Davidson, M.; Collingwood, J.F.; Mikhaylova, A.; et al. In The bioCAT microprobe for X-ray fluorescence imaging, microxafs and microdiffraction studies on biological samples. In Proceedings of the 8th International Conference on X-ray Microscopy, Himeji, Japan, 26–30 July 2005. [Google Scholar]
- Madhurapantula, R.S.; Krell, G.; Morfin, B.; Roy, R.; Lister, K.; Orgel, J. Advanced Methodology and Preliminary Measurements of Molecular and Mechanical Properties of Heart Valves under Dynamic Strain. Int. J. Mol. Sci. 2020, 21, 763. [Google Scholar] [CrossRef]
- Rainey, J.K.; Goh, M.C. A statistically derived parameterization for the collagen triple-helix. Protein Sci. 2002, 11, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Christopher, J.A.; Swanson, R.; Baldwin, T.O. Algorithms for finding the axis of a helix: Fast rotational and parametric-least squared methods. Comput. Chem. 1996, 20, 339. [Google Scholar] [CrossRef]
- Orgel, J.P.; Antipova, O.; Sagi, I.; Bitler, A.; Qiu, D.; Wang, R.; Xu, Y.; San Antonio, J.D. Collagen fibril surface displays a constellation of sites capable of promoting fibril assembly, stability, and hemostasis. Connect. Tissue Res. 2011, 52, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.A.; Onley, D.J.; Jarvis, G.E.; O’Connor, M.N.; Knight, C.G.; Herr, A.B.; Ouwehand, W.H.; Farndale, R.W. Structural basis for the platelet-collagen interaction: The smallest motif within collagen that recognizes and activates platelet Glycoprotein VI contains two glycine-proline-hydroxyproline triplets. J. Biol. Chem. 2007, 282, 1296–1304. [Google Scholar] [CrossRef]
- Wienke, D.; MacFadyen, J.R.; Isacke, C.M. Identification and characterization of the endocytic transmembrane glycoprotein Endo180 as a novel collagen receptor. Mol. Biol. Cell 2003, 14, 3592–3604. [Google Scholar] [CrossRef]
- Horii, K.; Kahn, M.L.; Herr, A.B. Structural basis for platelet collagen responses by the immune-type receptor glycoprotein VI. Blood 2006, 108, 936–942. [Google Scholar] [CrossRef]
- Haywood, J.; Qi, J.; Chen, C.C.; Lu, G.; Liu, Y.; Yan, J.; Shi, Y.; Gao, G.F. Structural basis of collagen recognition by human osteoclast-associated receptor and design of osteoclastogenesis inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 1038–1043. [Google Scholar] [CrossRef]
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006, 127, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J.; Fertala, A. Inhibition of the self-assembly of collagen I into fibrils with synthetic peptides. Demonstration that assembly is driven by specific binding sites on the monomers. J. Biol. Chem. 1998, 273, 15598–15604. [Google Scholar] [CrossRef]
- Rosenberg, K.; Olsson, H.; Morgelin, M.; Heinegard, D. Cartilage oligomeric matrix protein shows high affinity zinc-dependent interaction with triple helical collagen. J. Biol. Chem. 1998, 273, 20397–20403. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, A.; Okano-Kosugi, H.; Yamazaki, C.M.; Koide, T. Pigment epithelium-derived factor (PEDF) shares binding sites in collagen with heparin/heparan sulfate proteoglycans. J. Biol. Chem. 2011, 286, 26364–26374. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Madhurapantula, R.S.; Kalyanasundaram, A.; Sabharwal, T.; Antipova, O.; Bishnoi, S.W.; Orgel, J. Ultrastructural Location and Interactions of the Immunoglobulin Receptor Binding Sequence within Fibrillar Type I Collagen. Int. J. Mol. Sci. 2020, 21, 4166. [Google Scholar] [CrossRef] [PubMed]
- Orgel, J.P.; Eid, A.; Antipova, O.; Bella, J.; Scott, J.E. Decorin core protein (decoron) shape complements collagen fibril surface structure and mediates its binding. PLoS ONE 2009, 4, e7028. [Google Scholar] [CrossRef]
- Perumal, S.; Antipova, O.; Orgel, J.P. Collagen fibril architecture, domain organization, and triple-helical conformation govern its proteolysis. Proc. Natl. Acad. Sci. USA 2008, 105, 2824–2829. [Google Scholar] [CrossRef]
- Orgel, J.P.; San Antonio, J.D.; Antipova, O. Molecular and structural mapping of collagen fibril interactions. Connect. Tissue Res. 2011, 52, 2–17. [Google Scholar] [CrossRef]
- Ames, J.J.; Contois, L.; Caron, J.M.; Tweedie, E.; Yang, X.; Friesel, R.; Vary, C.; Brooks, P.C. Identification of an Endogenously Generated Cryptic Collagen Epitope (XL313) That May Selectively Regulate Angiogenesis by an Integrin Yes-associated Protein (YAP) Mechano-transduction Pathway. J. Biol. Chem. 2016, 291, 2731–2750. [Google Scholar] [CrossRef]
- Postlethwaite, A.E.; Kang, A.H. Collagen-and collagen peptide-induced chemotaxis of human blood monocytes. J. Exp. Med. 1976, 143, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.J.; Bhatnagar, R.S. Enhanced cell attachment to anorganic bone mineral in the presence of a synthetic peptide related to collagen. J. Biomed. Mater. Res. 1996, 31, 545–554. [Google Scholar] [CrossRef]
- Rong, Y.H.; Zhang, G.A.; Wang, C.; Ning, F.G. Quantification of type I and III collagen content in normal human skin in different age groups. Zhonghua Shao Shang Za Zhi 2008, 24, 51–53. [Google Scholar] [PubMed]
- Ottani, V.; Raspanti, M.; Ruggeri, A. Collagen structure and functional implications. Micron 2001, 32, 251–260. [Google Scholar] [CrossRef]
- Lehto, M.; Sims, T.J.; Bailey, A.J. Skeletal muscle injury--molecular changes in the collagen during healing. Res. Exp. Med. 1985, 185, 95–106. [Google Scholar] [CrossRef]
- Oliveira, H.C.; Popi, A.F.; Bachi, A.L.; Nonogaki, S.; Lopes, J.D.; Mariano, M. B-1 cells modulate the kinetics of wound-healing process in mice. Immunobiology 2010, 215, 215–222. [Google Scholar] [CrossRef]
- Leeming, D.J.; Karsdal, M.A. Type V Collagen. In Biochemistry of Collagens, Laminin, and Elastin; Karsdal, M., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 51–57. [Google Scholar]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Brinckmann, J.; Notbohm, H.; Tronnier, M.; Acil, Y.; Fietzek, P.P.; Schmeller, W.; Muller, P.K.; Batge, B. Overhydroxylation of lysyl residues is the initial step for altered collagen cross-links and fibril architecture in fibrotic skin. J. Investig. Dermatol. 1999, 113, 617–621. [Google Scholar] [CrossRef][Green Version]
- Pingel, J.; Lu, Y.; Starborg, T.; Fredberg, U.; Langberg, H.; Nedergaard, A.; Weis, M.; Eyre, D.; Kjaer, M.; Kadler, K.E. 3-D ultrastructure and collagen composition of healthy and overloaded human tendon: Evidence of tenocyte and matrix buckling. J. Anat. 2014, 224, 548–555. [Google Scholar] [CrossRef]
- Parkin, J.D.; San Antonio, J.D.; Persikov, A.V.; Dagher, H.; Dalgleish, R.; Jensen, S.T.; Jeunemaitre, X.; Savige, J. The collαgen III fibril has a “flexi-rod” structure of flexible sequences interspersed with rigid bioactive domains including two with hemostatic roles. PLoS ONE 2017, 12, e0175582. [Google Scholar] [CrossRef]
- Persikov, A.V.; Brodsky, B. Unstable molecules form stable tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 1101–1103. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.; Brodsky, B. Collagen model peptides: Sequence dependence of triple-helix stability. Biopolymers 2000, 55, 436–450. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.; Brodsky, B. Prediction of collagen stability from amino acid sequence. J. Biol. Chem. 2005, 280, 19343–19349. [Google Scholar] [CrossRef] [PubMed]
- Werkmeister, J.A.; Ramshaw, J.A.; Ellender, G. Characterisation of a monoclonal antibody against native human type I collagen. Eur. J. Biochem. 1990, 187, 439–443. [Google Scholar] [CrossRef]
- Boraschi-Diaz, I.; Wang, J.; Mort, J.S.; Komarova, S.V. Collagen type I as a ligand for receptor-mediated signaling. Front. Phys. 2017, 5. [Google Scholar] [CrossRef]
- Chung, H.J.; Steplewski, A.; Chung, K.Y.; Uitto, J.; Fertala, A. Collagen fibril formation. A new target to limit fibrosis. J. Biol. Chem. 2008, 283, 25879–25886. [Google Scholar] [CrossRef]
- Steplewski, A.; Fertala, J.; Beredjiklian, P.K.; Abboud, J.A.; Wang, M.L.Y.; Namdari, S.; Barlow, J.; Rivlin, M.; Arnold, W.V.; Kostas, J.; et al. Blocking collagen fibril formation in injured knees reduces flexion contracture in a rabbit model. J. Orthop. Res. 2017, 35, 1038–1046. [Google Scholar] [CrossRef]
- Putman, R.K.; Hatabu, H.; Araki, T.; Gudmundsson, G.; Gao, W.; Nishino, M.; Okajima, Y.; Dupuis, J.; Latourelle, J.C.; Cho, M.H.; et al. Association Between Interstitial Lung Abnormalities and All-Cause Mortality. JAMA 2016, 315, 672–681. [Google Scholar] [CrossRef]
- Lim, X.; Tateya, I.; Tateya, T.; Munoz-Del-Rio, A.; Bless, D.M. Immediate inflammatory response and scar formation in wounded vocal folds. Ann. Otol. Rhinol. Laryngol. 2006, 115, 921–929. [Google Scholar] [CrossRef]
- Fertala, J.; Rivlin, M.; Wang, M.L.; Beredjiklian, P.K.; Steplewski, A.; Fertala, A. Collagen-rich deposit formation in the sciatic nerve after injury and surgical repair: A study of collagen-producing cells in a rabbit model. Brain Behav. 2020, e01802. [Google Scholar] [CrossRef]
- McKay, T.B.; Hutcheon, A.E.K.; Zieske, J.D. Biology of corneal fibrosis: Soluble mediators, integrins, and extracellular vesicles. Eye 2020, 34, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Ihn, H. Scleroderma, fibroblasts, signaling, and excessive extracellular matrix. Curr. Rheumatol. Rep. 2005, 7, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Bateman, E.D.; Turner-Warwick, M.; Adelmann-Grill, B.C. Immunohistochemical study of collagen types in human foetal lung and fibrotic lung disease. Thorax 1981, 36, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Shayegan, M.; Altindal, T.; Kiefl, E.; Forde, N.R. Intact Telopeptides Enhance Interactions between Collagens. Biophys. J. 2016, 111, 2404–2416. [Google Scholar] [CrossRef]
- Fertala, J.; Kostas, J.; Hou, C.; Steplewski, A.; Beredjiklian, P.; Abboud, J.; Arnold, W.V.; Williams, G.; Fertala, A. Testing the anti-fibrotic potential of the single-chain Fv antibody against the alpha2 C-terminal telopeptide of collagen I. Connect. Tissue Res. 2014, 55, 115–122. [Google Scholar] [CrossRef]
- Tomasini-Johansson, B.R.; Zbyszynski, P.W.; Toraason, I.; Peters, D.M.; Kwon, G.S. PEGylated pUR4/FUD peptide inhibitor of fibronectin fibrillogenesis decreases fibrosis in murine Unilateral Ureteral Obstruction model of kidney disease. PLoS ONE 2018, 13, e0205360. [Google Scholar] [CrossRef]
- Rasheeda, K.; Muvva, C.; Fathima, N.N. Governing the Inhibition of Reconstituted Collagen Type I Assemblies Mediated Through Noncovalent Forces of (+/-)-alpha Lipoic Acid. Langmuir 2019, 35, 980–989. [Google Scholar] [CrossRef]
- Rasheeda, K.; Fathima, N.N. Trigonelline hydrochloride: A promising inhibitor for type I collagen fibrillation. Colloids Surf. B Biointerfaces 2018, 170, 273–279. [Google Scholar] [CrossRef]
- Knuppel, L.; Ishikawa, Y.; Aichler, M.; Heinzelmann, K.; Hatz, R.; Behr, J.; Walch, A.; Bachinger, H.P.; Eickelberg, O.; Staab-Weijnitz, C.A. A Novel Antifibrotic Mechanism of Nintedanib and Pirfenidone. Inhibition of Collagen Fibril Assembly. Am. J. Respir. Cell Mol. Biol. 2017, 57, 77–90. [Google Scholar] [CrossRef]
- Romanic, A.M.; Adachi, E.; Kadler, K.E.; Hojima, Y.; Prockop, D.J. Copolymerization of pNcollagen III and collagen I. pNcollagen III decreases the rate of incorporation of collagen I into fibrils, the amount of collagen I incorporated, and the diameter of the fibrils formed. J. Biol. Chem. 1991, 266, 12703–12709. [Google Scholar]
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type V collagen controls the initiation of collagen fibril assembly. J. Biol. Chem. 2004, 279, 53331–53337. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Stromblad, S.; Cheresh, D.A. Cell adhesion and angiogenesis. Trends Cell Biol. 1996, 6, 462–468. [Google Scholar] [CrossRef]
- Eliceiri, B.P.; Cheresh, D.A. The role of alphav integrins during angiogenesis: Insights into potential mechanisms of action and clinical development. J. Clin. Investig. 1999, 103, 1227–1230. [Google Scholar] [CrossRef]
- Levene, C.I.; Bartlet, C.; Heale, G. Phenotypic changes in morphology and collagen polymorphism of cultured bovine and porcine aortic endothelium. Atherosclerosis 1984, 52, 59–71. [Google Scholar] [CrossRef]
- Cotta-Pereira, G.; Sage, H.; Bornstein, P.; Ross, R.; Schwartz, S. Studies of morphologically atypical (“sprouting”) cultures of bovine aortic endothelial cells. Growth characteristics and connective tissue protein synthesis. J. Cell Physiol. 1980, 102, 183–191. [Google Scholar] [CrossRef]
- Fouser, L.; Iruela-Arispe, L.; Bornstein, P.; Sage, E.H. Transcriptional activity of the alpha 1(I)-collagen promoter is correlated with the formation of capillary-like structures by endothelial cells in vitro. J. Biol. Chem. 1991, 266, 18345–18351. [Google Scholar]
- Iruela-Arispe, M.L.; Hasselaar, P.; Sage, H. Differential expression of extracellular proteins is correlated with angiogenesis in vitro. Lab. Investig. 1991, 64, 174–186. [Google Scholar]
- Nicosia, R.F.; Ottinetti, A. Growth of microvessels in serum-free matrix culture of rat aorta. A quantitative assay of angiogenesis in vitro. Lab. Investig. 1990, 63, 115–122. [Google Scholar]
- Ingber, D.; Folkman, J. Inhibition of angiogenesis through modulation of collagen metabolism. Lab. Investig. 1988, 59, 44–51. [Google Scholar]
- Sweeney, S.M.; DiLullo, G.; Slater, S.J.; Martinez, J.; Iozzo, R.V.; Lauer-Fields, J.L.; Fields, G.B.; San Antonio, J.D. Angiogenesis in collagen I requires alpha2beta1 ligation of a GFP*GER sequence and possibly p38 MAPK activation and focal adhesion disassembly. J. Biol. Chem. 2003, 278, 30516–30524. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.R.; Adams, C.; Staelens, S.; Deckmyn, H.; San Antonio, J. Crucial Role for Endothelial Cell alpha2beta1 Integrin Receptor Clustering in Collagen-Induced Angiogenesis. Anat. Rec. 2020, 303, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Seandel, M.; Noack-Kunnmann, K.; Zhu, D.; Aimes, R.T.; Quigley, J.P. Growth factor-induced angiogenesis in vivo requires specific cleavage of fibrillar type I collagen. Blood 2001, 97, 2323–2332. [Google Scholar] [CrossRef] [PubMed]
- Chlenski, A.; Liu, S.; Baker, L.J.; Yang, Q.; Tian, Y.; Salwen, H.R.; Cohn, S.L. Neuroblastoma angiogenesis is inhibited with a folded synthetic molecule corresponding to the epidermal growth factor-like module of the follistatin domain of SPARC. Cancer Res. 2004, 64, 7420–7425. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chlenski, A.; Liu, S.; Guerrero, L.J.; Yang, Q.; Tian, Y.; Salwen, H.R.; Zage, P.; Cohn, S.L. SPARC expression is associated with impaired tumor growth, inhibited angiogenesis and changes in the extracellular matrix. Int. J. Cancer 2006, 118, 310–316. [Google Scholar] [CrossRef]
- Jarvelainen, H.; Sainio, A.; Wight, T.N. Pivotal role for decorin in angiogenesis. Matrix Biol. 2015, 43, 15–26. [Google Scholar] [CrossRef]
- Mirochnik, Y.; Aurora, A.; Schulze-Hoepfner, F.T.; Deabes, A.; Shifrin, V.; Beckmann, R.; Polsky, C.; Volpert, O.V. Short pigment epithelial-derived factor-derived peptide inhibits angiogenesis and tumor growth. Clin. Cancer Res. 2009, 15, 1655–1663. [Google Scholar] [CrossRef]
- Kotch, F.W.; Raines, R.T. Self-assembly of synthetic collagen triple helices. Proc. Natl. Acad. Sci. USA 2006, 103, 3028–3033. [Google Scholar] [CrossRef]
- O’Leary, L.E.; Fallas, J.A.; Bakota, E.L.; Kang, M.K.; Hartgerink, J.D. Multi-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel. Nat. Chem. 2011, 3, 821–828. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Homo sapiens * | GGGYDFGYDGDFYRA |
| Oryctolagus cuniculus * | GGGYDFGYDGDFYRA |
| Macaca nemestrina * | GGGYDFGYDGDFYRA |
| Rattus norvegicus | GGGYDFGFEGGFYRA |
| Mus musculus | GGGYDFGFEGDFYRA |
| Gallus gallus | GGGYEVGFDAEYYRA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
San Antonio, J.D.; Jacenko, O.; Fertala, A.; Orgel, J.P.R.O. Collagen Structure-Function Mapping Informs Applications for Regenerative Medicine. Bioengineering 2021, 8, 3. https://doi.org/10.3390/bioengineering8010003
San Antonio JD, Jacenko O, Fertala A, Orgel JPRO. Collagen Structure-Function Mapping Informs Applications for Regenerative Medicine. Bioengineering. 2021; 8(1):3. https://doi.org/10.3390/bioengineering8010003
Chicago/Turabian StyleSan Antonio, James D., Olena Jacenko, Andrzej Fertala, and Joseph P.R.O. Orgel. 2021. "Collagen Structure-Function Mapping Informs Applications for Regenerative Medicine" Bioengineering 8, no. 1: 3. https://doi.org/10.3390/bioengineering8010003
APA StyleSan Antonio, J. D., Jacenko, O., Fertala, A., & Orgel, J. P. R. O. (2021). Collagen Structure-Function Mapping Informs Applications for Regenerative Medicine. Bioengineering, 8(1), 3. https://doi.org/10.3390/bioengineering8010003