Brain Organoids—A Bottom-Up Approach for Studying Human Neurodevelopment



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Recapitulation of In Vivo Neurodevelopment
2.1. Neuronal Induction and Patterning
2.2. Cortical Expansion and the Subventricular Zone
2.3. Neurogenesis and Cortical Layers Formation
2.4. Neuronal Maturation and Activity
3. Organoids for Neurodevelopmental Disease Modeling
3.1. Genome Engineering of Stem Cells for Organoids
3.2. Modeling Genetic Diseases Associated with Brain Structure
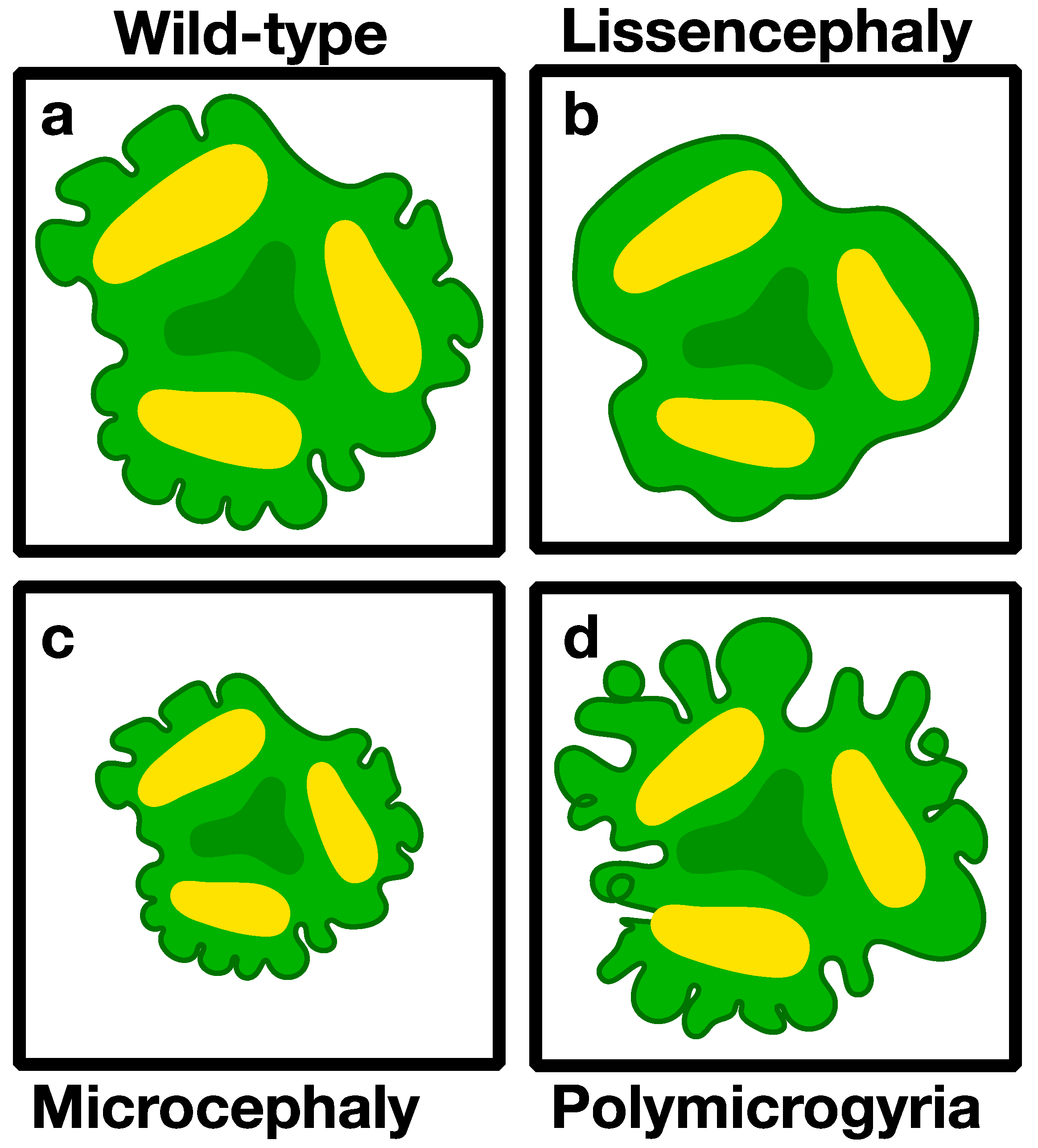
3.2.1. Microcephaly (Small Brains)
3.2.2. Macrocephaly (Large Brains)
3.2.3. Lissencephaly (Smooth Brain)
4. Bioengineering Challenges and Opportunities in Brain Organoids
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sasai, Y. Cytosystems dynamics in self-organization of tissue architecture. Nature 2013, 493, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Kelava, I.; Lancaster, M.A. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev. Biol. 2016, 420, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P. The rise of three-dimensional human brain cultures. Nature 2018, 553, 437–445. [Google Scholar] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Kelava, I.; Lancaster, M.A. Stem Cell Models of Human Brain Development. Cell Stem Cell 2016, 18, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Quadrato, G.; Brown, J.; Arlotta, P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat. Med. 2016, 22, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Karzbrun, E.; Kshirsagar, A.; Cohen, S.R.; Hanna, J.H.; Reiner, O. Human Brain Organoids on a Chip Reveal the Physics of Folding. Nat. Phys. 2018, 14, 515–522. [Google Scholar] [CrossRef]
- McMahon, J.A.; Takada, S.; Zimmerman, L.B.; Fan, C.M.; Harland, R.M.; McMahon, A.P. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 1998, 12, 1438–1452. [Google Scholar] [CrossRef]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Ten Berge, D.; Koole, W.; Fuerer, C.; Fish, M.; Eroglu, E.; Nusse, R. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 2008, 3, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed]
- Quadrato, G. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S. Apical contractility in growing epithelium supports robust maintenance of smooth curvatures against cell-division-induced mechanical disturbance. J. Biomech. 2013, 46, 1705–1713. [Google Scholar] [CrossRef]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef]
- Sloan, S.A. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 2017, 95, 779–790. [Google Scholar] [CrossRef]
- Qian, X.; Jacob, F.; Song, M.M.; Nguyen, H.N.; Song, H.; Ming, G.L. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 2018, 13, 565–580. [Google Scholar] [CrossRef]
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Kriks, S. Dopamine Neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.J.; Govindaiah, G.; Roselaar, N. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and InterNeuron Migration. Cell Stem Cell 2017, 21, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Sakaguchi, H.; Miyamoto, S.; Takahashi, J. Three-dimensional induction of dorsal, intermediate and ventral spinal cord tissues from human pluripotent stem cells. Development 2018, 145, dev162214. [Google Scholar] [CrossRef] [PubMed]
- Koehler, K.R. Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Nasu, M.; Takata, N.; Danjo, T.; Sakaguchi, H.; Kadoshima, T.; Futaki, S. Robust formation and maintenance of continuous stratified cortical neuroepithelium by laminin-containing matrix in mouse ES cell culture. PLoS ONE 2012, 7, e53024. [Google Scholar] [CrossRef]
- Demers, C.J. Development-on-chip: In vitro neural tube patterning with a microfluidic device. Development 2016, 143, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, E.; Quinkler, T.; de Renzis, S. Guided morphogenesis through optogenetic activation of Rho signalling during early Drosophila embryogenesis. Nat. Commun. 2018, 9, 2366. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Amourda, C.; Zhang, S.; Tolwinski, N.S.; Saunders, T.E. Decoding temporal interpretation of the morphogen Bicoid in the early Drosophila embryo. Elife 2017, 6, e26258. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; Rakic, P. Cortical evolution: Judge the brain by its cover. Neuron 2013, 80, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Rakic, P. Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 2009, 10, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Florio, M.; Huttner, W.B. Neural progenitors, neurogenesis and the evolution of the neocortex. Development 2014, 141, 2182–2194. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. The human brain in numbers: A linearly scaled-up primate brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.; Dicke, U. Evolution of the brain and intelligence. Trends Cogn. Sci. 2005, 9, 250–257. [Google Scholar] [CrossRef]
- Heide, M.; Huttner, W.B.; Mora-Bermudez, F. Brain organoids as models to study human neocortex development and evolution. Curr. Opin. Cell Biol. 2018, 55, 8–16. [Google Scholar] [CrossRef]
- Giandomenico, S.L.; Lancaster, M.A. Probing human brain evolution and development in organoids. Curr. Opin. Cell Biol. 2017, 44, 36–43. [Google Scholar] [CrossRef]
- Workman, A.D. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef]
- Mora-Bermudez, F.; Badsha, F.; Kanton, S.; Camp, J.G.; Vernot, B.; Köhler, K. Differences and similarities between human and chimpanzee neural progenitors during cerebral cortex development. Elife 2016, 5, e18683. [Google Scholar] [CrossRef]
- Fietz, S.A. Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self-renewal. Proc. Natl. Acad. Sci. USA 2012, 109, 11836–11841. [Google Scholar] [CrossRef] [PubMed]
- Otani, T. 2D and 3D Stem Cell Models of Primate Cortical Development Identify Species-Specific Differences in Progenitor Behavior Contributing to Brain Size. Cell Stem Cell 2016, 18, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kirwan, P.; Smith, J.; Robinson, H.P.; Livesey, F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 2012, 15, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Taverna, E.; Gotz, M.; Huttner, W.B. The cell biology of neurogenesis: Toward an understanding of the development and evolution of the neocortex. Annu. Rev. Cell Dev. Biol. 2014, 30, 465–502. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef]
- Dehay, C.; Kennedy, H.; Kosik, K.S. The outer subventricular zone and primate-specific cortical complexification. Neuron 2015, 85, 683–694. [Google Scholar] [CrossRef]
- Bayatti, N.; Moss, J.A.; Sun, L.; Ambrose, P.; Ward, J.F.; Lindsay, S.; Clowry, G.J. A molecular neuroanatomical study of the developing human neocortex from 8 to 17 postconceptional weeks revealing the early differentiation of the subplate and subventricular zone. Cereb. Cortex 2008, 18, 1536–1548. [Google Scholar] [CrossRef]
- Vasung, L.; Lepage, C.; Radoš, M.; Pletikos, M.; Goldman, J.S.; Richiardi, J. Quantitative and Qualitative Analysis of Transient Fetal Compartments during Prenatal Human Brain Development. Front. Neuroanat. 2016, 10, 11. [Google Scholar] [CrossRef]
- Fietz, S.A.; Huttner, W.B. Cortical progenitor expansion, self-renewal and neurogenesis-a polarized perspective. Curr. Opin. Neurobiol. 2011, 21, 23–35. [Google Scholar] [CrossRef]
- Molnar, Z. Evolution of cerebral cortical development. Brain Behav. Evol. 2011, 78, 94–107. [Google Scholar] [CrossRef]
- Thomsen, E.R. Fixed single-cell transcriptomic characterization of human radial glial diversity. Nat. Methods 2016, 13, 87–93. [Google Scholar] [CrossRef]
- Pollen, A.A. Molecular identity of human outer radial glia during cortical development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Mota, B.; Herculano-Houzel, S. BRAIN STRUCTURE. Cortical folding scales universally with surface area and thickness, not number of Neurons. Science 2015, 349, 74–77. [Google Scholar] [CrossRef]
- Sun, T.; Hevner, R.F. Growth and folding of the mammalian cerebral cortex: From molecules to malformations. Nat. Rev. Neurosci. 2014, 15, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Cerda, E.; Mahadevan, L. Geometry and physics of wrinkling. Phys. Rev. Lett. 2003, 90, 074302. [Google Scholar] [CrossRef] [PubMed]
- Tallinen, T. Gyrification from constrained cortical expansion. Proc. Natl. Acad. Sci. USA 2014, 111, 12667–12672. [Google Scholar] [CrossRef]
- Budday, S.; Raybaud, C.; Kuhl, E. A mechanical model predicts morphological abnormalities in the developing human brain. Sci. Rep. 2014, 4, 5644. [Google Scholar] [CrossRef]
- Bayly, P.V.; Okamoto, R.J.; Xu, G.; Shi, Y.; Taber, L.A. A cortical folding model incorporating stress-dependent growth explains gyral wavelengths and stress patterns in the developing brain. Phys. Biol. 2013, 10, 016005. [Google Scholar] [CrossRef]
- Karzbrun, E.; Tshuva, R.Y.; Reiner, O. An On-Chip Method for Long-Term Growth and Real-Time Imaging of Brain Organoids. Curr. Protoc. Cell Biol. 2018, 81, e62. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396. [Google Scholar] [CrossRef]
- Kang, H.J. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.L. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 2014, 83, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat. Neurosci. 2015, 18, 154–161. [Google Scholar] [CrossRef]
- Mariani, J. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef]
- Siegenthaler, J.A. Retinoic acid from the meninges regulates cortical Neuron generation. Cell 2009, 139, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V. Non-epithelial stem cells and cortical inter Neuron production in the human ganglionic eminences. Nat. Neurosci. 2013, 16, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Ma, T. Subcortical origins of human and monkey neocortical interNeurons. Nat. Neurosci. 2013, 16, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Rep. 2017, 8, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, P. Development and function of human cerebral cortex neural networks from pluripotent stem cells in vitro. Development 2015, 142, 3178–3187. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, M. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 2018, 15, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Franke, F.; Jäckel, D.; Dragas, J.; Müller, J.; Radivojevic, M.; Bakkum, D.; Hierlemann, A. High-density microelectrode array recordings and real-time spike sorting for closed-loop experiments: An emerging technology to study neural plasticity. Front. Neural Circuits 2012, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, D.A. Controlling bursting in cortical cultures with closed-loop multi-electrode stimulation. J. Neurosci. 2005, 25, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, K.; Clevers, H. Organoids: Modeling Development and the Stem Cell Niche in a Dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef]
- Quadrato, G.; Arlotta, P. Present and future of modeling human brain development in 3D organoids. Curr. Opin. Cell Biol. 2017, 49, 47–52. [Google Scholar] [CrossRef]
- Takahashi, K. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Cho, S.W. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Mali, P. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, H.; Kadoshima, T.; Soen, M.; Narii, N.; Ishida, Y.; Ohgushi, M. Generation of functional hippocampal Neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 2015, 6, 8896. [Google Scholar] [CrossRef]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Bond, J.; Enard, W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef]
- Megraw, T.L.; Sharkey, J.T.; Nowakowski, R.S. Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol. 2011, 21, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Kaindl, A.M. Many roads lead to primary autosomal recessive microcephaly. Prog. Neurobiol. 2010, 90, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, E. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016, 35, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.W. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 2010, 42, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Thornton, G.K.; Woods, C.G. Primary microcephaly: Do all roads lead to Rome? Trends Genet. 2009, 25, 501–510. [Google Scholar] [CrossRef]
- Parrini, E. Genetic Basis of Brain Malformations. Mol. Syndromol. 2016, 7, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Raff, J.W. Centrioles, centrosomes, and cilia in health and disease. Cell 2009, 139, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Walsh, C.A. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 2004, 44, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Bond, J. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 2005, 37, 353–355. [Google Scholar] [CrossRef]
- Bettencourt-Dias, M. Centrosomes and cilia in human disease. Trends Genet. 2011, 27, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.R.; Kilmartin, J.V.; Gergely, F. CDK5RAP2 functions in centrosome to spindle pole attachment and DNA damage response. J. Cell Biol. 2010, 189, 23–39. [Google Scholar] [CrossRef]
- Bakircioglu, M. The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am. J. Hum. Genet. 2011, 88, 523–535. [Google Scholar] [CrossRef]
- Alkuraya, F.S. Human mutations in NDE1 cause extreme microcephaly with lissencephaly. Am. J. Hum. Genet. 2011, 88, 536–547. [Google Scholar] [CrossRef]
- Bond, J. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 2002, 32, 316–320. [Google Scholar] [CrossRef]
- Bond, J. Protein-truncating mutations in ASPM cause variable reduction in brain size. Am. J. Hum. Genet. 2003, 73, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sun, L.; Fang, A.; Li, P.; Wu, Q.; Wang, X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell 2017, 8, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.F. Katanin p80 Regulates Human Cortical Development by Limiting Centriole and Cilia Number. Neuron 2014, 84, 1240–1257. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Pomp, O.; Shinoda, T.; Toba, S.; Torisawa, T.; Furuta, K.Y. Katanin p80, NuMA and cytoplasmic dynein cooperate to control microtubule dynamics. Sci. Rep. 2017, 7, 39902. [Google Scholar] [CrossRef]
- Lai, E.C. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef]
- Fiddes, I.T.; Lodewijk, G.A.; Mooring, M.; Bosworth, C.M.; Ewing, A.D.; Mantalas, G.L. Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell 2018, 173, 1356–1369. [Google Scholar] [CrossRef]
- Suzuki, I.K.; Gacquer, D.; Van Heurck, R.; Kumar, D.; Wojno, M.; Bilheu, A. Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell 2018, 173, 1370–1384. [Google Scholar] [CrossRef]
- Bizzotto, S.; Walsh, C.A. Making a Notch in the Evolution of the Human Cortex. Dev. Cell 2018, 45, 548–550. [Google Scholar] [CrossRef]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef]
- Lee, D.Y. Roles of mTOR Signaling in Brain Development. Exp. Neurobiol. 2015, 24, 177–185. [Google Scholar] [CrossRef]
- Striano, P.; Zara, F. Genetics: Mutations in mTOR pathway linked to megalencephaly syndromes. Nat. Rev. Neurol. 2012, 8, 542–544. [Google Scholar] [CrossRef] [PubMed]
- San Yeung, K.; Tso, W.W.Y.; Ip, J.J.K.; Mak, C.C.Y.; Leung, G.K.C.; Tsang, M.H.Y. Identification of mutations in the PI3K-AKT-mTOR signalling pathway in patients with macrocephaly and developmental delay and/or autism. Mol. Autism 2017, 8, 66. [Google Scholar] [CrossRef]
- D’Gama, A.M. Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias. Cell Rep. 2017, 21, 3754–3766. [Google Scholar] [CrossRef]
- Butler, M.G. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Marchese, M.; Conti, V.; Valvo, G.; Moro, F.; Muratori, F.; Tancredi, R. Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening. BMC Med. Genet. 2014, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Reiner, O. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 1993, 364, 717–721. [Google Scholar] [CrossRef]
- Reiner, O.; Sapir, T. LIS1 functions in normal development and disease. Curr. Opin. Neurobiol. 2013, 23, 951–956. [Google Scholar] [CrossRef]
- Bershteyn, M. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 2017, 20, 435–449. [Google Scholar] [CrossRef]
- Iefremova, V. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017, 19, 50–59. [Google Scholar] [CrossRef]
- Long, K.R. Extracellular Matrix Components HAPLN1, Lumican, and Collagen I Cause Hyaluronic Acid-Dependent Folding of the Developing Human Neocortex. Neuron 2018, 99, 702–719. [Google Scholar] [CrossRef] [PubMed]
- Yin, X. Engineering Stem Cell Organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y. 3D spherical microtissues and microfluidic technology for multi-tissue experiments and analysis. J. Biotechnol. 2015, 205, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Picollet-D’hahan, N. A 3D Toolbox to Enhance Physiological Relevance of Human Tissue Models. Trends Biotechnol. 2016, 34, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Song, H.G. Vascular Tissue Engineering: Progress, Challenges, and Clinical Promise. Cell Stem Cell 2018, 22, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.G.; Niklason, L.E. A short discourse on vascular tissue engineering. NPJ Regen. Med. 2017, 2, 7. [Google Scholar] [CrossRef]
- Kolesky, D.B. 3D bioprinting of vascularized, heterogeneous cell-laden tissue constructs. Adv. Mater. 2014, 26, 3124–3130. [Google Scholar] [CrossRef] [PubMed]
- Vollert, I. In vitro perfusion of engineered heart tissue through endothelialized channels. Tissue Eng. Part A 2014, 20, 854–863. [Google Scholar] [CrossRef]
- Tang, M.D.; Golden, A.P.; Tien, J. Fabrication of Collagen Gels That Contain Patterned, Micrometer-Scale Cavities. Adv. Mater. 2004, 16, 1345–1348. [Google Scholar] [CrossRef]
- Zhang, B. Biodegradable scaffold with built-in vasculature for organ-on-a-chip engineering and direct surgical anastomosis. Nat. Mater. 2016, 15, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W. Direct 3D bioprinting of prevascularized tissue constructs with complex microarchitecture. Biomaterials 2017, 124, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A. Microfluidic techniques for development of 3D vascularized tissue. Biomaterials 2014, 35, 7308–7325. [Google Scholar] [CrossRef] [PubMed]
- Kolesky, D.B. Three-dimensional bioprinting of thick vascularized tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 3179–3184. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.T. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.M.; Weber, R.J.; Gartner, Z.J. Dissecting the stem cell niche with organoid models: An engineering-based approach. Development 2017, 144, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Todhunter, M.E. Programmed synthesis of three-dimensional tissues. Nat. Methods 2015, 12, 975–981. [Google Scholar] [CrossRef]
- Lee, G.H.; Lee, J.S.; Lee, G.H.; Joung, W.Y.; Kim, S.H.; Lee, S.H. Networked concave microwell arrays for constructing 3D cell spheroids. Biofabrication 2017, 10, 015001. [Google Scholar] [CrossRef]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef]
- Khazipov, R.; Sirota, A.; Leinekugel, X.; Holmes, G.L.; Ben-Ari, Y.; Buzsáki, G. Early motor activity drives spindle bursts in the developing somatosensory cortex. Nature 2004, 432, 758–761. [Google Scholar] [CrossRef]
- Rao, L.; Qian, Y.; Khodabukus, A.; Ribar, T.; Bursac, N. Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat. Commun. 2018, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Happe, C.L.; Tenerelli, K.P.; Gromova, A.K.; Kolb, F.; Engler, A.J. Mechanically patterned neuromuscular junctions-in-a-dish have improved functional maturation. Mol. Biol. Cell 2017, 28, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, C.; Rich, M.H.; Raman, R.; Kong, H.; Bashir, R. A 3D-printed platform for modular neuromuscular motor units. Microsyst. Nanoeng. 2017, 3, 17015. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karzbrun, E.; Reiner, O. Brain Organoids—A Bottom-Up Approach for Studying Human Neurodevelopment. Bioengineering 2019, 6, 9. https://doi.org/10.3390/bioengineering6010009
Karzbrun E, Reiner O. Brain Organoids—A Bottom-Up Approach for Studying Human Neurodevelopment. Bioengineering. 2019; 6(1):9. https://doi.org/10.3390/bioengineering6010009
Chicago/Turabian StyleKarzbrun, Eyal, and Orly Reiner. 2019. "Brain Organoids—A Bottom-Up Approach for Studying Human Neurodevelopment" Bioengineering 6, no. 1: 9. https://doi.org/10.3390/bioengineering6010009
APA StyleKarzbrun, E., & Reiner, O. (2019). Brain Organoids—A Bottom-Up Approach for Studying Human Neurodevelopment. Bioengineering, 6(1), 9. https://doi.org/10.3390/bioengineering6010009