Engineering Pathways in Central Carbon Metabolism Help to Increase Glycan Production and Improve N-Type Glycosylation of Recombinant Proteins in E. coli

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. PCR, DNA Cloning and Vectors
2.2. Cell Surface Representation of Glycans
2.3. Colony Analysis
2.4. Bacterial Growth and Protein Expression
2.5. Periplasmic Protein Extraction
2.6. SDS-PAGE and Western Blot Analysis
3. Results and Discussion
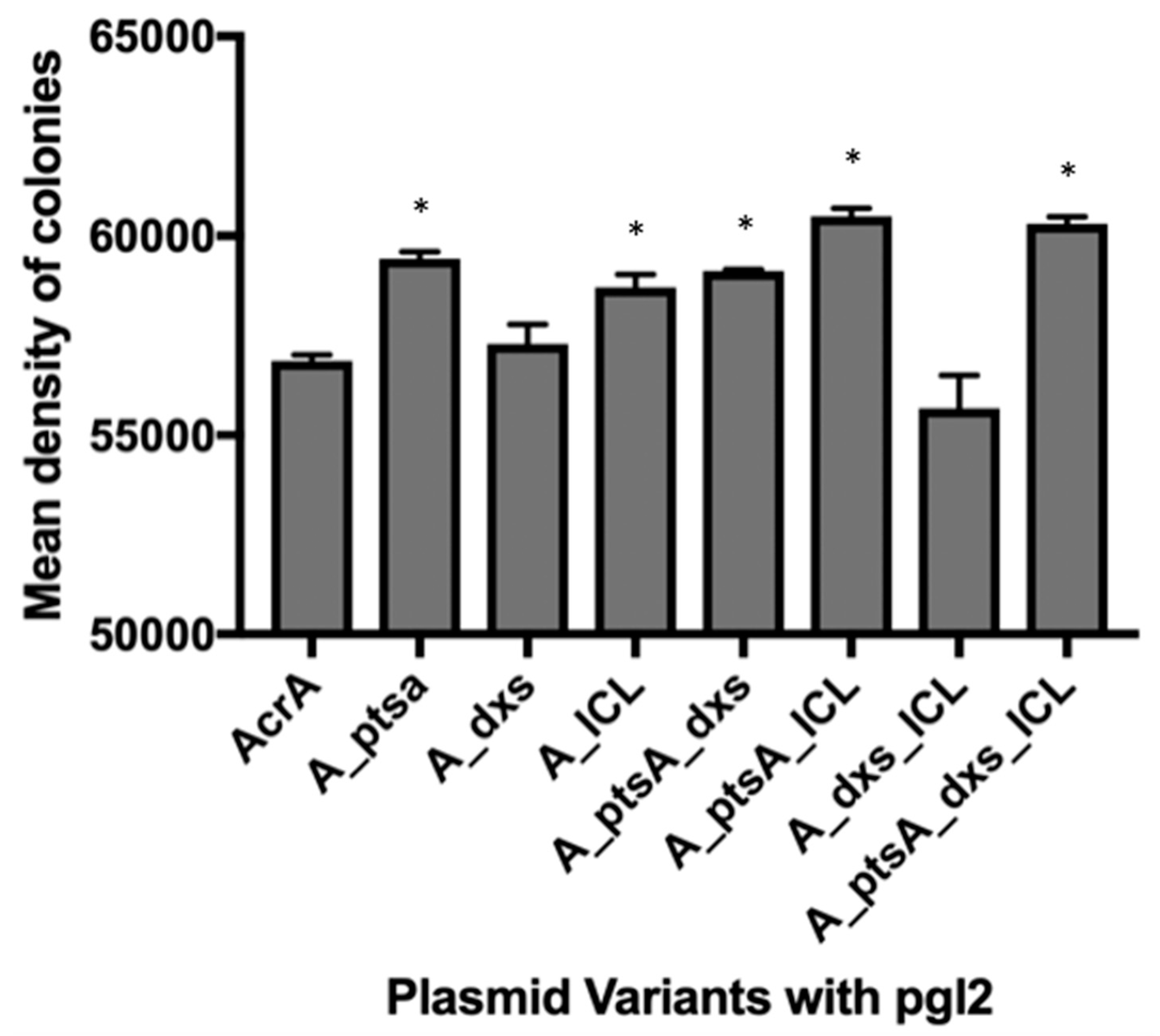
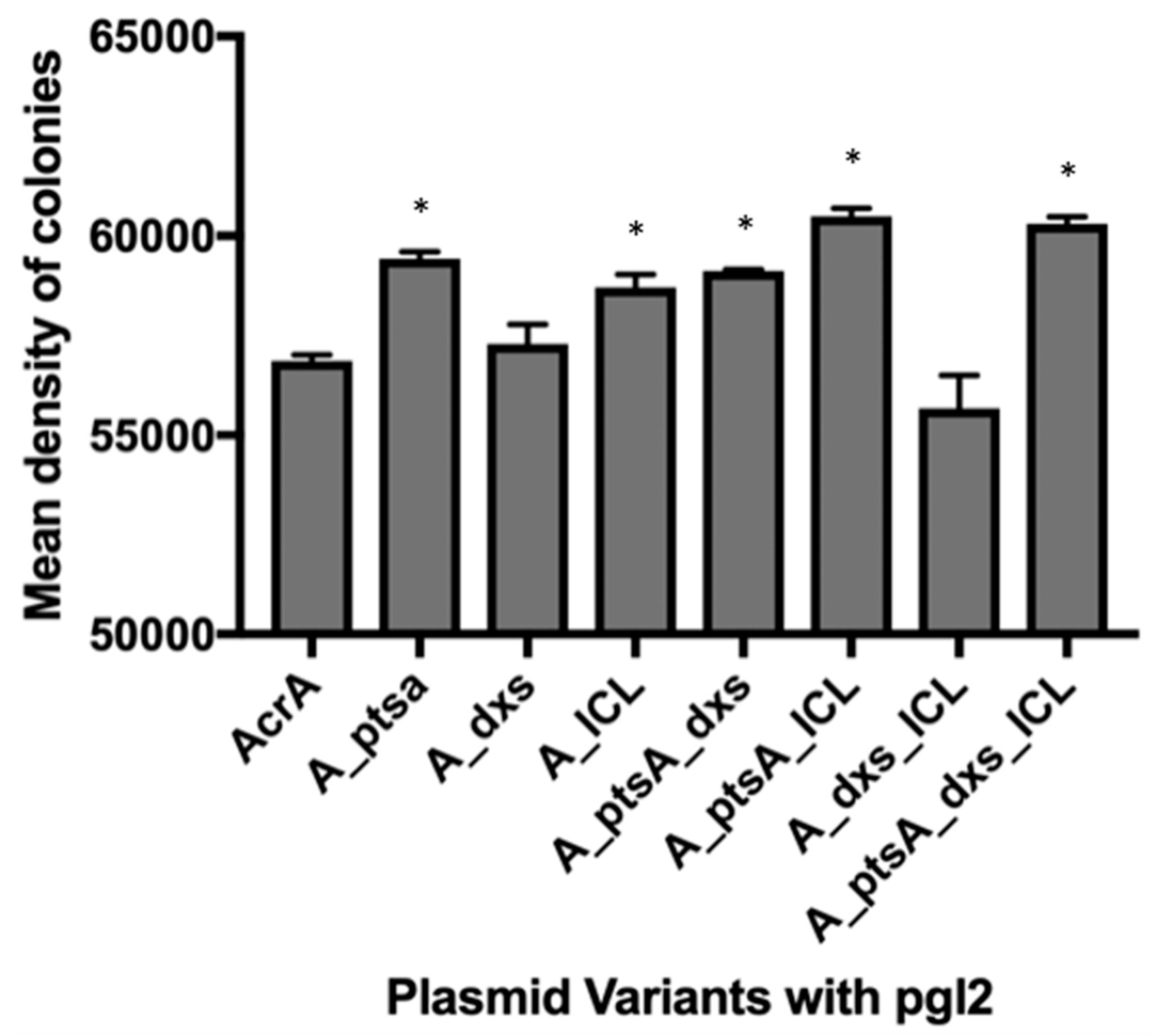
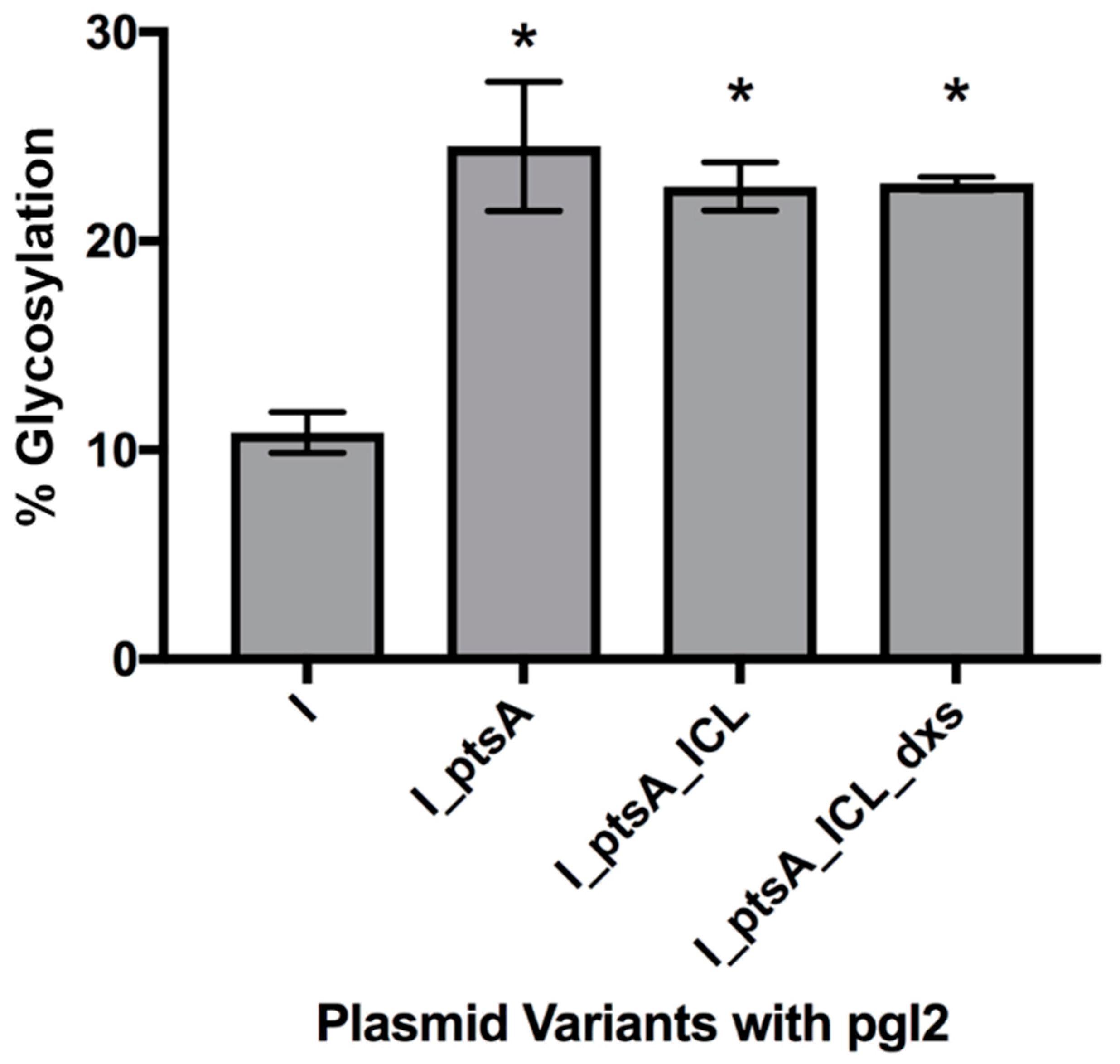
3.1. MC4100 Cell Surface Representation of Glycans
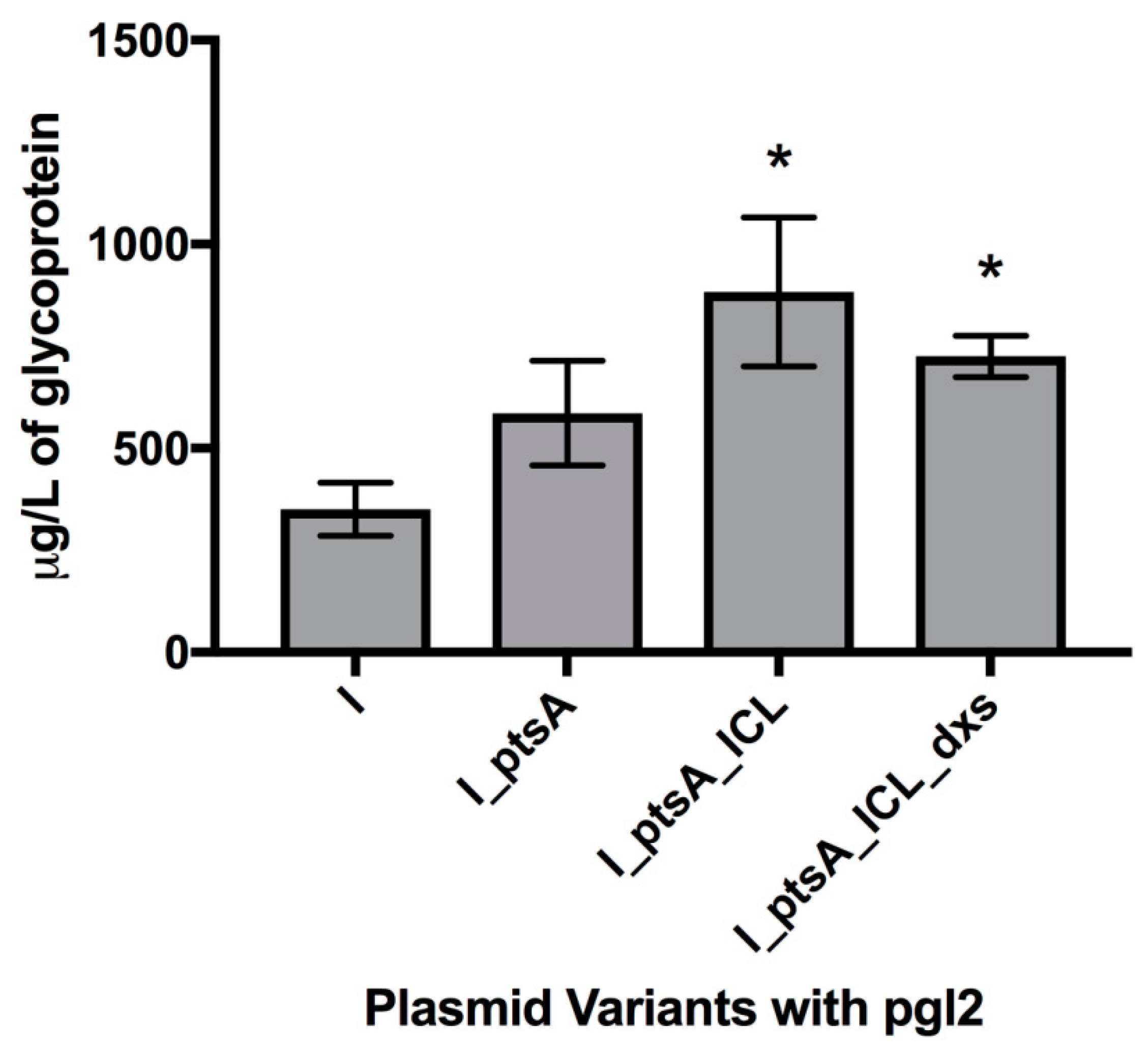
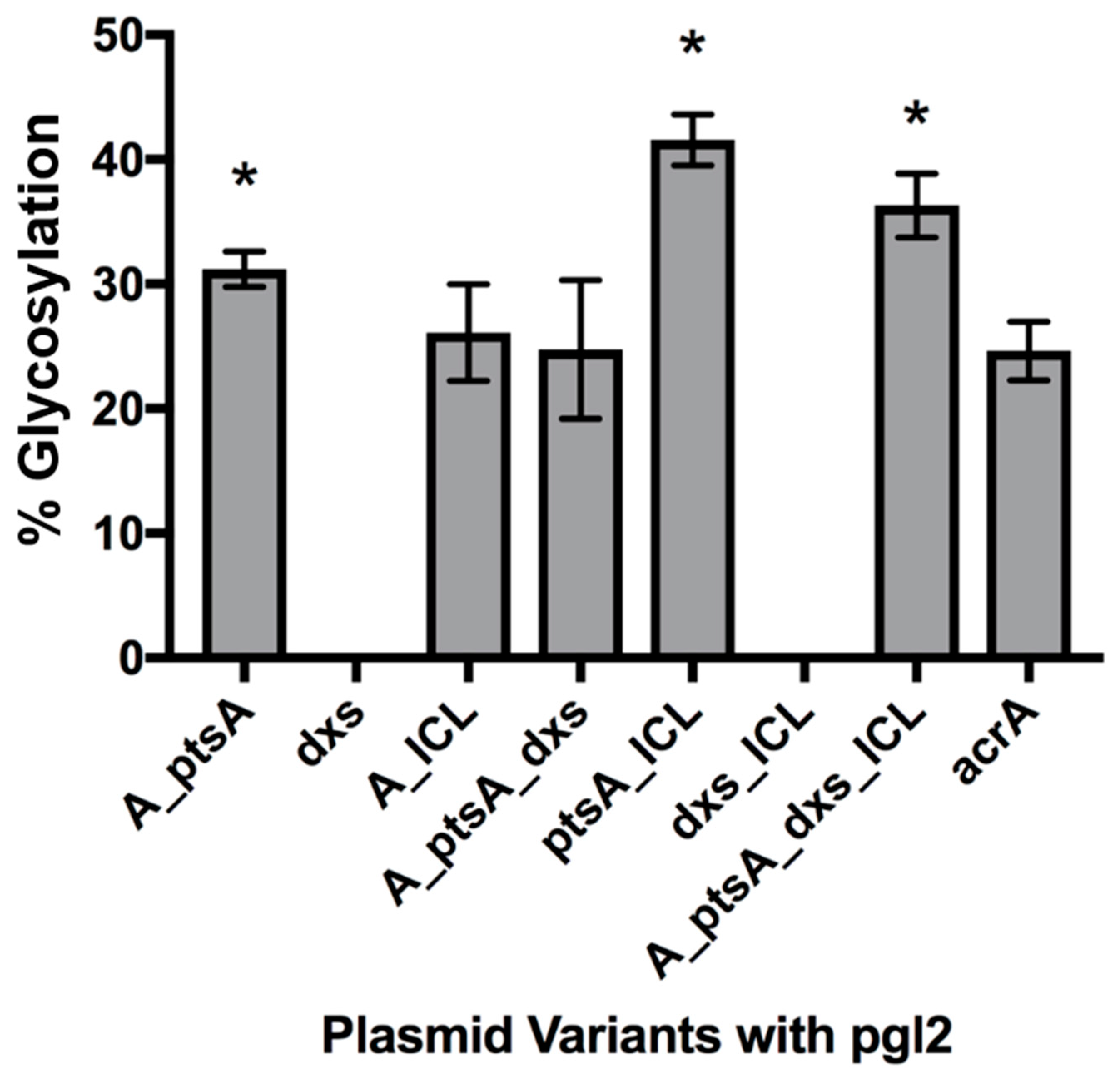
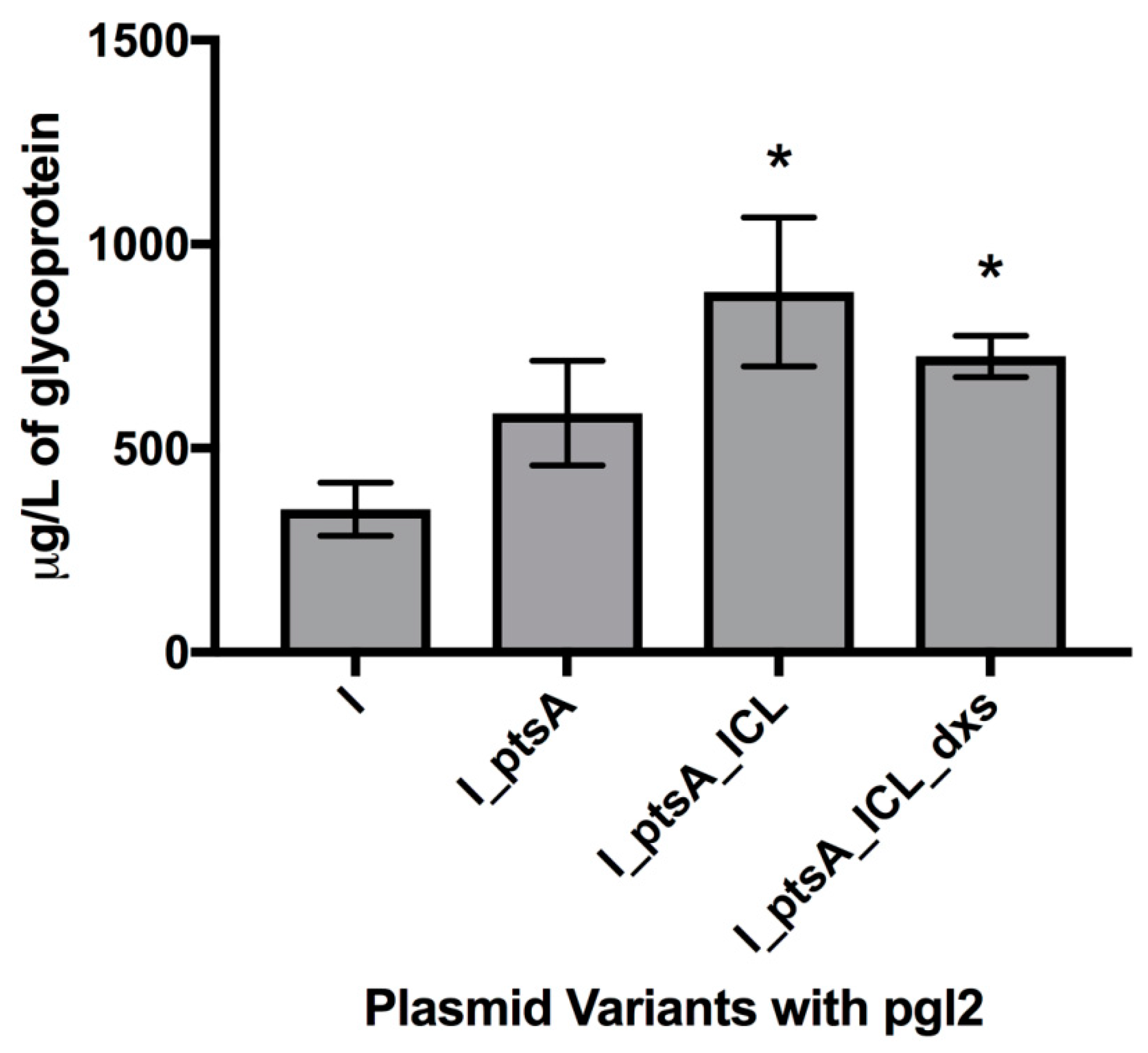
3.2. Measuring Glycoprotein Production Capability
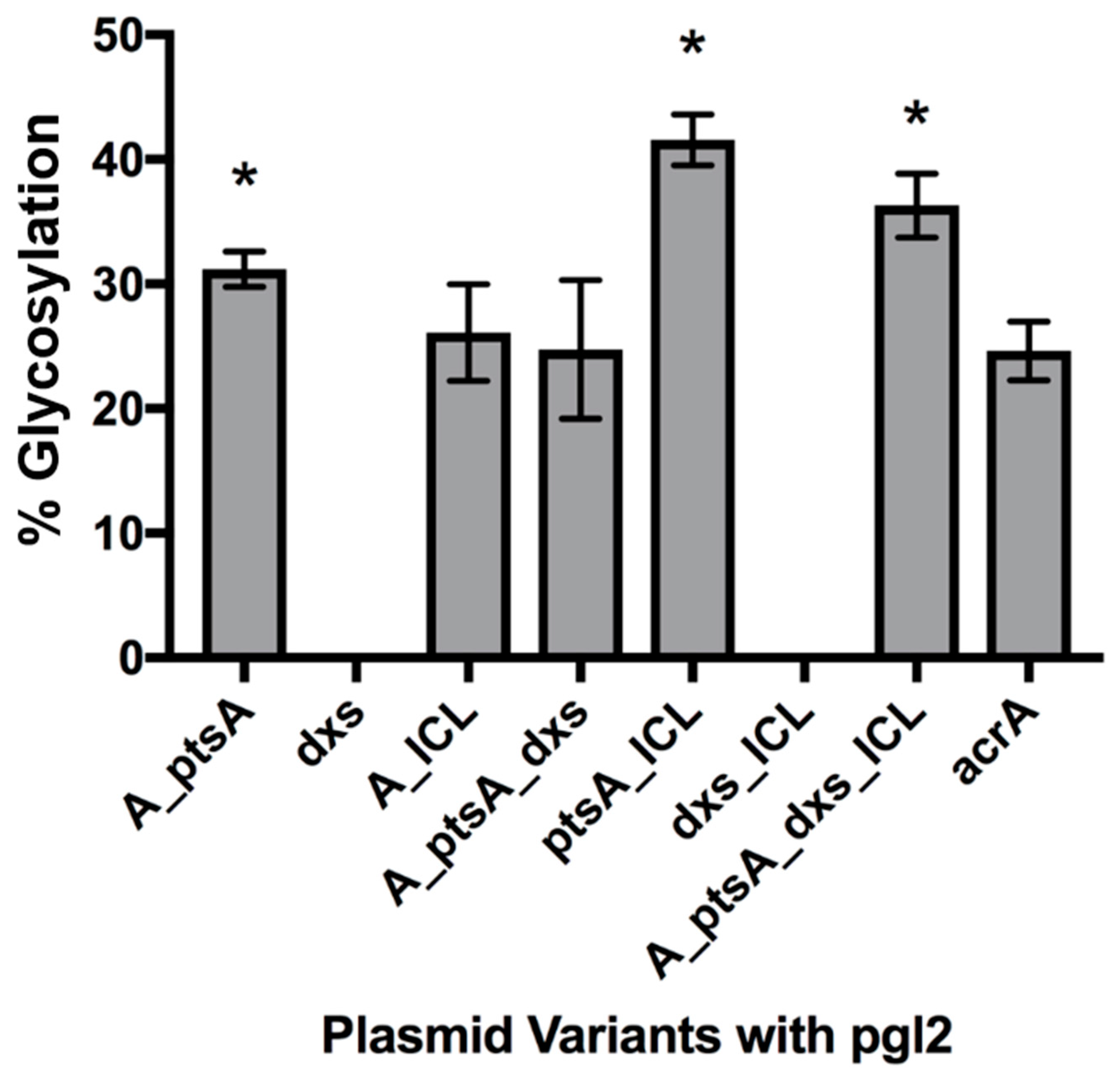
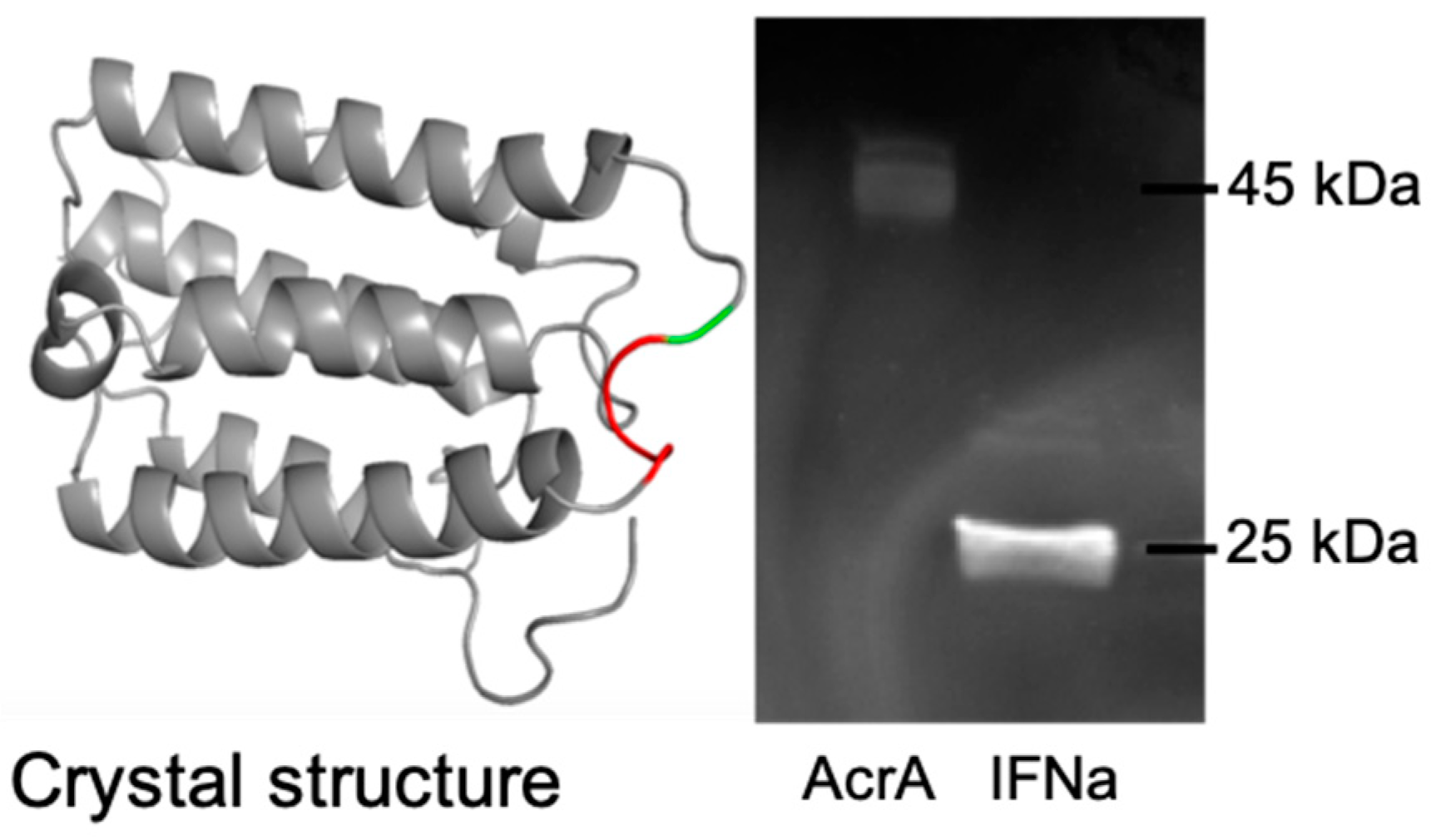
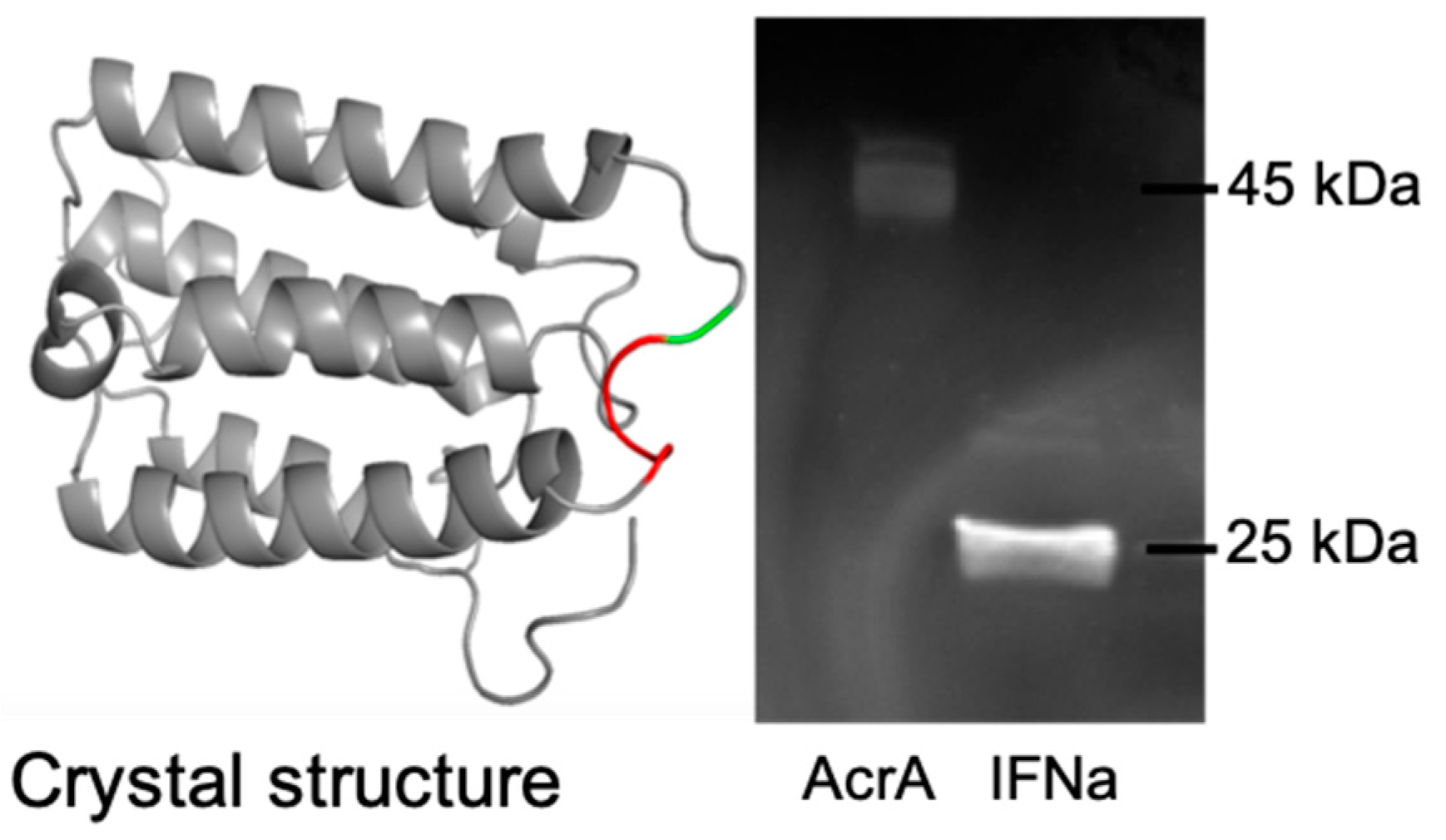
3.2.1. Expression of Glycosylated AcrA in E. coli Cells, CLM24
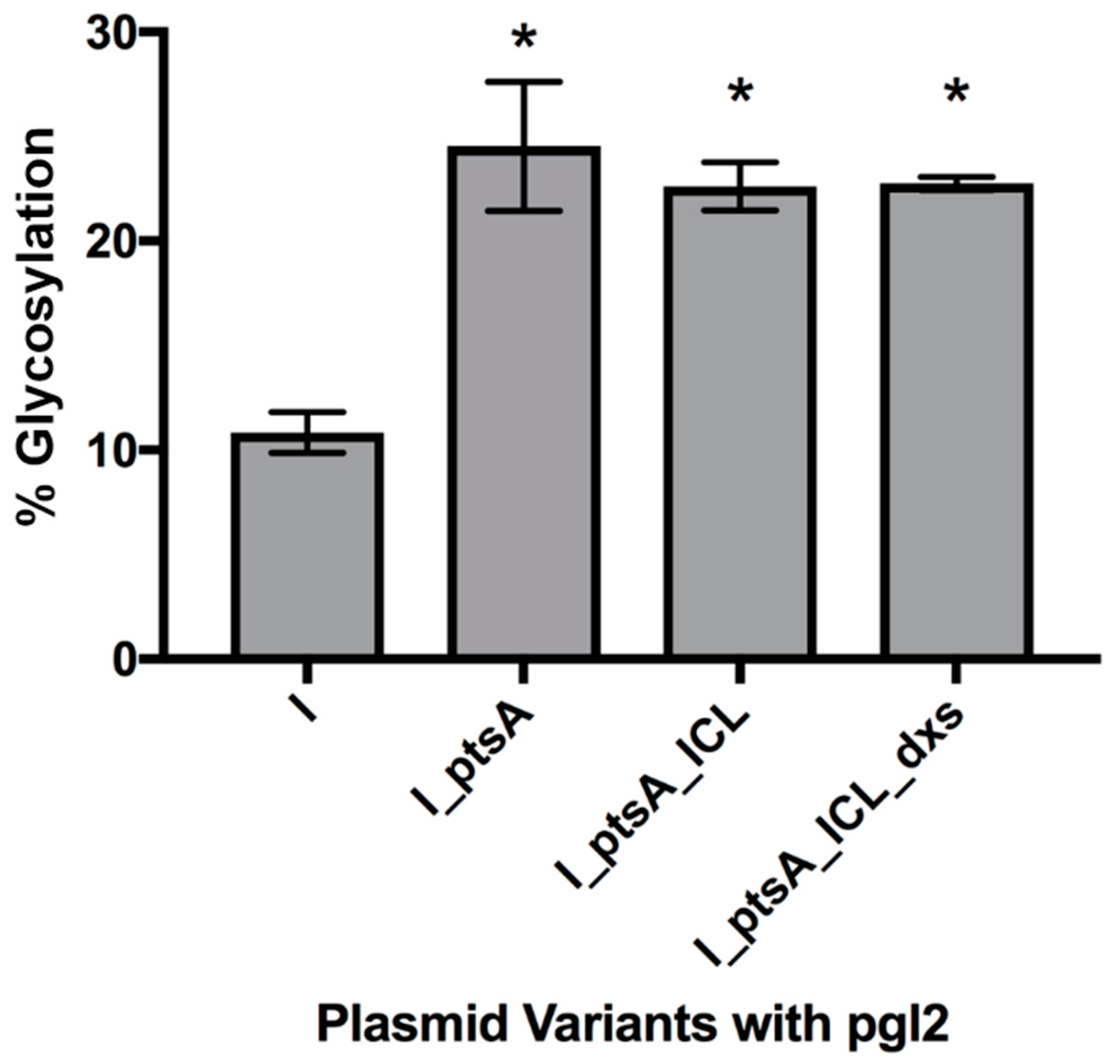
3.2.2. Expression of Glycosylated IFNα2b in E. coli Cells, CLM24
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the swiss-prot database. Biochim. Biophys. Acta Gen. Subj. 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Wagner, S.; Klepsch, M.M.; Schlegel, S.; Appel, A.; Draheim, R.; Tarry, M.; Hogbom, M.; van Wijk, K.J.; Slotboom, D.J.; Persson, J.O.; et al. Tuning Escherichia coli for membrane protein overexpression. Proc. Natl. Acad. Sci. USA 2008, 105, 14371–14376. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.F.R.O.; Robinson, C.; Alanen, H.I.; Prus, P.; Uchida, Y.; Ruddock, L.W.; Freedman, R.B.; Keshavarz-Moore, E. Efficient export of prefolded, disulfide-bonded recombinant proteins to the periplasm by the tat pathway in Escherichia coli cydisco strains. Biotechnol. Prog. 2014, 30, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Hatahet, F.; Nguyen, V.D.; Salo, K.E.H.; Ruddock, L.W. Disruption of reducing pathways is not essential for efficient disulfide bond formation in the cytoplasm of e. Coli. Microb. Cell Fact. 2010, 9, 67. [Google Scholar] [PubMed]
- Gaciarz, A.; Veijola, J.; Uchida, Y.; Saaranen, M.J.; Wang, C.G.; Horkko, S.; Ruddock, L.W. Systematic screening of soluble expression of antibody fragments in the cytoplasm of e. Coli. Microb. Cell Fact. 2016, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Gaciarz, A.; Khatri, N.K.; Velez-Suberbie, M.L.; Saaranen, M.J.; Uchida, Y.; Keshavarz-Moore, E.; Ruddock, L.W. Efficient soluble expression of disulfide bonded proteins in the cytoplasm of Escherichia coli in fed-batch fermentations on chemically defined minimal media. Microb. Cell Fact. 2017, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, C.; Messner, P. Emerging facets of prokaryotic glycosylation. FEMS Microbiol. Rev. 2017, 41, 49–91. [Google Scholar] [CrossRef]
- Nothaft, H.; Szymanski, C.M. Bacterial protein n-glycosylation: New perspectives and applications. J. Biol. Chem. 2013, 288, 6912–6920. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.; Linton, D.; Hitchen, P.G.; Nita-Lazar, M.; Haslam, S.M.; North, S.J.; Panico, M.; Morris, H.R.; Dell, A.; Wren, B.W.; et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into e. Coli. Science 2002, 298, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Pandhal, J.; Woodruff, L.B.A.; Jaffe, S.; Desai, P.; Ow, S.Y.; Noirel, J.; Gill, R.T.; Wright, P.C. Inverse metabolic engineering to improve Escherichia coli as an n-glycosylation host. Biotechnol. Bioeng. 2013, 110, 2482–2493. [Google Scholar] [CrossRef] [PubMed]
- Lizak, C.; Fan, Y.Y.; Weber, T.C.; Aebi, M. N-linked glycosylation of antibody fragments in Escherichia coli. Bioconjug. Chem. 2011, 22, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Valderrama-Rincon, J.D.; Fisher, A.C.; Merritt, J.H.; Fan, Y.Y.; Reading, C.A.; Chhiba, K.; Heiss, C.; Azadi, P.; Aebi, M.; DeLisa, M.P. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat. Chem. Biol. 2012, 8, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Srichaisupakit, A.; Ohashi, T.; Misaki, R.; Fujiyama, K. Production of initial-stage eukaryotic n-glycan and its protein glycosylation in Escherichia coli. J. Biosci. Bioeng. 2015, 119, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Imperiali, B. Bacterial n-glycosylation efficiency is dependent on the structural context of target sequons. J. Biol. Chem. 2016, 291, 22001–22010. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Yang, C.G.; Sun, S.X.; Han, L.C.; Ruan, Y.; Guo, L.H.; Hu, X.J.; Zhang, J.N. Increased glycosylation efficiency of recombinant proteins in Escherichia coli by auto-induction. Biochem. Biophys. Res. Commun. 2017, 485, 138–143. [Google Scholar] [CrossRef]
- Kornfeld, R.; Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985, 54, 631–664. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yan, Q.; Jones, J.A.; Tang, Y.J.J.; Fong, S.S.; Koffas, M.A.G. Metabolic burden: Cornerstones in synthetic biology and metabolic engineering applications. Trends Biotechnol. 2016, 34, 652–664. [Google Scholar] [CrossRef]
- Pandhal, J.; Desai, P.; Walpole, C.; Doroudi, L.; Malyshev, D.; Wright, P.C. Systematic metabolic engineering for improvement of glycosylation efficiency in Escherichia coli. Biochem. Biophys. Res. Commun. 2012, 419, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Strutton, B.; Jaffe, S.; Pandhal, J.; Wright, P.C. Producing a glycosylation host strain of Escherichia coli: The placement of the bacterial oligosaccharyl transferase pglb onto the genome. Biochem. Biophys. Res. Commun. 2017, 495, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Pandhal, J.; Ow, S.Y.; Noirel, J.; Wright, P.C. Improving n-glycosylation efficiency in Escherichia coli using shotgun proteomics, metabolic network analysis, and selective reaction monitoring. Biotechnol. Bioeng. 2011, 108, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Poolman, B.; Knol, J.; Mollet, B.; Nieuwenhuis, B.; Sulter, G. Regulation of bacterial sugar-h+ symport by phosphoenolpyruvate-dependent enzyme i/hpr-mediated phosphorylation. Proc. Natl. Acad. Sci. USA 1995, 92, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Garrett, D.S.; Seok, Y.-J.; Peterkofsky, A.; Clore, G.M.; Gronenborn, A.M. Identification by nmr of the binding surface for the histidine-containing phosphocarrier protein hpr on the n-terminal domain of enzyme i of the Escherichia coli phosphotransferase system. Biochemistry 1997, 36, 4393–4398. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, G.A.; Schörken, U.; Wiegert, T.; Grolle, S.; De Graaf, A.A.; Taylor, S.V.; Begley, T.P.; Bringer-Meyer, S.; Sahm, H. Identification of a thiamin-dependent synthase in Escherichia coli required for the formation of the 1-deoxy-d-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proc. Natl. Acad. Sci. USA 1997, 94, 12857–12862. [Google Scholar] [CrossRef] [PubMed]
- Brammer, L.A.; Smith, J.M.; Wade, H.; Meyers, C.F. 1-deoxy-d-xylulose 5-phosphate synthase catalyzes a novel random sequential mechanism. J. Biol. Chem. 2011, 286, 36522–36531. [Google Scholar] [CrossRef]
- Kuzuyama, T.; Takagi, M.; Takahashi, S.; Seto, H. Cloning and characterization of 1-deoxy-d-xylulose 5-phosphate synthase from streptomyces sp. Strain cl190, which uses both the mevalonate and nonmevalonate pathways for isopentenyl diphosphate biosynthesis. J. Bacteriol. 2000, 182, 891–897. [Google Scholar]
- Feldman, M.F.; Wacker, M.; Hernandez, M.; Hitchen, P.G.; Marolda, C.L.; Kowarik, M.; Morris, H.R.; Dell, A.; Valvano, M.A.; Aebi, M. Engineering n-linked protein glycosylation with diverse o antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. USA 2005, 102, 3016–3021. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, F.; Huang, W.; Li, C.; Schulz, B.; Lizak, C.; Palumbo, A.; Numao, S.; Neri, D.; Aebi, M.; Wang, L. A combined method for producing homogeneous glycoproteins with eukaryotic n-glycosylation. Nat. Chem. Biol. 2010, 6, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Bairamashvili, D.I.; Rabinovich, M.L. Russia through the prism of the world biopharmaceutical market. Biotechnol. J. 2007, 2, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-W.; Keasling, J.D. Metabolic engineering of the nonmevalonate isopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol. Bioeng. 2001, 72, 408–415. [Google Scholar] [CrossRef]
- Alper, H.; Fischer, C.; Nevoigt, E.; Stephanopoulos, G. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. USA 2005, 102, 12678–12683. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, V.E.B.; Maldonado, L.M.T.P.; Rivero, E.M.; de la Rosa, A.P.B.; Jimenez-Bremont, J.F.; Acevedo, L.G.O.; Rodriguez, A.D.L. Periplasmic expression and recovery of human interferon gamma in Escherichia coli. Protein Expr. Purif. 2008, 59, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Morowvat, M.H.; Babaeipour, V.; Rajabi-Memari, H.; Vahidi, H.; Maghsoudi, N. Overexpression of recombinant human beta interferon (rhinf-beta) in periplasmic space of Escherichia coli. Iran. J. Pharm. Res. 2014, 13, 151–160. [Google Scholar] [PubMed]
- Varki, A.; Cummings, R.; Esko, J.; Freeze, H.; Stanley, P.; Bertozzi, C.; Hart, G.; Etzler, M. Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1999. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strutton, B.; Jaffe, S.R.; Evans, C.A.; Fowler, G.J.; Dobson, P.D.; Pandhal, J.; Wright, P.C. Engineering Pathways in Central Carbon Metabolism Help to Increase Glycan Production and Improve N-Type Glycosylation of Recombinant Proteins in E. coli. Bioengineering 2019, 6, 27. https://doi.org/10.3390/bioengineering6010027
Strutton B, Jaffe SR, Evans CA, Fowler GJ, Dobson PD, Pandhal J, Wright PC. Engineering Pathways in Central Carbon Metabolism Help to Increase Glycan Production and Improve N-Type Glycosylation of Recombinant Proteins in E. coli. Bioengineering. 2019; 6(1):27. https://doi.org/10.3390/bioengineering6010027
Chicago/Turabian StyleStrutton, Benjamin, Stephen RP Jaffe, Caroline A Evans, Gregory JS Fowler, Paul D Dobson, Jagroop Pandhal, and Phillip C Wright. 2019. "Engineering Pathways in Central Carbon Metabolism Help to Increase Glycan Production and Improve N-Type Glycosylation of Recombinant Proteins in E. coli" Bioengineering 6, no. 1: 27. https://doi.org/10.3390/bioengineering6010027
APA StyleStrutton, B., Jaffe, S. R., Evans, C. A., Fowler, G. J., Dobson, P. D., Pandhal, J., & Wright, P. C. (2019). Engineering Pathways in Central Carbon Metabolism Help to Increase Glycan Production and Improve N-Type Glycosylation of Recombinant Proteins in E. coli. Bioengineering, 6(1), 27. https://doi.org/10.3390/bioengineering6010027