1. Introduction
Analysis of immunohistochemistry (IHC) images for measuring cell structures and proteins is a tedious, manual, and subjective process. Existing commercial software programs for analysis (such as Neurolucida and Imaris) are very expensive and primarily developed for nuances of analysis in brain tissue. These platforms are difficult to use for isolating somas and analyzing protein expression of motoneurons in the spinal cord, where protein structures are more varied in size and somas are much larger and less-densely packed. Spinal motoneuron protein analysis with existing software tools requires manual cell-by-cell threshold adjustments and cluster-size tuning based on subjective visual interpretations of the cell bodies and protein structures. These manual adjustments are qualitative, subject to bias, and largely untraceable, contributing to lack of transparency and lack of reproducibility of resulting measurements. Additionally, as the number of cells analyzed increases, so does the heavy manual analyzer burden and the difficulty for maintaining consistency in the subjective tuning. As a result, researchers have resorted to custom 2D analysis methods [
1,
2,
3,
4,
5,
6] to ease the analysis burden, more tightly control the approach, and increase the number of cells analyzed. However, these approaches remain time-consuming and inefficient, plagued by similar subjectivity and additional stereological cluster sampling limitations that hinder reproducibility [
2] and reduce sensitivity to detecting biological changes. Critically, small methodological decisions in these analyses can drastically alter study outcomes—even reversing conclusions [
2]. Despite these challenges, obtaining accurate and reproducible measurements is essential for detecting subtle changes in protein expression, which may help uncover previously unknown mechanisms driving neuronal vulnerability, aging, and degeneration.
To meet the demands for analytical efficiency, specificity, objectivity, and reproducibility, we developed an automated pipeline that streamlines key components of IHC image analysis, with an emphasis on spinal tissue analysis. Our algorithm generates 3D reconstructions of motoneuron somas and quantifies 100% of the protein labeling on the somatic membrane. Using Canny edge detection and a custom Cartesian reconstruction approach, the algorithm produces accurate 3D soma reconstructions, enabling highly precise and consistent soma size measurements. These reconstructions also define the precise 3D image coordinates for analyzing total somatic protein expression and for automatically identifying and quantifying all membrane-associated protein clusters. All cluster analysis settings are quantified and automatically tracked, with built-in setting sensitivity analysis and automatic threshold adjustments per cell. In this validation study, we present a series of performance assessments and comparisons to existing methods to evaluate the accuracy and robustness of our approach. Due to the absence of definitive ground-truth labels for protein clusters in these noisy biological images, we prioritized a validation strategy centered on geometric benchmarking, cross-condition consistency, robustness to parameter (mainly threshold) shifts, and reproducibility across users over empirical accuracy metrics for image segmentation. This strategy directly reflects the operational goals of the algorithm in its intended biological context. Although the algorithm is adaptable for analyzing any membrane-associated protein, we used the C-bouton—an example of a macro-clustering synaptic protein—as the primary case study for algorithm validation.
2. Materials and Methods
2.1. Three-Dimensional Cartesian Reconstructions
We used Olympus FV1000 (.oib) 60× IHC (Olympus Corporation, Tokyo, Japan) confocal images of quadruple-labeled mouse lumbar spinal tissue collected for various study controls over the last decade in our lab. In the example macro-analysis for this validation study, we used Nissl (NeuroTrace, Life Technologies, cat #: N21479, Thermo Fisher Scientific, Carlsbad, CA, USA) or NeuN (Millipore-Sigma, cat#ABN90) as our soma label, and VAChT (Millipore-Sigma, cat #: ABN100, Sigma Aldrich, St. Louis, MO, USA) or ChAT (Novus Biologicals, cat #: NBP246620, Toronto, ON, Canada) as our alpha motoneuron-specific indicator and label for our protein of interest, the C-bouton.
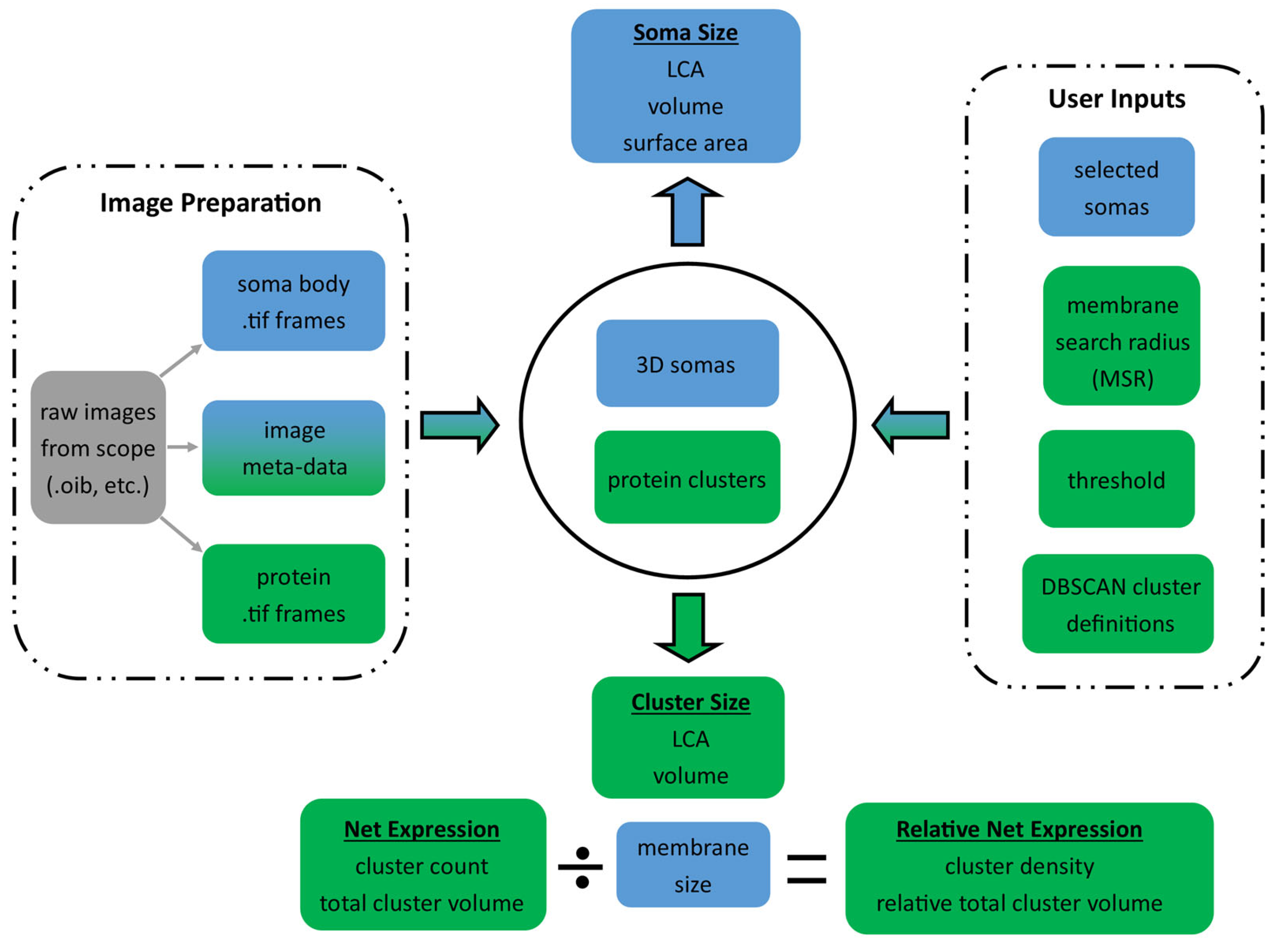
Figure 1 shows the overall flow of the algorithm from the data inputs and user inputs (shown in the dashed boxes) to the soma and cluster measurement outputs. First, the algorithm converts the .oib or .oir Olympus images to standard .tif frames for each of the 4 labels. Then, the user loads the soma body label frames into our GUI, which uses a triangle-algorithm [
7] threshold approach with custom-built Canny edge-detection to automatically outline the somas. The user scrolls through the frames to identify cells of interest and then queues the algorithm to batch-process the reconstructions. The algorithm automatically finds the outline of the identified motoneuron somas through the stack of images (
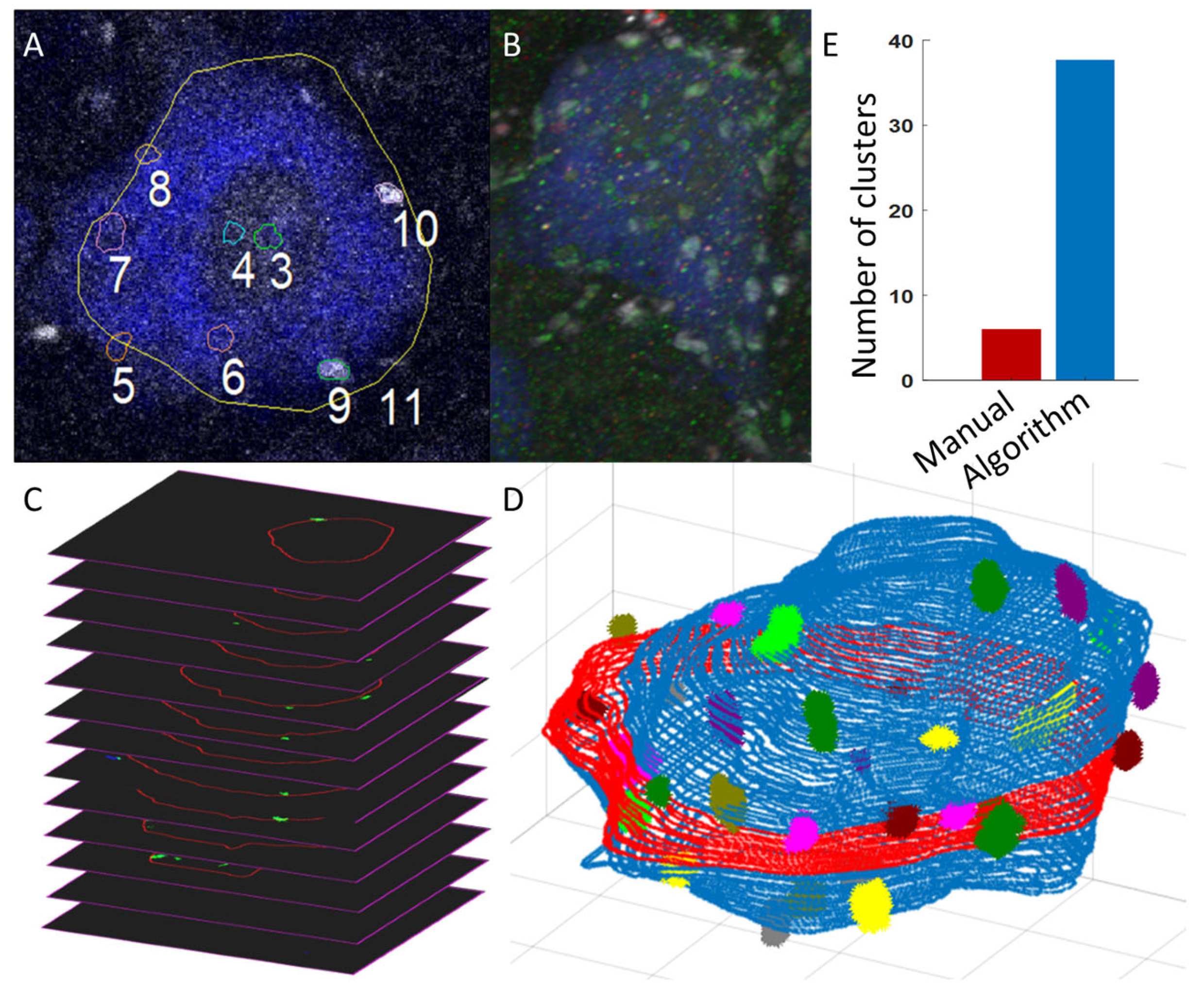
Figure 2C) and generates 3D region-of-interest (ROI) Cartesian matrices indicating the location of the soma edge. These Cartesian matrices are loaded into our visualizer and displayed as clean and minimalist 3D soma reconstructions (blue 3D soma with colored protein clusters in
Figure 2D), a marked improvement over the clunky and background-cluttered visualizations offered by current software like Neurolucida (
Figure 2B). The reconstructions are then converted to a list of X,Y,Z coordinates, defining each soma membrane and thus enabling the algorithm to perform automatic soma size and somatic protein expression analysis in large batches, with minimal human oversight required.
2.2. Somatic Morphology Measurements
Traditionally, spinal motoneuron soma size has been estimated by scrolling through stacks of 2D image frames and manually tracing the visually identified largest cross-sectional area (LCA) of the somas (see
Figure 2A). Similarly, but with more accuracy and consistency, our algorithm counts the pixels contained inside the automatic traces of our 3D Cartesian reconstructions and returns the true LCA in the z plane in μm
2. Additionally, our algorithm measures the 3D volume in μm
3 and surface area in μm
2 of the soma reconstructions, using similar voxel (3D pixel) counting.
2.3. Protein Expression Analysis
The XYZ coordinates of the 3D soma reconstruction are used to create a “shell” of the soma membrane that includes pixels within a given membrane search radius (MSR) from the defined 3D soma edge (default is 2 μm). This search radius must be small enough to avoid protein expression on neighboring structures, yet large enough to capture the entire volume of expression on the soma membrane. Once the membrane shell is defined, the .tif frames for the labeled protein of interest are then loaded into MATLAB (R2023A), and the 4-dimensional (X,Y,Z, intensity) data is extracted for coordinates in the soma membrane shell. Next, the protein intensity values are used to calculate a label threshold. Pixels above the calculated threshold intensity are considered labeled for that protein, and their spatial distributions are analyzed to identify distinct protein macro-clusters with our custom-built density-based spatial-clustering application with noise (DBSCAN)-clustering algorithm.
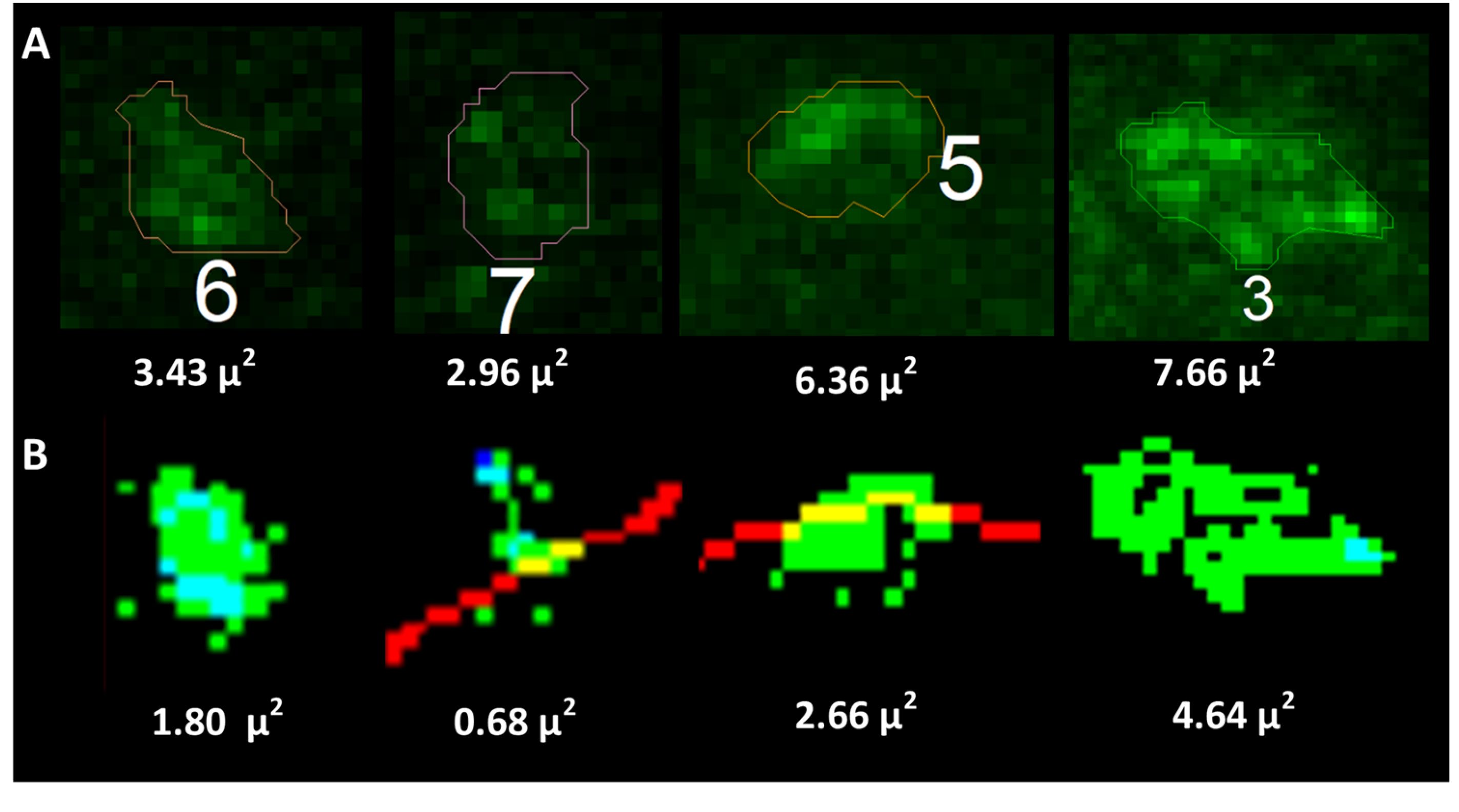
Specifically, labeled pixels (those brighter than the threshold) with enough labeled neighbors are assigned to a cluster using our DBSCAN-clustering algorithm, and then clumps of clusters are combined where appropriate. DBSCAN is a density-based approach for clustering nearby points that does not require a predetermined number of clusters, which is ideal for our application, where the number of clusters is initially unknown. Following cluster identification, the algorithm then generates a simplified 3D visualization of the clusters on the soma membrane (
Figure 2D) and measures each cluster’s LCA and volume. Further, the algorithm calculates 1-net expression of the protein clusters per cell as the total number of clusters and total volume of clusters, and 2-relative net expression as the cluster density (# of clusters per membrane size) and the relative total cluster volume (total cluster volume per membrane size).
2.4. Validation: Reproducibility and Sensitivity
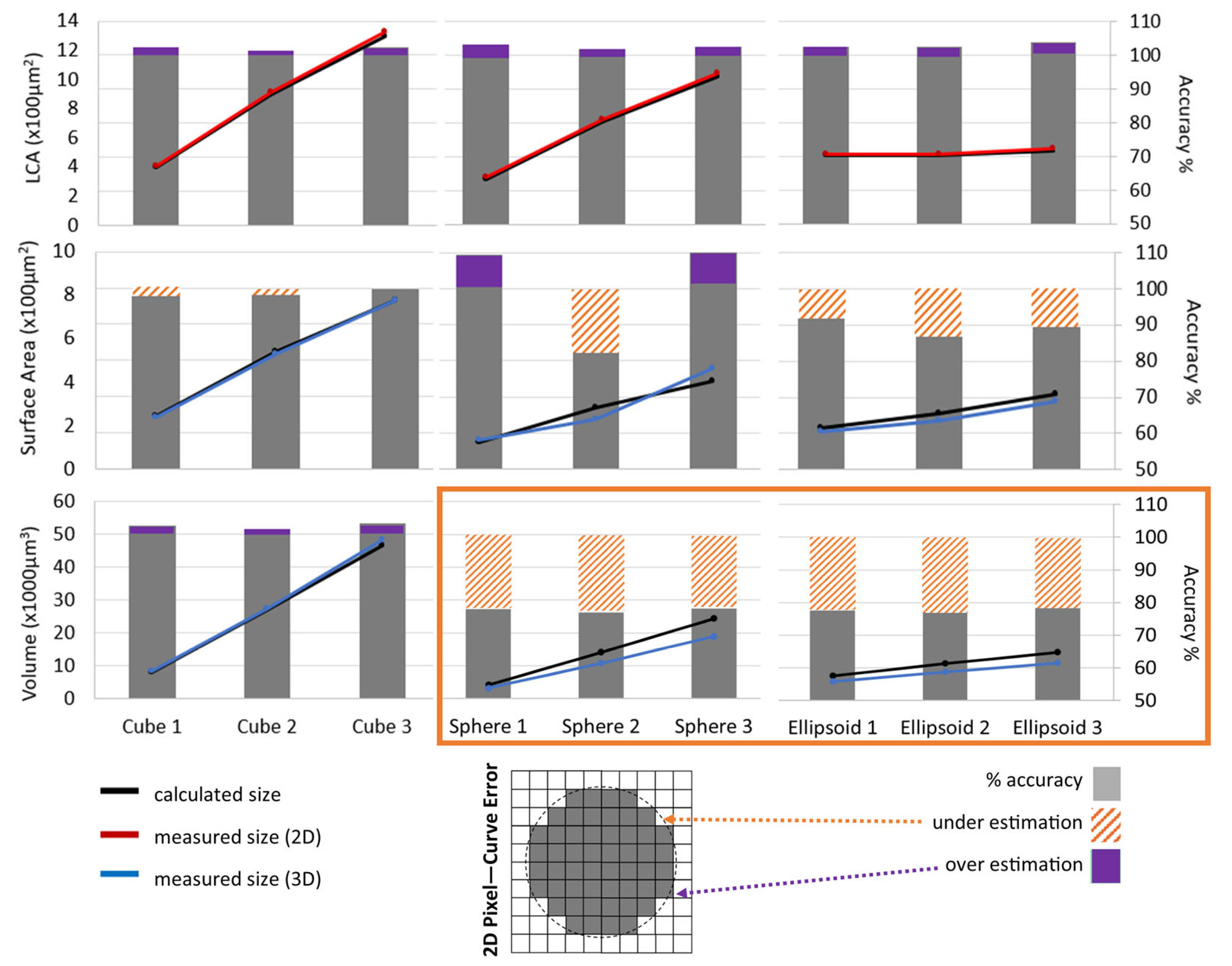
Soma size measurements were validated (see
Figure 3) with quantified accuracy using known shapes of three different sizes (small, medium, and large), with different levels of curvature: cube (no curvature), sphere (symmetric curvature), and ellipsoid (asymmetric curvature). The three sizes were selected to encompass the range of physiological motoneuron soma sizes. The algorithm-measured soma LCA, surface area, and volume were compared to the mathematically calculated sizes (
Figure 3). Further, we compared the algorithm’s sampling to manual methods by measuring the % of membrane analyzed for cluster density and the number of clusters measured for size (
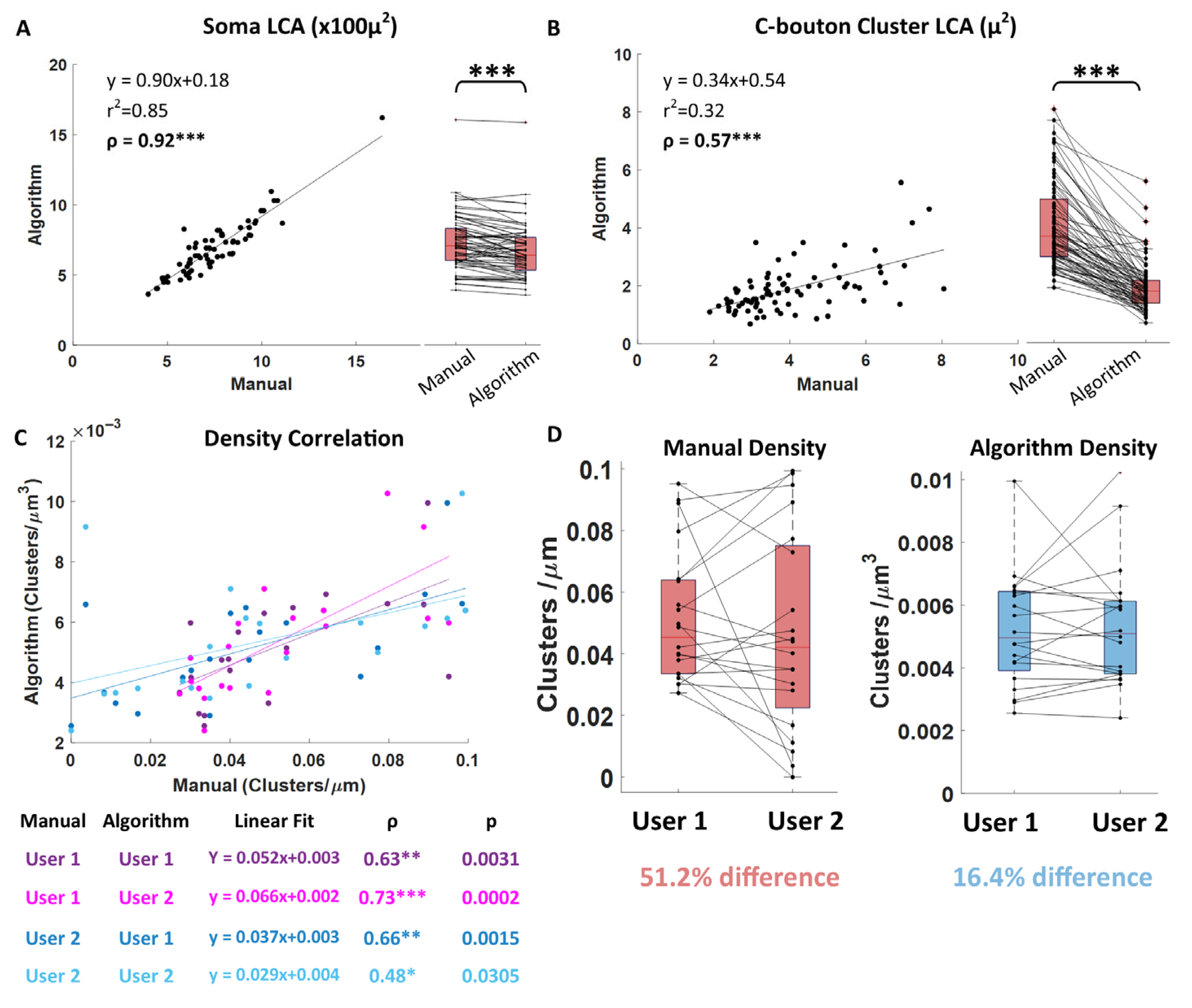
Figure 2E). We also compared algorithm measurements directly to standard manual measurements on the same cells: soma LCA, cluster LCA, and cluster density (
Figure 4 and
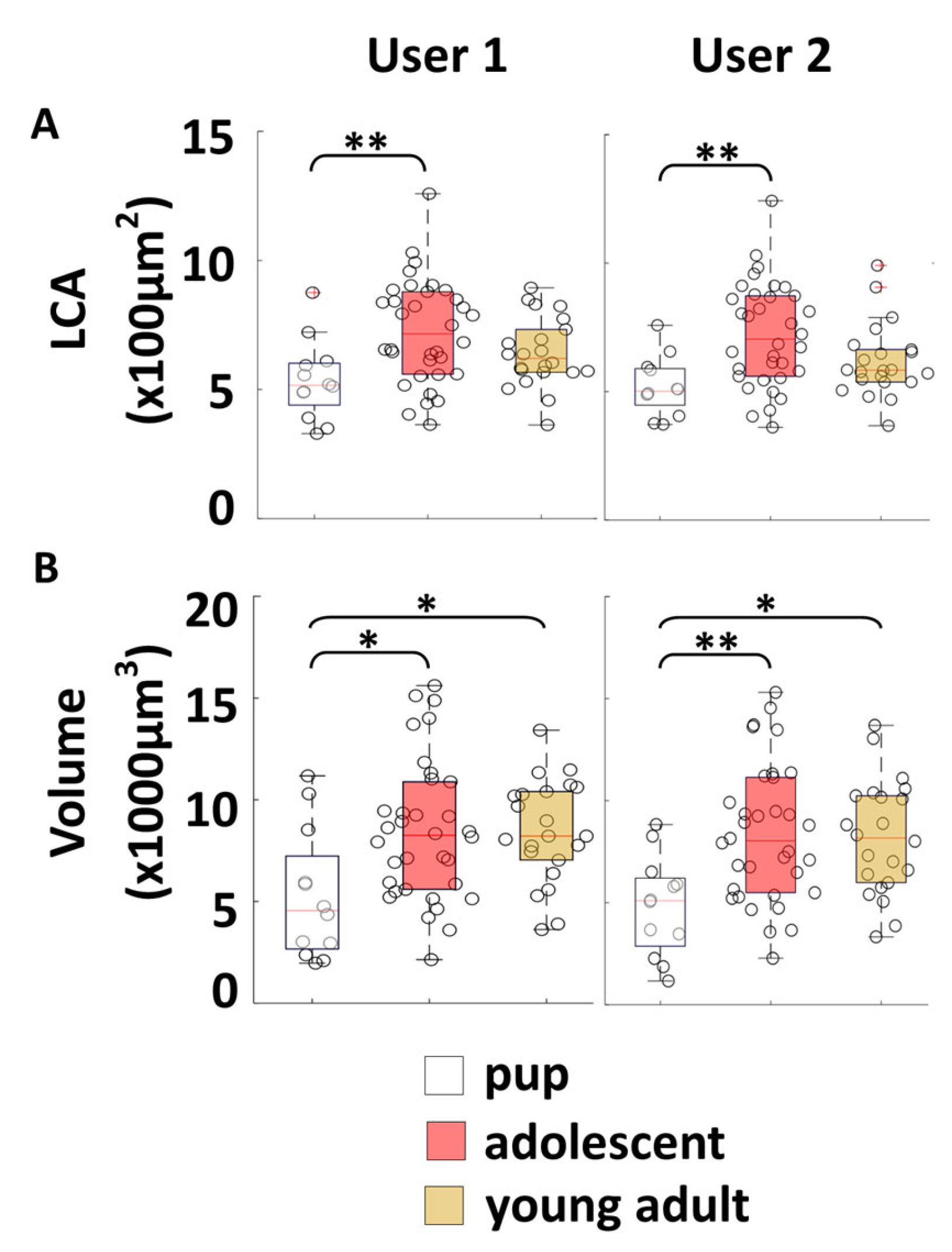
Figure 5). The reproducibility of our new algorithmic approach for soma measurements was tested by comparing results of 2 users who independently reconstructed and measured somas on the same set of cells from various control mice at different ages (
Figure 6). We visually verified our protein labeling We likewise tested the reproducibility of our algorithm’s cluster measurements by comparing the results of 2 independent users (
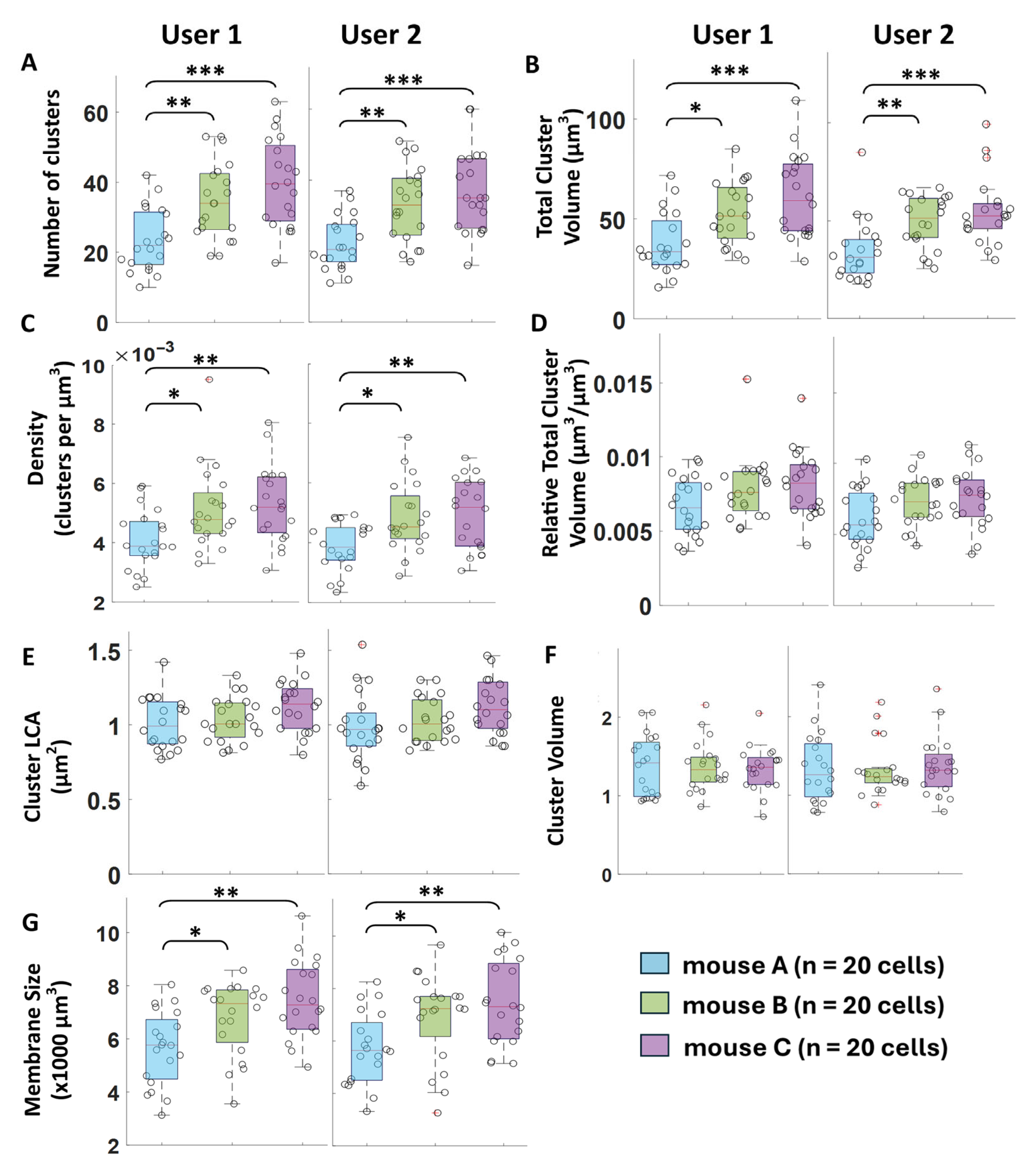
Figure 7). However, we eliminated external sources of variability in our cluster comparisons to strengthen our validation and determine the most appropriate protein cluster measurements. We narrowed our subject pool to 3 control animals from the same animal line, age, and sex, collected in a single randomized and batch-controlled study with highly consistent antibody batches and labeling methodology. Then, we characterized how our algorithm captures biological variability by comparing the measured protein expression between the 3 control animals (
Figure 7).
2.5. Thresholding
Thresholding is a vital part of our analysis, as it could greatly impact the measurements of the algorithm. Complicating the issue, image background and off-target binding artifact can be highly variable due to the nature of IHC labeling. Nonetheless, it is important that the chosen threshold represents protein expression accurately and can fairly represent label across study groups with a variety of image-background conditions inherent to the IHC process. We initially picked our thresholding method by visually minimizing background and maximizing visually identified “true” protein label during our algorithm-tuning process for the C-bouton macro-clusters. We used a randomly selected subset of cells from each study group for this tuning. Our chosen thresholding method is a custom implementation of the triangle algorithm [
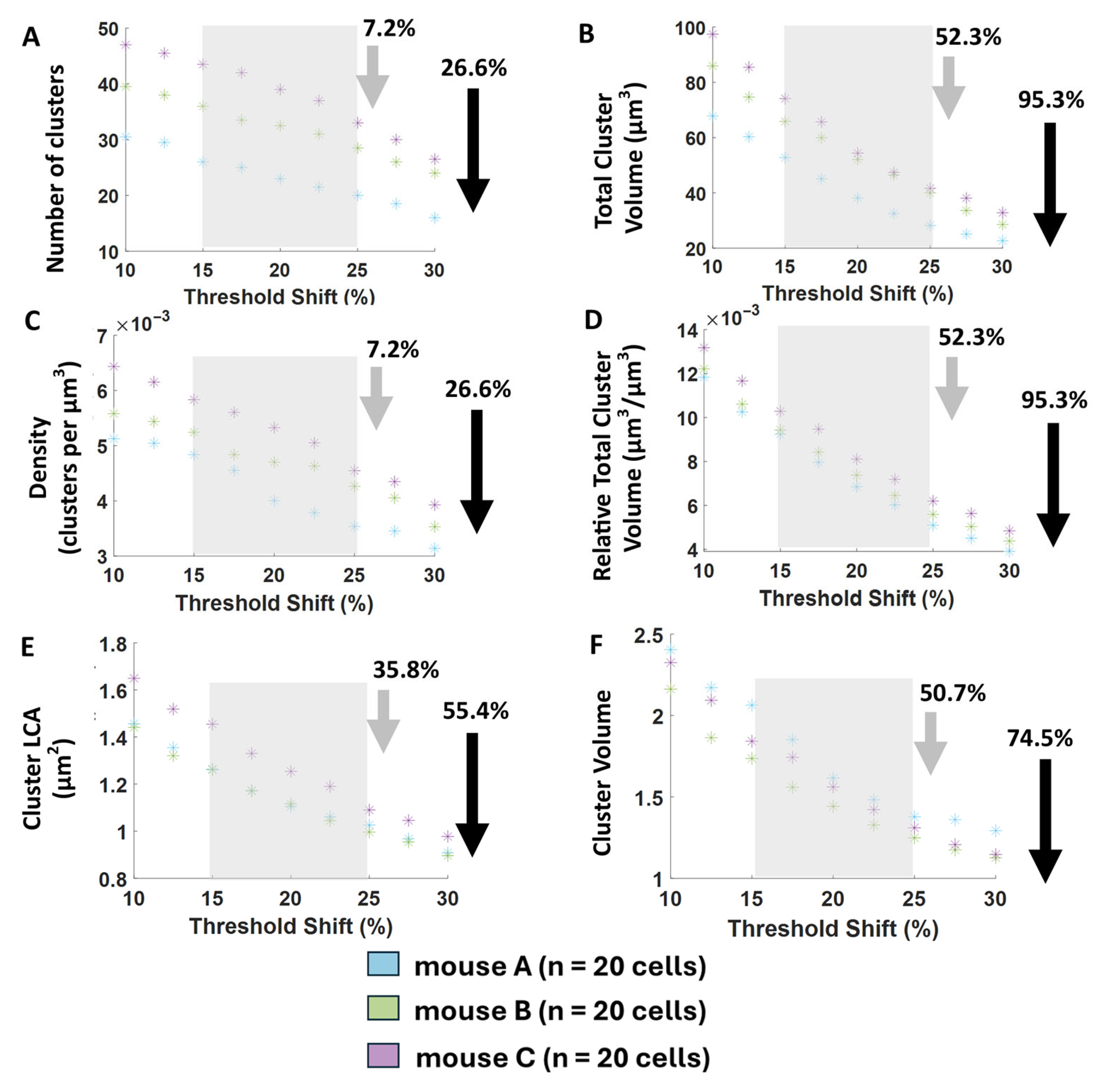
7], with a default shift of 10%. We applied this thresholding algorithm on the membrane shell separately for each 2D image frame, since we found that label intensity and background vary most in the z-direction due to the normal gradient of antibody penetration. Though we were visually satisfied with our chosen threshold shift of 10%, we wanted to objectively determine if the threshold level would impact our findings. Thus, we performed a threshold-sensitivity analysis to explore how each output parameter of the algorithm is changed over a large range of threshold shifts (see
Figure 8).
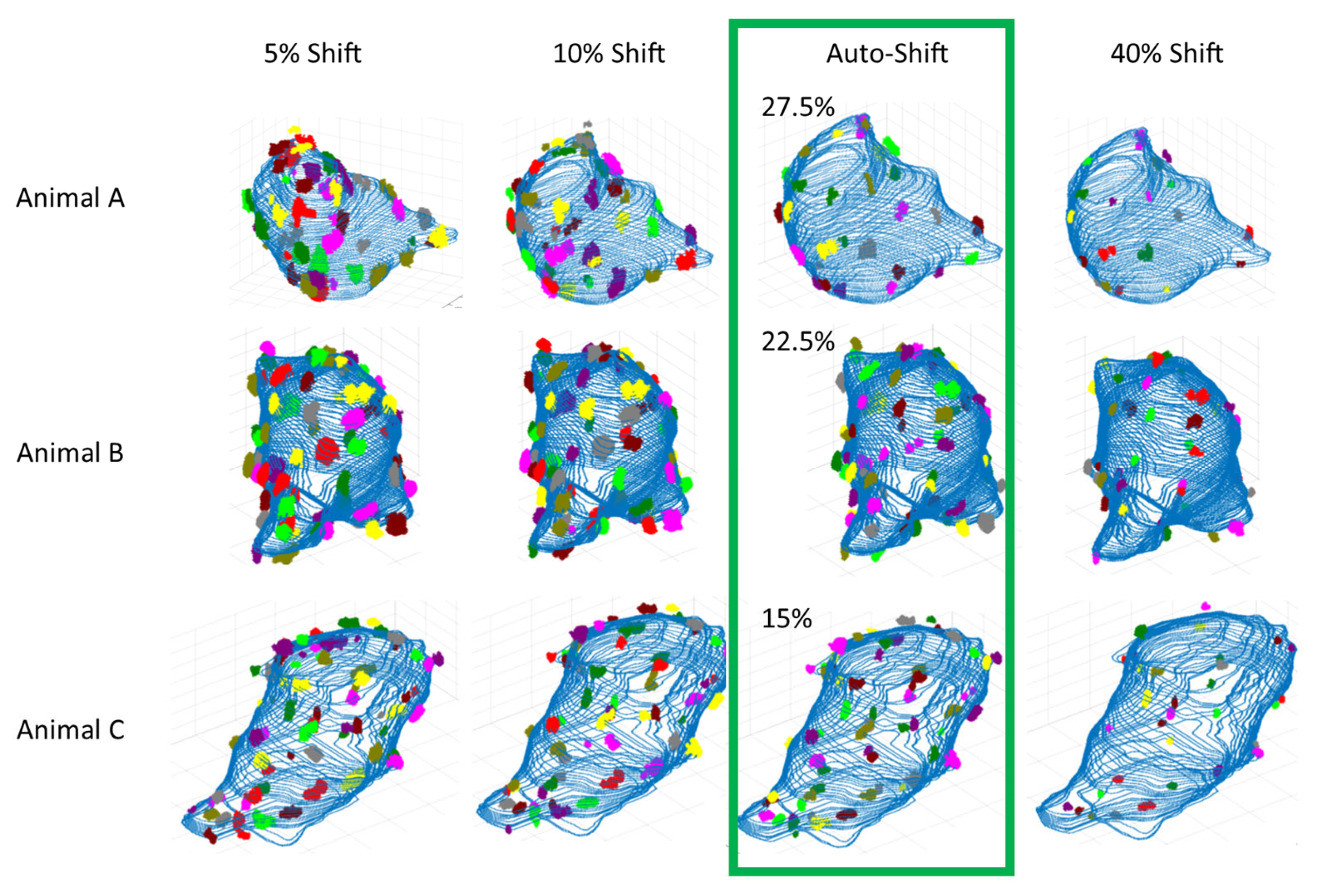
Furthermore, to simplify the thresholding process for future studies implementing our algorithm, we used our threshold-sensitivity results and our extensive and methodical manual observations of the visualized clusters at different thresholds to develop threshold stability-range criteria. The stability range was then used to automatically calculate an optimum threshold-shift suggestion per cell to maximize the stability, reasonability, and reproducibility of our protein cluster measures (see
Figure 9).
2.6. Statistics
All data was analyzed in MATLAB as follows. All groups were tested for normality using Shapiro–Wilk’s test (swtest). All groups were tested for equal variance with their comparison groups, using Bartlett’s equal variance test (vartestn). Parameters with no groups that violated normality and with equal variance between groups were analyzed by ANOVA (anovan) and post hoc Tukey’s for ANOVA (multcompare). Most parameters violated either normality in at least one group or equal variance between groups, and so they were compared by the Kruskal–Wallis non-parametric test (kruskalwallis) with post hoc Tukey’s for Kruskal–Wallis (multcompare). Since the non-parametric tests were used most often, all data is reported as box plots with median line, with boxes indicating the inner-quartile range, and whiskers indicating the range of non-outliers. Correlation coefficients (ρ) were Pearson’s (corrcoef). Paired tests were performed with Wilcoxon signed-rank test (signrank) since non-normality was indicated. All tests were performed with an alpha of 0.05. One asterisk indicates p < 0.05, two asterisks indicate p < 0.01, and three asterisks indicate p < 0.001.
4. Discussion
Characterizing soma size and protein expression patterns is critical for linking motoneuron structure to function under both normal and pathological conditions. This is especially important in neurodegenerative research, such as studies of aging and motoneuron disease, where understanding structural changes in somas and their associated proteins under altered electrical activity may reveal mechanisms of degeneration and identify potential targets for therapeutic intervention aimed at preserving motoneuron function and preventing or slowing down cell death.
Immunohistochemistry (IHC) is a powerful tool that enables the visualization of motoneuron somas and associated proteins for structural analysis. However, despite its strength, IHC is often underutilized as a source of primary quantitative data due to challenges in objectively interpreting and quantifying protein expression, as IHC images often exhibit high variability in label intensity and background noise. These inconsistencies make fair and consistent thresholding across images particularly difficult. Furthermore, existing analysis tools are primarily designed for analysis of brain tissue and typically require extensive manual effort to identify and measure protein clusters, leading to subjective sampling, limited throughput, and poor reproducibility. These issues contribute to inconsistent findings between studies—and even within the same dataset analyzed by the same individual at different times [
2].
To address these limitations, we developed a custom software tool, independent of costly commercial platforms, specially designed to facilitate consistent comparisons of soma size and protein expression across IHC images in spinal tissue. Importantly, our automated analysis is inherently blind to study groups and is order-agnostic, meaning the results are independent of the order in which the cells are analyzed (removing the need for order randomization).
Soma size: Our Cartesian-based approach enables precise 3D measurement of soma size at the highest possible voxel resolution. This represents a significant improvement over many commercial tools, which often rely on adaptive cylindrical compartment models with varying sizes and, consequently, inconsistent resolution. In contrast to these tools—which do not disclose the accuracy of their volume or surface area calculations—we transparently report a consistent 23% underestimation in volume. This known and reproducible bias allows for accurate comparisons of soma size across experimental groups and offers the option to apply a correction factor to estimate true physiological soma volume.
Importantly, our results show that volume is the most appropriate and reliable metric for assessing soma size. We also show that surface area is not a reliable measure at this resolution, due to its inability to accurately capture complex 3D geometry. Our automated edge detection method enhances the efficiency, objectivity, and physiological relevance of both 2D and 3D soma reconstructions. Additionally, we confirm that 3D volume is more sensitive than 2D LCA in detecting subtle changes in soma size.
Protein expression: We developed an automated approach to replace the traditionally tedious and subjective manual analysis of protein expression, significantly improving both objectivity and consistency. This automation also greatly enhances analytical efficiency by eliminating the need for manual protein cluster sampling and tracing, making it feasible to process large datasets with high cell counts in a fraction of the time previously required.
Furthermore, all algorithm settings are user-accessible numerical values, allowing for transparent control and easy reporting—critical for ensuring reproducibility. For the first time, our algorithm enables efficient and systematic exploration of various threshold and cluster definition scenarios, allowing us to track measurement sensitivity and verify the robustness of our results.
With this new level of reliability and rigor, IHC-based protein expression data can now serve as primary quantitative results in studies—yielding reproducible, biologically meaningful insights that advance our mechanistic understanding of motoneuron function.
Limitations and other insights: One major limitation of this analysis is the necessary assumption that the tissue, images, and label are all in fair condition and reasonably represent the physiology being studied. We learned, from our extensive testing during development, that directly comparing protein expressions from images taken in separate experiments, antibody batches, or under different protocols could easily confound and confuse the conclusions of a study. Notably, these limitations also exist in prior analysis methods, but are often “compensated for” by manual adjustments or corrections during analysis. Although these manual corrections are fairly common practice, these adjustments introduce great bias and subjectivity, and are likely a large contributor to the lack of rigor and reproducibility that plague the field. Our algorithm should be used to analyze images from tightly controlled experimental conditions, where all groups in a study are processed in batches, i.e., each batch of processing contains tissue from all study groups. The algorithm does not perform corrections by filtering or adjusting brightness levels to meet visual assumptions of how the label is expected to look. Rather, our auto-threshold adjustments and DBSCAN parameters are applied objectively, mathematically, traceably, and consistently to all cells in the study.
Another limitation to consider is image resolution. For this validation study on motoneuron somas and somatic C-boutons, our sampling resolutions were 0.172 microns/pixel and 0.3 microns/slice. It should be noted that our reported 23% volume underestimation was based on this resolution, and could vary at alternative resolutions. We would expect a smaller underestimation for higher resolutions, and a larger underestimation for lower resolutions. For analyzing motoneuron somatic protein structure, we do not recommend lower resolutions given the small size of the proteins.
Future work: In this study, our analysis of protein expression focused on the C-bouton, a structure known to form distinct macro-clusters on the motoneuron somatic membrane. However, our algorithm is also capable of analyzing proteins that exhibit more diffuse or less distinct clustering patterns, including those with variable levels of phosphorylation. Determining the most effective methods to quantify these proteins—both in terms of phosphorylation state and total expression—across different conditions is a key focus of our ongoing research. Additionally, our algorithm is also equipped to study proteins in other key regions of spinal or brain tissue, either isolated to cell geometry areas or over entire image regions. We are continuing to expand the scope through additional features and workflows that can be customized for a variety of applications.
Broader implications and perspectives: Although this study was focused on spinal tissue and motoneuron analysis, accurate segmentation and quantification remain significant challenges in other basic research and clinical diagnostic applications involving immunohistochemical imaging [
10,
11]. Our algorithm improves on several subjectivity, usability, and reproducibility issues that plague alternative analysis algorithms and graphical user interface (GUI)-based qualitative approaches. Our specialized level of automation and rigorous stability testing are among the features that make our algorithm unique. Alternatively, while not yet utilized for motoneuron analysis, deep learning is commonly leveraged in other IHC applications to perform desired image segmentations. While conceptually promising, these deep learning techniques have proven to have limited accuracy and generalizability so far, with no clear model consistently outperforming other models across datasets [
10,
11]. Future studies and generalizations of our algorithm are needed to compare performance to such State-of-the-Art (SOTA) deep learning models outside the context of motoneuron analysis.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}