1. Introduction
In silico vaccine designs provide a cost-effective method for identifying potential immunogenic epitopes prior to clinical testing. The development of these vaccine designs has been steadily increasing due to the ability of vaccines to safely boost the immune system’s innate defense mechanisms against a broad number of diseases and pathogens. Some of the latest findings involving in silico vaccine designs include potential methods of protection against different viruses, bacteria, and some single-celled organisms [
1,
2,
3,
4,
5]. However, there is a lack of powerful in silico epitope vaccine designs specifically targeting cancer. Furthermore, there is a demand for user-friendly graphical software to encourage clinical researchers in adopting in silico methods for cancer vaccine clinical trials. Some of our current methods, alongside other recent studies, manually filter a large set of epitope data using a set of strict exclusion criteria [
6,
7,
8,
9,
10]. While these cancer vaccine designs identify immunogenic epitopes with favorable characteristics, much time is spent on data collection and epitope sorting. This limits the time available for further epitope analysis or preclinical testing. In one of our previous studies, we aimed to combat this issue by integrating multiple deep learning tools into a machine learning-based multivalent vaccine design called IntegralVac [
11]. This comprehensive method reduces the time spent on data collection and drastically improves immunogenicity and binding affinity predictions [
11]. In contrast, a study by Stranzl et al. focused on a different approach to MHC Class I epitope prediction. They introduced a ranking model named NetCTL, which is trained on known Class I ligands [
12]. NetCTL ranks epitopes based on proteasomal cleavage and other epitope characteristics, offering a more efficient way to prioritize epitopes for further analysis. However, this model is limited in the number of characteristics it considers and cannot be used to predict MHC Class II epitopes.
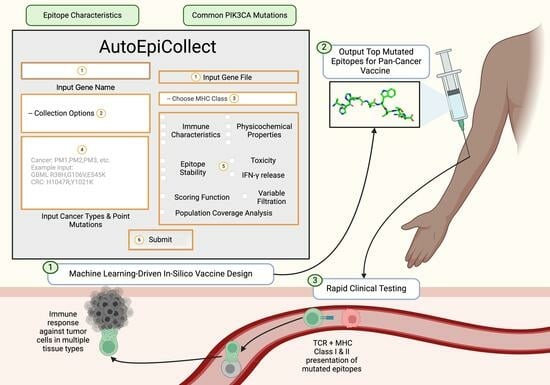
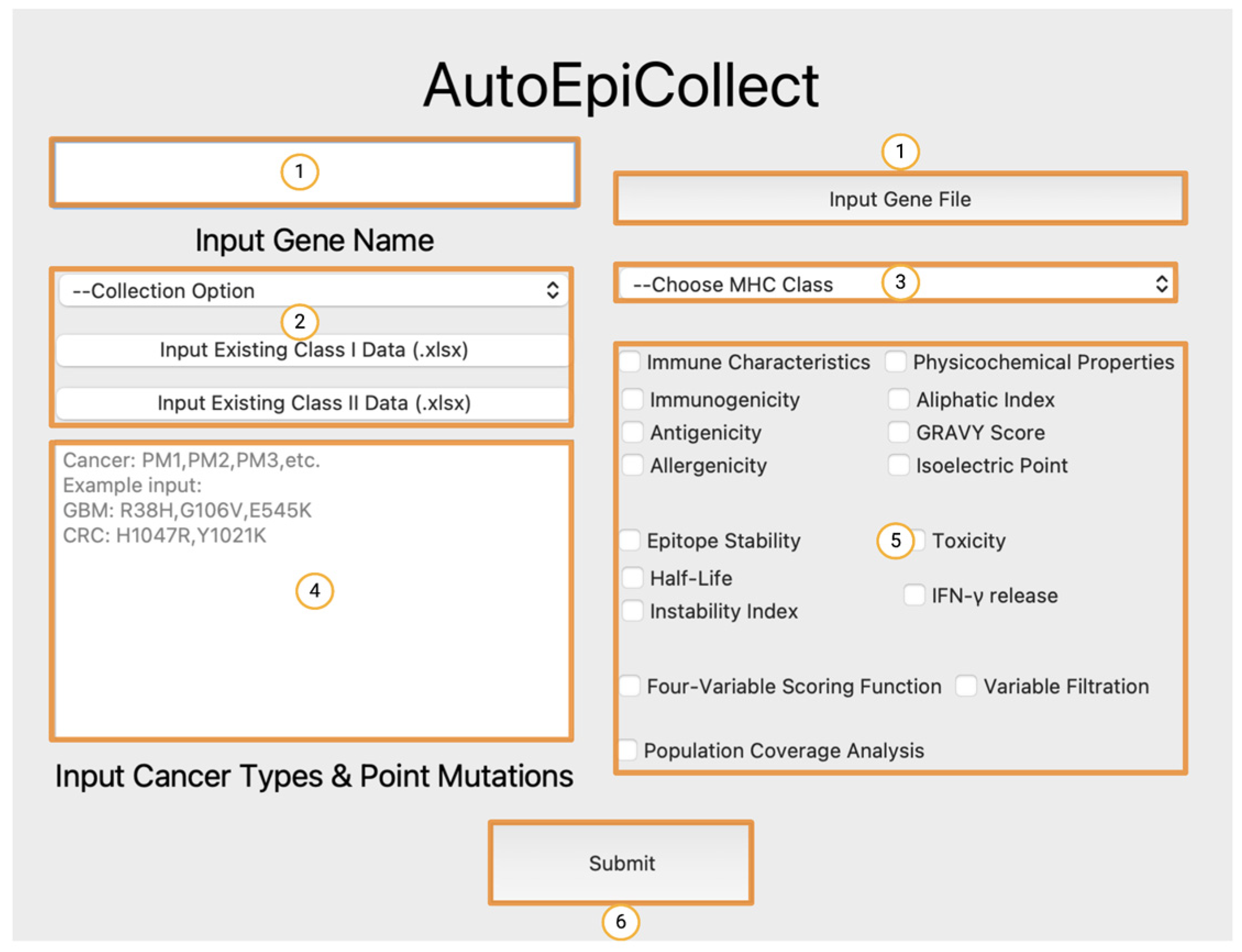
We developed AutoEpiCollect, an automated epitope selection software that integrates a novel machine learning-based epitope ranking model with individual variable filtration. AutoEpiCollect features a user-friendly graphical user interface (GUI) designed to identify potential MHC Class I and II T-cell eliciting epitopes, specifically tailored for peptide-based cancer vaccines. The software requires only two inputs: the name of the target oncogene and a list of point mutations associated with prevalent cancer subtypes affected by the mutated gene. AutoEpiCollect then gathers mutated epitope characteristics and employs a machine learning model to rank epitopes based on their immunogenic potential. To ensure the selection of safe and stable epitopes, we incorporated individual variable filtration using exclusion criteria. Additionally, we used our PCOptim and PCOptim-CD programs to optimize the final list of epitopes before evaluating the population coverage of a vaccine containing the predicted top epitopes [
6,
7]. In this study, we applied AutoEpiCollect’s vaccine design process to develop a pan-cancer epitope-based vaccine targeting PIK3CA-mutated cancers.
Pan-cancer epitope vaccines target epitopes from multiple cancer types. Epitopes taken from tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs) are delivered via vaccination, where antigen-presenting cells (APCs) uptake them. The epitopes are presented on major histocompatibility complex molecules as markers for T-cells to activate. As a result, cytokines are released and regulate immune responses towards the epitopes [
13]. While a pan-cancer epitope vaccine targeting various PIK3CA gene mutations has not been developed yet, an in vitro study focused on the H1047R point mutation, using PBMCs from healthy individuals, induced T-cell responses [
14]. Specifically, 27mers peptides from the PIK3CA H1047R point mutation stimulated CD4
+ and CD8
+ T-cell responses in 16% and 4.0% of donors, despite the study’s limited sample size.
However, this study focused on a few epitopes due to the extended duration required to obtain epitopes from an extensive set of point mutations in the PI3KCA gene. Pan-cancer vaccines target a greater number of mutations, thereby increasing the number of epitopes involved. Furthermore, they facilitate safer, more precise immune responses against mutated epitopes. Cafri et al. designed an mRNA cancer vaccine containing mutated sequences from TP53, KRAS, and PIK3CA genes, among others [
15]. This vaccine was capable of eliciting T-cell responses against selected neoantigens in metastatic gastrointestinal cancer patients. No significant clinical responses were observed in the patients treated with the vaccine during the trial; however, Cafri et al. were able to identify T-cell responses against in silico predicted neoantigens [
15]. This study highlights the immunogenic potential of a cancer vaccine consisting of virtually predicted epitopes targeting missense mutations in PIK3CA. Based on the studies discussed above, we decided to research common PIK3CA point mutations in an effort to develop a pan-cancer vaccine with in silico tools [
9,
10].
Prevalent PIK3CA missense mutations found in colorectal adenocarcinoma (CRC), breast cancer (BC), endometrial carcinoma (EC), glioblastoma multiforme (GBM), and meningioma are primarily seen in exons 9 and 20 [
16,
17,
18,
19,
20,
21,
22]. These mutations affect the helical and kinase domains of the p110ɑ protein, leading to increased cell proliferation and tumor metastasis [
17]. Current treatment options aiming to prevent metastatic recurrence include endocrine therapy for HR+ tumors, aromatase inhibitors, tamoxifen (associated with a 50% reduced recurrence rate), and chemotherapy for the unfavorable prognosis of triple-negative BC [
23]. Several small-molecule inhibitors targeting the PI3K protein have also demonstrated efficacy in inducing tumor regression in cells harboring PIK3CA mutations. In a study by Janku et al., the FDA-approved drug Alpelisib was used to target PIK3CA-mutated BC, resulting in improved clinical responses among patients receiving the inhibitors [
24]. Despite these promising outcomes, small molecule inhibitors are characterized by their limited pathway targeting, potentially leading to restricted effectiveness. Furthermore, they may induce off-target cytotoxic effects, further complicating their use in clinical settings [
25].
Due to the limited success of PIK3CA-directed treatments, patients with the cancers discussed above could potentially experience improved clinical outcomes through a pan-cancer epitope vaccine targeting prevalent PIK3CA point mutations. Our study considered various clinical variables based on prior studies that served as primary benchmarks when developing our novel epitope selection process [
6,
7,
26,
27]. These variables confirmed that AutoEpiCollect’s top predicted epitopes were immunogenic, safe, and stable. Our automated AutoEpiCollect GUI software is the first machine learning-based epitope selection method to target PIK3CA missense mutations. With its ability to streamline a significant portion of the preclinical vaccine development process, AutoEpiCollect has the potential to reduce days of preclinical epitope selection procedures to just a few hours.
4. Discussion
AutoEpiCollect was developed to accurately predict safe and potentially immunogenic MHC Class I and II epitope sequences. This tool is specifically designed for the expedited development of pan-cancer vaccines targeting any oncogene of interest. AutoEpiCollect aims to accelerate the tedious task of epitope collection and analysis, with our vaccine design ultimately allowing the validation of a greater number of pan-cancer vaccines in clinical settings. Previous cancer vaccine designs subjected mutated epitopes targeting one cancer subtype of interest to manual variable filtration based on a predetermined set of exclusion criteria [
6,
7,
8,
9,
10]. However, these methods are time-consuming and cannot efficiently analyze a large set of mutated epitopes. It is crucial that vaccines are capable of targeting a broad spectrum of mutations, since tumors mutate rapidly. We attempted to mitigate this issue with our deep learning-based method, IntegralVac. While IntegralVac is a powerful machine learning-based program that drastically increases the accuracy of MHC Class I in silico predictions, it is missing a comprehensive Class II prediction feature. This restricts the applicability of vaccines predicted with IntegralVac [
11]. Stranzl et al. also tried to improve upon individual variable filtration through a tool called NetCTL. NetCTL identifies epitopes and ranks them according to their MHC Class I binding affinity, Transporter Associated with Antigen Processing transport efficiency, and proteasomal cleavage [
12]. Although this tool prioritizes the overall effectiveness of an epitope’s binding affinity, transport, and ability to be broken down, it does not consider immunogenicity, antigenicity, allergenicity, stability, and toxicity. These variables play a major role in predicting whether or not an epitope will induce a strong immune response without triggering an allergic reaction [
29,
30].
AutoEpiCollect’s mutated epitope selection process incorporates a combination of machine learned-based ranking and individual variable filtration, as demonstrated by the studies discussed above [
6,
7,
8,
9,
10,
11,
12]. We subjected epitopes to a novel ranking system using a probabilistic logistic regression model that was trained on experimentally validated epitopes. Unlike a conventional binary logistic regression classification of immunogenic or non-immunogenic, our model ranked epitopes with a probabilistic logistic regression function based on the probability of triggering a safe immune response. This prioritization is rooted in clinically relevant factors, including immunogenicity, antigenicity, allergenicity, and binding affinity, all of which are crucial determinants for the safety and efficacy of cancer vaccines [
29,
30]. The choice of a ranking algorithm over a classification approach stems from the understanding that epitope selection is a nuanced process, ultimately determined by researchers conducting clinical trials for vaccine testing. Instead of studying a definitive set of epitopes, researchers can use the epitopes generated by AutoEpiCollect as a starting point to identify the epitopes with the highest likelihood of eliciting an immune response.
However, before finalizing the epitope ranking algorithm, we compared the performances of linear regression scoring functions and probabilistic logistic regression scoring functions to determine which regression model would be the best to incorporate into AutoEpiCollect. The Class I and II linear regression scoring functions had R
2 values of 0.060 and 0.028, respectively. The probabilistic logistic regression scoring function for Class I epitopes performed at an accuracy of 76% (AUC = 0.73), while the function for Class II epitopes performed at an accuracy of 56% (AUC = 0.62) with the test data. Based on these metrics, we realized that the linear regression scoring functions were not viable for epitope prediction. The low R
2 values for the linear regression functions may be attributed to the lack of experimentally procured epitope characteristics. The immunogenicity, antigenicity, allergenicity, and binding affinity scores for each training data epitope are based on in silico prediction tools that use the epitope’s peptide sequence in some way to calculate scores. The training data from the IEDB solely reports experimentally validated T-cell assay data. As a result, the training data matches up experimental T-cell data with virtual epitope characteristics. In addition, we utilized DeepImmuno’s beta–binomial distribution approach to compute the probability of an epitope eliciting a T-cell response based on the number of successful and unsuccessful experimental T-cell assays [
40]. This was done with the hope of placing a higher priority on training data epitopes that elicited more positive T-cell responses. However, this virtual computation combined with the virtual epitope characteristics may have built-in inconsistencies within the training data for the linear regression models, leading to a lack of fit for both the Class I and II scoring functions.
While the metrics for the probabilistic logistic regression scoring functions were a significant improvement over the linear regression metrics, there were still some discrepancies in accuracy observed between Class I and II epitopes. This can be traced back to distinctions in the in silico prediction tools used for each class. Notably, we collected Class I and II binding affinity, immunogenicity, and allergenicity epitope data from different tools, leading to inherent differences in the training datasets. In essence, while operating within the same program, the Class I and II training data collection processes are distinct tools, and the discrepancy in model accuracy emerges from all the differences in the two data collection approaches. In addition, IEDB’s CD4 Immunogenicity tool only calculates the immunogenicity of 15mers peptides. Thus, we limited the training data to only 15mers when training our Class II scoring function. This reduced diversity of the Class II training dataset led to homogeneity, potentially contributing to lower model accuracy. Despite these limitations, our ranking algorithm outputs the relative immunogenic potential of an epitope. This approach allows for a more flexible and informed selection process during the early stages of vaccine development.
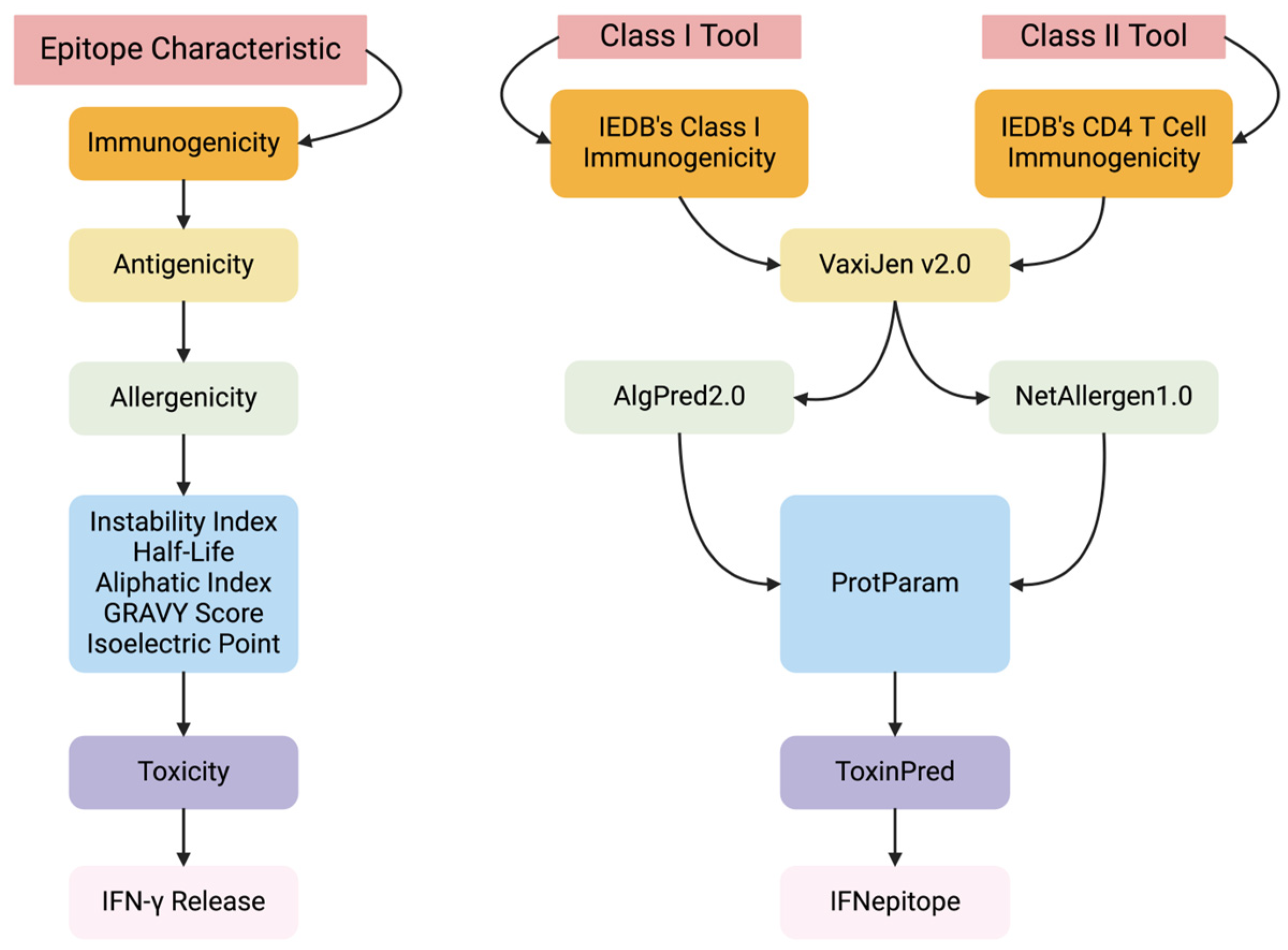
Following ranking, Class I and II epitopes underwent individual variable filtration following the same process we previously used, based on half-life, instability, toxicity, and IFN-γ release [
6,
7]. This filtration method relied on exclusion criteria rather than a ranking system due to the crucial nature of these variables. The half-life and instability of an epitope are indicative of the effectiveness of the elicited T-cell response. Regardless of an epitope’s immunogenic ability, a low half-life and unstable nature will significantly decrease the sustained immune response, necessitating higher doses and frequent administration. Furthermore, higher doses may cause more potent side effects, contradicting one of the primary objectives of this study. Toxicity and IFN-γ release predictions are binary variables, each characterized as either positive or negative. Similar to half-life and instability, toxicity is important when considering the potential side effects of vaccine administration. Our decision to include solely non-toxic epitopes was driven by the imperative to establish pan-cancer epitope-based vaccines as a powerful immunotherapeutic option for a diverse patient population without compromising on tolerability. We took IFN-γ release predictions into account for MHC Class II epitopes due to the fact that the IFNepitope server is only trained on Class II data. Activated CD8
+ T-cells, prompted by MHC Class I molecules, spearhead the primary antitumor response, while CD4
+ helper T-cells, activated by Class II molecules, play a complementary role by enhancing and prolonging CD8
+ effector function. The secretion of IFN-γ by CD4
+ T-cells triggers the expression of chemokines necessary for recruiting CD8
+ T-cells. Furthermore, CD4
+ T-cells exhibit direct tumor suppression through various IFN-γ functionalities. Consequently, MHC Class II epitopes capable of inducing IFN-γ release prove instrumental in stimulating antitumor effects through multiple mechanisms [
53]. Considering this information, we decided to exclusively include predicted IFN-γ-releasing Class II epitopes, aiming to amplify the immunogenic effects of the vaccine.
After finalizing our ranking and individual filtration algorithms, as well as the rest of AutoEpiCollect, we proceeded to test our machine learning-driven vaccine design on a pan-cancer vaccine targeting a common oncogene. Our study focused on the PIK3CA gene due to its pivotal role in the PI3K/Akt/mTOR intracellular signaling pathway, as illustrated in
Figure 4. This pathway governs vital cellular processes such as growth, proliferation, and differentiation [
50]. Mutations within the PIK3CA gene impact the p110ɑ catalytic subunit of the PI3K protein, disrupting the signaling pathway and leading to immune disorders, cardiovascular diseases, and cancer in various tissue types [
50]. After inputting prevalent PIK3CA point mutations into AutoEpiCollect, we received Class I epitopes targeting 34 mutations and Class II epitopes targeting 11 mutations from the total 49 mutations initially input. These Class I and II epitopes demonstrate predicted attributes of robust immunogenicity, safety, stability, and IFN-γ release. Notably, 10 mutations yielded overlapping epitopes for both MHC Class I and II, strengthening our vaccine design. This overlap holds significant potential for eliciting heightened immunogenic responses by activating both CD8
+ cytotoxic T-cells and CD4
+ helper T-cells. As illustrated in
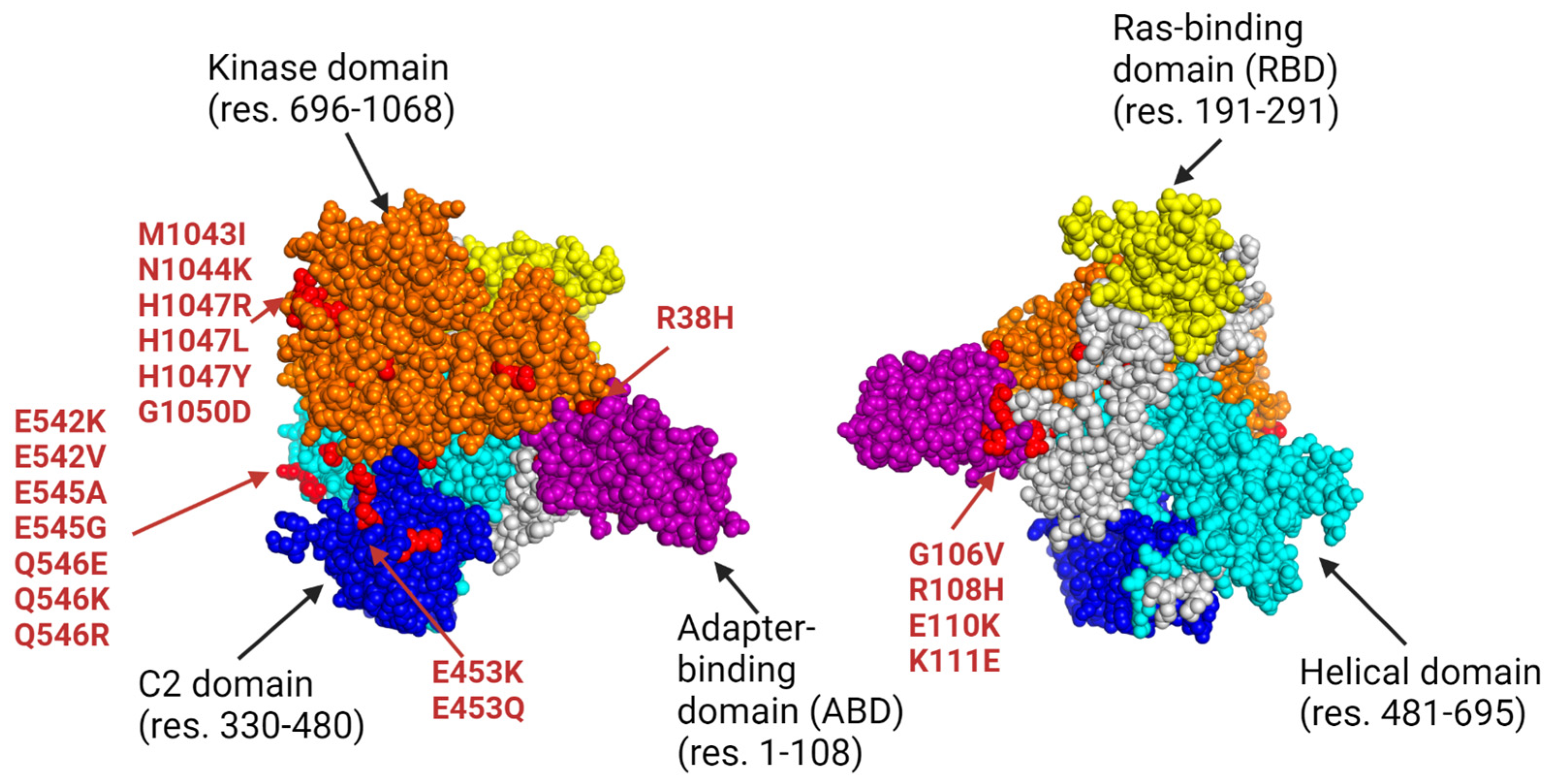
Figure 2 and
Table 6, a substantial portion of the epitopes in the final datasets are directed at mutations found in either the kinase or helical domain of the p110ɑ protein. These domains are recognized as hotspot regions prone to prevalent gain-of-function mutations in the PI3K kinase protein and underlie many different subtypes of cancer [
12]. The targeting of these hotspot regions by a large number of our final epitopes enhances our vaccine’s utility and emphasizes its potential efficacy in addressing the major molecular drivers of PIK3CA-mutated cancers.
Following the optimization of MHC Class I and II epitope datasets using PCOptim and PCOptim-CD, AutoEpiCollect input the epitope/HLA allele pairs into IEDB’s Population Coverage Analysis tool, resulting in world population coverages of 98.09% and 81.81%, respectively. The high Class I epitope coverage underscores the potential for our PIK3CA vaccine to impact various patients worldwide. In addition, it confirms that AutoEpiCollect’s epitope selection method generated an extensive dataset. However, both Class I and II coverages are not uniformly distributed across regions. Population coverage is contingent on the prevalence of specific HLA alleles in certain global regions and the corresponding epitopes that they can bind to. Europe, North America, and East Asia demonstrated the highest population coverages, since our predicted immunogenic epitopes bind to HLA alleles common in these areas. In contrast, Central America exhibited lower population coverages, at 7.76% for MHC Class I alleles and 49.91% for MHC Class II alleles. This discrepancy arises from the tool’s limitation in accounting for the diverse array of HLA alleles prevalent in Central America. Future vaccine designs might consider prioritizing epitopes that bind to alleles prevalent in regions historically characterized by low coverage, thereby maximizing the vaccine’s applicability. Before calculating population coverage for the Class II dataset, many epitope/HLA allele pairs had to be excluded due to constraints within IEDB’s allele dataset. These excluded alleles are detailed in
Table 4. As a result, the population coverage for Class II epitope/HLA allele pairs was based on a restricted dataset and may not accurately reflect the coverage that would be attained with the inclusion of all potentially immunogenic epitope/HLA allele pairs.
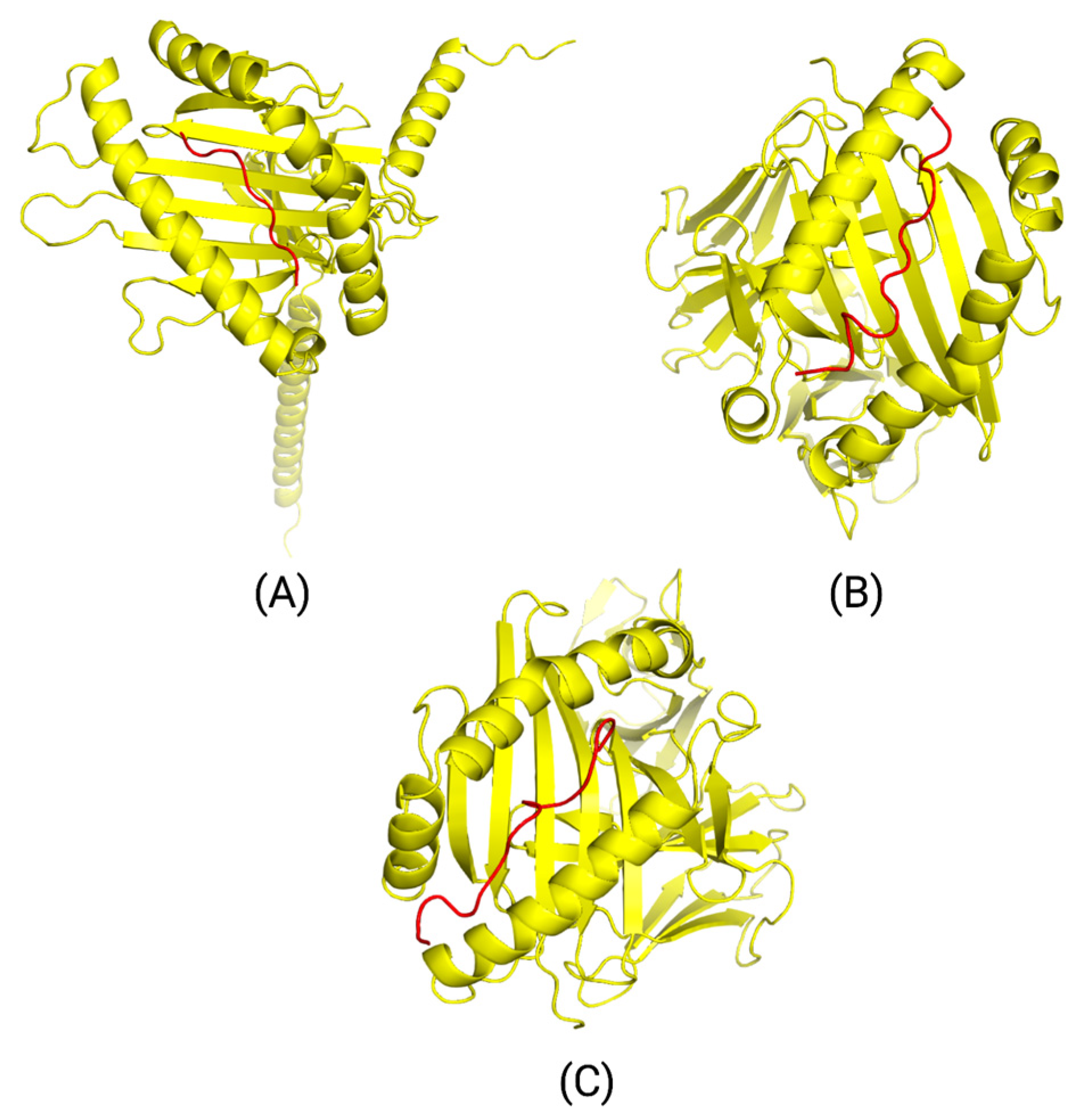
We modeled the interactions between three top peptides, HLA alleles, and T-cell receptors through TCRmodel and input them into MolProbity to analyze the overall structures. Additionally, we conducted a Ramachandran plot analysis to determine the Ramachandran favored and outlier regions along with the Rama z-score. Ramachandra plot analysis is a representation of the dihedral angles of each amino acid residue. Both phi (Φ) and psi (ψ) dihedral angle combinations were analyzed to determine Ramachandran favored and outlier regions [
54]. Shown in
Supplemental Tables S23–S25, Models B and C had ideal Ramachandran favored and outlier regions of less than 0.05% and greater than 98%, illustrating energetically favored dihedral angle combinations. Model A had a slightly higher amount of dihedral angle combinations that were energetically unfavorable. However, since the absolute value of the Rama z-score for all three models met the goal of less than 2, this demonstrated that no significant structural issues were present and that the proteins were properly folded. Furthermore, all models had an optimal clashing score at the 90th percentile or higher. Analyzing the structure through MolProbity not only allows for a more accurate assessment of the structure but also allows for greater precision when correcting issues related to the backbone or sidechain of the structure through the use of refinement and energy minimization software [
55].
Looking at the binding affinity, immunogenicity, and antigenicity of the final PIK3CA Class I and II epitopes, we can see that the top-ranked epitopes are characterized by strong binders with high immunogenic and antigenic potential. Therefore, AutoEpiCollect’s pan-cancer epitope-based vaccine design is predicted to elicit a strong, sustained immune response in patients with PIK3CA-mutated cancer cells. In addition, the high world population coverage for Class I epitopes shows that our pan-cancer vaccine has the potential to impact a large percentage of the population. These results support the applicability of AutoEpiCollect in developing the starting point for a pan-cancer vaccine, drastically reducing the time it takes it obtain epitopes for in vitro or in vivo studies. However, while AutoEpiCollect is a powerful tool for developing a pan-cancer vaccine, it ultimately cannot replace results validated by experimental or clinical data. AutoEpiCollect and our PIK3CA pan-cancer vaccine data should be used in future murine model trials to test the efficacy of our novel vaccine design and strengthen future epitope selection methods.
5. Limitations
AutoEpiCollect produced ranked Class I and II epitopes through a probabilistic logistic regression scoring function. While the Class I function demonstrated relatively high rates of accuracy at 76%, the Class II function had a significantly lower rate of 56%, due to epitope size constraints placed by in silico tools when collecting the training data. The model was ultimately trained based on limited experimental T-cell assays that were filtered to only include 15mers. This contributed to a lower diversity, as a significant portion of training data epitopes were removed. The lack of experimental data was also an issue when training the linear regression scoring functions, leading to low R
2 values. Another limitation that affected the Class II dataset was the set of HLA alleles unaccounted for by IEDB’s population coverage analysis tool. Many of the top Class II epitopes output by AutoEpiCollect were restricted by the HLA alleles shown in
Table 4. As a result, they were not included in the Class II population coverage analysis, leading to an underestimated world population coverage of 81.81%.
As our study also primarily focused on missense point mutations, the epitopes chosen only included a small portion of mutations. Mutations such as frameshift and deletion mutations were not considered in AutoEpiCollect. However, as mutations in the PIK3CA gene generally result in mosaicism or missense mutations in somatic cells, the software we created allows for the most common type of mutation to be targeted [
56]. As we only focused on one particular gene to determine the quality and accuracy of AutoEpiCollect, it assumes homogeneity across different cancer types.
With the epitopes derived from point mutations prevalent in our targeted cancer populations, we identified three top epitopes to model the interaction between the epitopes, HLA alleles, and T-cell receptors. All interactions portrayed energetically favored contributions with minimal clashes, excluding model A. The scores of these interactions are shown in
Supplemental Tables S23–S25. Though model A was properly folded overall, it had less than optimal Ramachandran outlier regions. These regions demonstrate energetically unfavorable dihedral angle combinations, contributing to a slightly weakened backbone. To address this issue, macromolecular refinement software, such as Phenix, can be utilized, along with energy minimization software [
52]. Analysis of these structures in MolProbity allows for more precise refinement. Moreover, it is crucial to highlight that modeling of the interactions between HLA alleles, epitopes, and T-cell receptors cannot accurately illustrate actual interactions within patients. With heterogeneity across different cancers and immune systems, in vitro and in vivo studies would need to be performed to reveal authentic interactions.
7. Conclusions
This study aimed to develop a novel, machine learning-driven vaccine design using our GUI automation software, AutoEpiCollect. Our software automatically gathers MHC Class I and II epitopes and outputs a list of top immunogenic epitopes with the use of a novel probabilistic logistic regression scoring function. With the implementation of AutoEpiCollect, pre-clinical processes of epitope collection can be significantly streamlined, allowing more resources to be allocated toward gathering clinical data for cancer vaccines. These clinical data can facilitate the advancement of precision oncology by targeting the underlying mutated genetic signatures of cancer, instead of solely the symptoms. Along with the potential to be used in combination with conventional therapies to overcome tumor resistance mechanisms, AutoEpiCollect’s epitope pan-cancer vaccines are applicable on a global scale for early disease intervention. Our automated epitope selection software is an invaluable step for the development of future precision oncology treatments and cancer prevention.
We tested AutoEpiCollect’s pan-cancer vaccine design on common cancers containing prevalent PIK3CA mutations. Previous cancer vaccines targeting the PIK3CA gene primarily target small sets of missense mutations, thus reducing the scope and efficiency of the vaccine [
1,
2,
3,
4,
5]. Therefore, no clinical trials have been conducted with pan-cancer vaccines targeting the PIK3CA gene. However, AutoEpiCollect aims to strengthen the efficacy of a pan-cancer vaccine by collecting missense mutations and ranking a large set of epitopes. After inputting 49 point mutations into AutoEpiCollect, we found MHC Class I epitopes targeting 34 of these mutations and MHC Class II epitopes targeting 11 mutations. Both Class I and II epitopes were ranked based on immunogenicity, antigenicity, allergenicity, and binding affinity and were subsequently filtered based on other clinically relevant parameters. Optimized Class I epitopes resulted in 98.09% world population coverage, while optimized Class II epitopes resulted in 81.81% world population coverage. This indicates that AutoEpiCollect’s pan-cancer vaccine design was able to generate a widely applicable vaccine for Class I epitopes. Due to the HLA allele constraints of the IEDB population coverage analysis tool, further analysis must be done to validate the world coverage of Class II epitope/HLA combinations. Modeled interactions of peptides, HLA alleles, and T-cell receptors from
Figure 8 were analyzed with MolProbity to demonstrate properly folded structures with minimal clashing.
One challenge when developing AutoEpiCollect’s vaccine design process was when collecting training data for the Class II scoring function. The lack of available experimental data for 15mers epitopes significantly decreased the variety of epitopes used to train the function; as a result, the accuracy for the Class II scoring function was 56% (AUC = 0.62) when implemented on the test data. To address this issue, more experimental data and alternative in silico tools will be explored that include predictions for non-15mers epitopes. This will expand the size and diversity of the Class II scoring function training dataset, potentially leading to higher model accuracies. Epitope selection is a complex process that requires a more holistic and nuanced approach. The future architecture of AutoEpiCollect’s epitope selection algorithm will include a deep learning framework that considers a vast number of epitope characteristics before predicting top epitopes. The deep learning model will be able to handle more complex experimental training data as well, thereby representing a wider range of gold-standard epitopes for a cancer vaccine. These improvements to the selection algorithm are intended to increase confidence in using in silico methods among future clinical researchers when conducting experimental cancer vaccine trials.
AutoEpiCollect is a comprehensive Python-based GUI application. To access the installation and user guide, please visit this project’s GitHub repository at
https://github.com/mvsamudrala/AutoEpiCollect (accessed on 2 February 2024) and click on the link provided below the “About this project” section. This guide will provide detailed instructions on how to install and operate AutoEpiCollect.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}