

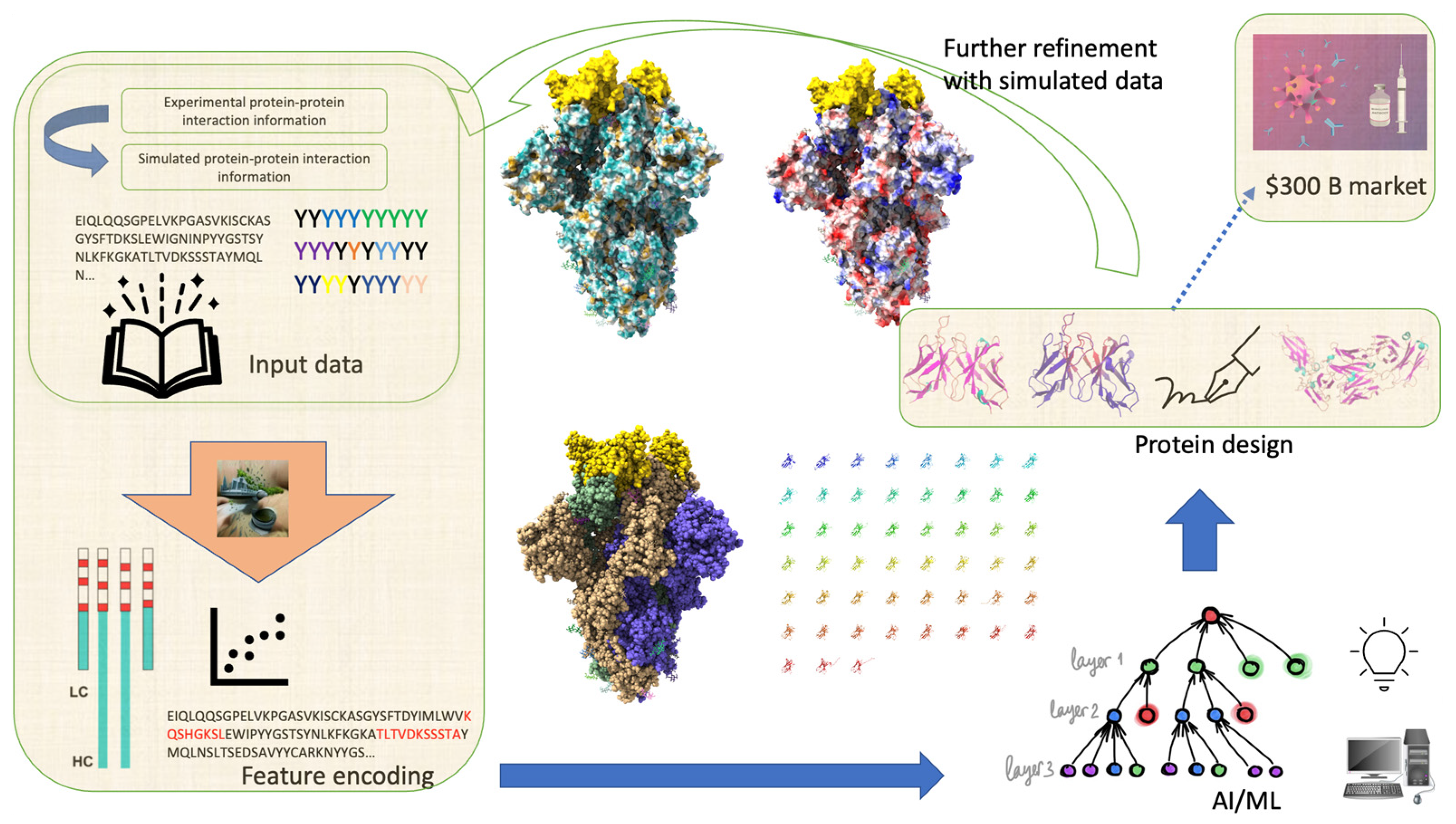
Leveraging Artificial Intelligence to Expedite Antibody Design and Enhance Antibody–Antigen Interactions


Abstract
1. Introduction
1.1. Historical Perspective and Rise of Deep Learning in the Biomedical Field
1.2. Sequence-Based and Structure-Based Approaches with Implications for Antibody Design
2. Antibody Design
2.1. Role of Antibodies in Mediating Protein–Protein Interactions
2.2. Generative Modeling for Antibody Sequences
2.2.1. Introduction to Gated Recurrent Units (GRUs) and Long Short-Term Memory (LSTM) Models
2.2.2. Introduction to Variational Autoencoders (VAEs)
2.2.3. Application of Generative Adversarial Networks (GANs)
2.2.4. Introduction to Autoregressive Method
3. Antibody Structural Modeling
3.1. Fragment Variable Structure and Predicting the Impacts of Mutations on the Structure and Function
3.2. Methods and Techniques in Screening for Binding Antibodies
3.3. Strategies in Designing Both Sequence and Structure in Optimizing Antibody Efficacy
3.4. Application of Diffusion Methods
3.5. Graph-Based Supervised Learning for Biophysical Property Prediction
3.6. Curation of Sequence and Structural Datasets to Develop Unsupervised Machine Learning Methods
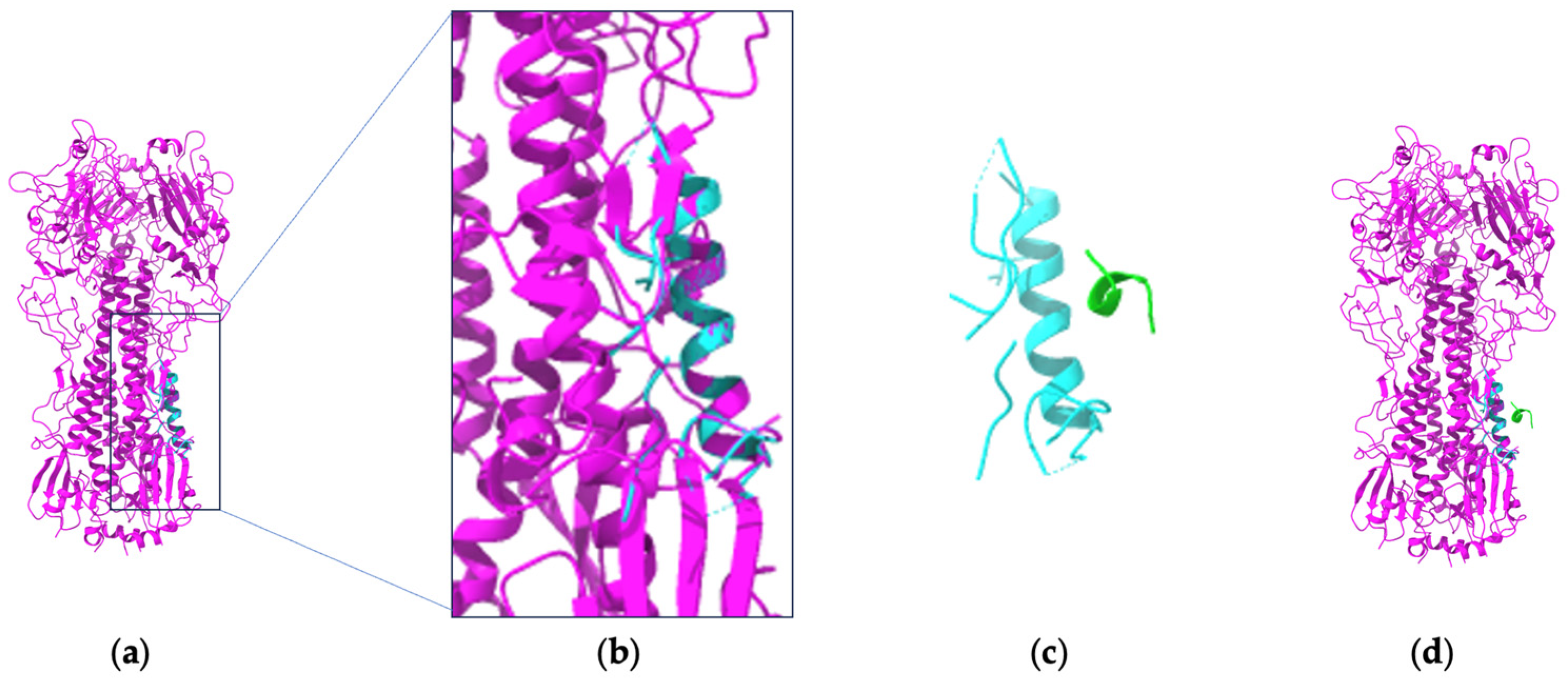
4. The Role of Antibodies and Deep Learning in the Fight against SARS-CoV-2
4.1. Understanding How SARS-CoV-2 Interacts with Host Cells
4.2. Overview of Experimental Datasets in Studying SARS-CoV-2
4.3. How Deep Learning Is Advancing Research on SARS-CoV-2
5. Conclusions and Future Directions
6. Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bailly, M.; Mieczkowski, C.; Juan, V.; Metwally, E.; Tomazela, D.; Baker, J.; Uchida, M.; Kofman, E.; Raoufi, F.; Motlagh, S.; et al. Predicting Antibody Developability Profiles through Early Stage Discovery Screening. mAbs 2020, 12, 1743053. [Google Scholar] [CrossRef] [PubMed]
- Wilman, W.; Wróbel, S.; Bielska, W.; Deszynski, P.; Dudzic, P.; Jaszczyszyn, I.; Kaniewski, J.; Młokosiewicz, J.; Rouyan, A.; Satława, T.; et al. Machine-designed biotherapeutics: Opportunities, feasibility and advantages of deep learning in computational antibody discovery. Brief. Bioinform. 2022, 23, bbac267. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsia, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Shaver, J.M.; Smith, J.; Amimeur, T. Deep Learning in Therapeutic Antibody Development. Methods Mol. Biol. 2022, 2390, 433–445. [Google Scholar] [PubMed]
- Graves, J.; Byerly, J.; Priego, E.; Makkapati, N.; Parish, S.V.; Medellin, B.; Berrondo, M. A Review of Deep Learning Methods for Antibodies. Antibodies 2020, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; Greiff, V.; Karatt-Vellatt, A.; Muyldermans, S.; Jenkins, T.P. Animal Immunization, in vitro Display Technologies, and Machine Learning for Antibody Discovery. Trends Biotechnol. 2021, 39, 1263–1273. [Google Scholar] [CrossRef]
- Greiff, V.; Yaari, G.; Cowell, L.G. Mining adaptive immune receptor repertoires for biological and clinical information using machine learning. Curr. Opin. Syst. Biol. 2020, 24, 109–119. [Google Scholar] [CrossRef]
- Kim, J.; McFee, M.; Fang, Q.; Abdin, O.; Kim, P.M. Computational and artificial intelligence-based methods for antibody development. Trends Pharmacol. Sci. 2023, 44, 175–189. [Google Scholar] [CrossRef]
- Mason, D.M.; Friedensohn, S.; Weber, C.R.; Jordi, C.; Wagner, B.; Meng, S.M.; Ehling, R.A.; Bonati, L.; Dahinden, J.; Gainza, P.; et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat. Biomed. Eng. 2021, 5, 600–612. [Google Scholar] [CrossRef]
- Deac, A.; VeliČković, P.; Sormanni, P. Attentive Cross-Modal Paratope Prediction. J. Comput. Biol. 2018, 26, 536–545. [Google Scholar] [CrossRef]
- Abanades, B.; Georges, G.; Bujotzek, A.; Deane, C.M. ABlooper: Fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinformatics 2022, 38, 1877–1880. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Sulam, J.; Gray, J.J. Antibody structure prediction using interpretable deep learning. Patterns 2022, 3, 100406. [Google Scholar] [CrossRef]
- Warszawski, S.; Katz, A.B.; Lipsh, R.; Khmelnitsky, L.; Nissan, G.B.; Javitt, G.; Dym, O.; Unger, T.; Knop, O.; Albeck, S.; et al. Optimizing antibody affinity and stability by the automated design of the variable light-heavy chain interfaces. PLoS Comput. Biol. 2019, 15, e1007207. [Google Scholar] [CrossRef]
- Koehler Leman, J.; Weitzner, B.D.; Renfrew, P.D.; Lewis, S.M.; Moretti, R.; Watkins, A.M.; Mulligan, V.K.; Lyskov, S.; Adolf-Bryfogle, J.; Labonte, J.W.; et al. Better together: Elements of successful scientific software development in a distributed collaborative community. PLoS Comput. Biol. 2020, 16, e1007507. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Luo, S.; Yang, Z.; Hong, J.; Su, Y.; Ding, F.; Fu, L.; Li, C.; Chen, P.; Ma, J.; et al. Deep learning guided optimization of human antibody against SARS-CoV-2 variants with broad neutralization. Proc. Natl. Acad. Sci. USA 2022, 119, e2122954119. [Google Scholar] [CrossRef]
- Huang, L.; Jiao, S.; Yang, S.; Zhang, S.; Zhu, X.; Guo, R.; Wang, Y. LGFC-CNN: Prediction of lncRNA-Protein Interactions by Using Multiple Types of Features through Deep Learning. Genes 2021, 12, 1689. [Google Scholar] [CrossRef] [PubMed]
- Knutson, C.; Bontha, M.; Bilbrey, J.A.; Kumar, N. Decoding the protein–ligand interactions using parallel graph neural networks. Sci. Rep. 2022, 12, 7624. [Google Scholar] [CrossRef]
- Bileschi, M.L.; Belanger, D.; Bryant, D.H.; Sanderson, T.; Carter, B.; Sculley, D.; Bateman, A.; DePristo, M.A.; Colwell, L.J. Using deep learning to annotate the protein universe. Nat. Biotechnol. 2022, 40, 932–937. [Google Scholar] [CrossRef]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.P.; Gebauer, N.W.A.; Bontha, M.; Khazaieli, M.; James, R.M.; Brown, J.B.; Kumar, N. 3D-Scaffold: A Deep Learning Framework to Generate 3D Coordinates of Drug-like Molecules with Desired Scaffolds. J. Phys. Chem. B 2021, 125, 12166–12176. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Conyne, M.; Lake, M.R.; Walter, K.A.; Min, J. In silico high throughput mutagenesis and screening of signal peptides to mitigate N-terminal heterogeneity of recombinant monoclonal antibodies. mAbs 2022, 14, 2044977. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Tyka, M.D.; Keedy, D.A.; André, I.; Dimaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef] [PubMed]
- McPartlon, M.; Xu, J. An end-to-end deep learning method for protein side-chain packing and inverse folding. Proc. Natl. Acad. Sci. USA 2023, 120, e2216438120. [Google Scholar] [CrossRef] [PubMed]
- Misiura, M.; Shroff, R.; Thyer, R.; Kolomeisky, A.B. DLPacker: Deep learning for prediction of amino acid side chain conformations in proteins. Proteins 2022, 90, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nakajima An, D.; Parks, J.M.; Skolnick, J. AF2Complex predicts direct physical interactions in multimeric proteins with deep learning. Nat. Commun. 2022, 13, 1744. [Google Scholar] [CrossRef]
- Basu, S.; Wallner, B. DockQ: A Quality Measure for Protein-Protein Docking Models. PLoS ONE 2016, 11, e0161879. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T. Antibody-Antigen Docking and Design via Hierarchical Structure Refinement. Proc. Mach. Learn. Res. 2022, 162, 10217. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762v7. [Google Scholar] [CrossRef]
- Barlow, D.J.; Edwards, M.S.; Thornton, J.M. Continuous and discontinuous protein antigenic determinants. Nature 1986, 322, 747–748. [Google Scholar] [CrossRef]
- Syrlybaeva, R.; Strauch, E.-M. Deep learning of Protein Sequence Design of Protein-protein Interactions. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wohlwend, J.; Barzilay, R.; Jaakkola, T. Iterative Refinement Graph Neural Network for Antibody Sequence-Structure Co-design. arXiv 2021, arXiv:2110.04624. [Google Scholar]
- Widrich, M.; Schäfl, B.; Ramsauer, H.; Pavlović, M.; Gruber, L.; Holzleitner, M.; Brandstetter, J.; Sandve, G.K.; Greiff, V.; Hochreiter, S.; et al. Modern Hopfield Networks and Attention for Immune Repertoire Classification. Adv. Neural Inf. Process. Syst. 2020, 33, 18832–18845. [Google Scholar]
- Goldenzweig, A.; Fleishman, S.J. Principles of Protein Stability and Their Application in Computational Design. Annu. Rev. Biochem. 2018, 87, 105–129. [Google Scholar] [CrossRef]
- Barlow, K.A.; Conchúir, S.Ó.; Thompson, S.; Suresh, P.; Lucas, J.E.; Heinonen, M.; Kortemme, T. Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein-Protein Binding Affinity upon Mutation. J. Phys. Chemistry. B 2018, 122, 5389–5399. [Google Scholar] [CrossRef] [PubMed]
- Dequeker, C.; Mohseni Behbahani, Y.; David, L.; Laine, E.; Carbone, A. From complete cross-docking to partners identification and binding sites predictions. PLoS Comput. Biol. 2022, 18, e1009825. [Google Scholar] [CrossRef] [PubMed]
- Saka, K.; Kakuzaki, T.; Metsugi, S.; Kashiwagi, D.; Yoshida, K.; Wada, M.; Tsunoda, H.; Teramoto, R. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci. Rep. 2021, 11, 5852. [Google Scholar] [CrossRef]
- Sher, G.; Zhi, D.; Zhang, S. DRREP: Deep ridge regressed epitope predictor. BMC Genom. 2017, 18, 676. [Google Scholar] [CrossRef]
- Akbar, R.; Bashour, H.; Rawat, P.; Robert, P.A.; Smorodina, E.; Cotet, T.S.; Flem-Karlsen, K.; Frank, R.; Mehta, B.B.; Vu, M.H.; et al. Progress and challenges for the machine learning-based design of fit-for-purpose monoclonal antibodies. mAbs 2022, 14, 2008790. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Schneider, C.; Raybould, M.I.J.; Deane, C.M. SAbDab in the age of biotherapeutics: Updates including SAbDab-nano, the nanobody structure tracker. Nucleic Acids Res. 2022, 50, D1368–D1372. [Google Scholar] [CrossRef]
- Ferdous, S.; Martin, A.C.R. AbDb: Antibody structure database—A database of PDB-derived antibody structures. Database J. Biol. Databases Curation 2018, 2018, bay040. [Google Scholar] [CrossRef]
- Sarkar, D.; Saha, S. Machine-learning techniques for the prediction of protein-protein interactions. J. Biosci. 2019, 44, 104. [Google Scholar] [CrossRef]
- Kuroda, D.; Gray, J.J. Shape complementarity and hydrogen bond preferences in protein-protein interfaces: Implications for antibody modeling and protein-protein docking. Bioinformatics 2016, 32, 2451–2456. [Google Scholar] [CrossRef] [PubMed]
- Greiff, V.; Menzel, U.; Miho, E.; Weber, C.; Riedel, R.; Cook, S.; Valai, A.; Lopes, T.; Radbruch, A.; Winkler, T.H.; et al. Systems Analysis Reveals High Genetic and Antigen-Driven Predetermination of Antibody Repertoires throughout B Cell Development. Cell Rep. 2017, 19, 1467–1478. [Google Scholar] [CrossRef]
- Elhanati, Y.; Sethna, Z.; Marcou, Q.; Callan, C.G., Jr.; Mora, T.; Walczak, A.M. Inferring processes underlying B-cell repertoire diversity. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2015, 370, 20140243. [Google Scholar] [CrossRef]
- Wang, M.; Cang, Z.; Wei, G.-W. A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nat. Mach. Intell. 2020, 2, 116–123. [Google Scholar] [CrossRef]
- Jankauskaitė, J.; Jiménez-García, B.; Dapkūnas, J.; Fernández-Recio, J.; Moal, I.H. SKEMPI 2.0: An updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 2019, 35, 462–469. [Google Scholar] [CrossRef]
- Biswas, S.; Khimulya, G.; Alley, E.C.; Esvelt, K.M.; Church, G.M. Low-N protein engineering with data-efficient deep learning. Nat. Methods 2021, 18, 389–396. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Whitehead, T.A.; Ekiert, D.C.; Dreyfus, C.; Corn, J.E.; Strauch, E.M.; Wilson, I.A.; Baker, D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 2011, 332, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Robert, P.A.; Akbar, R.; Frank, R.; Pavlović, M.; Widrich, M.; Snapkov, I.; Chernigovskaya, M.; Scheffer, L.; Slabodkin, A.; Mehta, B.B.; et al. One billion synthetic 3D-antibody-antigen complexes enable unconstrained machine-learning formalized investigation of antibody specificity prediction. bioRxiv 2021. [Google Scholar] [CrossRef]
- Joshi, R.P.; Kumar, N. Artificial intelligence for autonomous molecular design: A perspective. Molecules 2021, 26, 6761. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Ran, T.; Chen, H. De Novo Molecule Design through the Molecular Generative Model Conditioned by 3D Information of Protein Binding Sites. J. Chem. Inf. Model. 2021, 61, 3240–3254. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, S.; Huang, P.-S. Structure-based protein design with deep learning. Curr. Opin. Chem. Biol. 2021, 65, 136–144. [Google Scholar] [CrossRef]
- Wu, Z.; Johnston, K.E.; Arnold, F.H.; Yang, K.K. Protein sequence design with deep generative models. Curr. Opin. Chem. Biol. 2021, 65, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Defresne, M.; Barbe, S.; Schiex, T. Protein Design with Deep Learning. Int. J. Mol. Sci. 2021, 22, 11741. [Google Scholar] [CrossRef]
- Shin, J.-E.; Riesselman, A.J.; Kollasch, A.W.; McMahon, C.; Simon, E.; Sander, C.; Manglik, A.; Kruse, A.C.; Marks, D.S. Protein design and variant prediction using autoregressive generative models. Nat. Commun. 2021, 12, 2403. [Google Scholar] [CrossRef]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos, J.L., Jr.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef]
- Gruver, N.; Stanton, S.; Frey, N.C.; Rudner, T.G.J.; Hotzel, I.; Lafrance-Vanasse, J.; Rajpal, A.; Cho, K.; Wilson, A.G. Protein Design with Guided Discrete Diffusion. arXiv 2023, arXiv:2305.20009. [Google Scholar] [CrossRef]
- Shanehsazzadeh, A.; Bachas, S.; Kasun, G.; Sutton, J.M.; Steiger, A.K.; Shuai, R.; Kohnert, C.; Morehead, A.; Brown, A.; Chung, C.; et al. Unlocking de novo antibody design with generative artificial intelligence. bioRxiv 2023. [Google Scholar] [CrossRef]
- Murphy, G.S.; Sathyamoorthy, B.; Der, B.S.; Machius, M.C.; Pulavarti, S.V.; Szyperski, T.; Kuhlman, B. Computational de novo design of a four-helix bundle protein—DND_4HB. Protein Sci. A Publ. Protein Soc. 2015, 24, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J.; Hard, R.; Wang, W. The RESP AI model accelerates the identification of tight-binding antibodies. Nat. Commun. 2023, 14, 454. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zeng, H.; Mueller, J.; Carter, B.; Wang, Z.; Schilz, J.; Horny, G.; Birnbaum, M.E.; Ewert, S.; Gifford, D.K. Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics 2019, 36, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Akbar, R.; Robert, P.A.; Weber, C.R.; Widrich, M.; Frank, R.; Pavlović, M.; Scheffer, L.; Chernigovskaya, M.; Snapkov, I.; Slabodkin, A.; et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Hua, C.; Sentman, C.L.; Ackerman, M.E.; Bailey-Kellogg, C. Antibody humanization by structure-based computational protein design. mAbs 2015, 7, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Wollacott, A.M.; Xue, C.; Qin, Q.; Hua, J.; Bohnuud, T.; Viswanathan, K.; Kolachalama, V.B. Quantifying the nativeness of antibody sequences using long short-term memory networks. Protein Eng. Des. Sel. PEDS 2019, 32, 347–354. [Google Scholar] [CrossRef]
- Syrlybaeva, R.; Strauch, E.-M. One-sided design of protein-protein interaction motifs using deep learning. bioRxiv 2022. [Google Scholar] [CrossRef]
- Chaudhury, S.; Lyskov, S.; Gray, J.J. PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.; Ertelt, M.; Merkl, R.; Meiler, J. Rosetta design with co-evolutionary information retains protein function. PLoS Comput. Biol. 2021, 17, e1008568. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J.B.; Haddox, H.K.; Strickland, D.; Halabiya, S.F.; Coventry, B.; Griffin, J.R.; Pulavarti, S.V.S.R.K.; Cummins, M.; Thieker, D.F.; Klavins, E.; et al. Perturbing the energy landscape for improved packing during computational protein design. Proteins 2021, 89, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Stranges, P.B.; Kuhlman, B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013, 22, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Friedensohn, S.; Neumeier, D.; Khan, T.A.; Csepregi, L.; Parola, C.; de Vries, A.R.G.; Erlach, L.; Mason, D.M.; Reddy, S.T. Convergent selection in antibody repertoires is revealed by deep learning. bioRxiv 2020. [Google Scholar] [CrossRef]
- Davidsen, K.; Olson, B.J.; DeWitt, W.S., 3rd; Feng, J.; Harkins, E.; Bradley, P.; Matsen, F.A., 4th. Deep generative models for T cell receptor protein sequences. eLife 2019, 8, e46935. [Google Scholar] [CrossRef]
- Eguchi, R.R.; Anand, N.; Choe, C.A.; Huang, P.-S. IG-VAE: Generative Modeling of Immunoglobulin Proteins by Direct 3D Coordinate Generation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef]
- Brock, A.; Donahue, J.; Simonyan, K. Large Scale GAN Training for High Fidelity Natural Image Synthesis. arXiv 2018, arXiv:1809.11096. [Google Scholar]
- Amimeur, T.; Shaver, J.M.; Ketchem, R.R.; Taylor, J.A.; Clark, R.H.; Smith, J.; Van Citters, D.; Siska, C.C.; Smidt, P.; Sprague, M.; et al. Designing Feature-Controlled Humanoid Antibody Discovery Libraries Using Generative Adversarial Networks. bioRxiv 2020. [Google Scholar] [CrossRef]
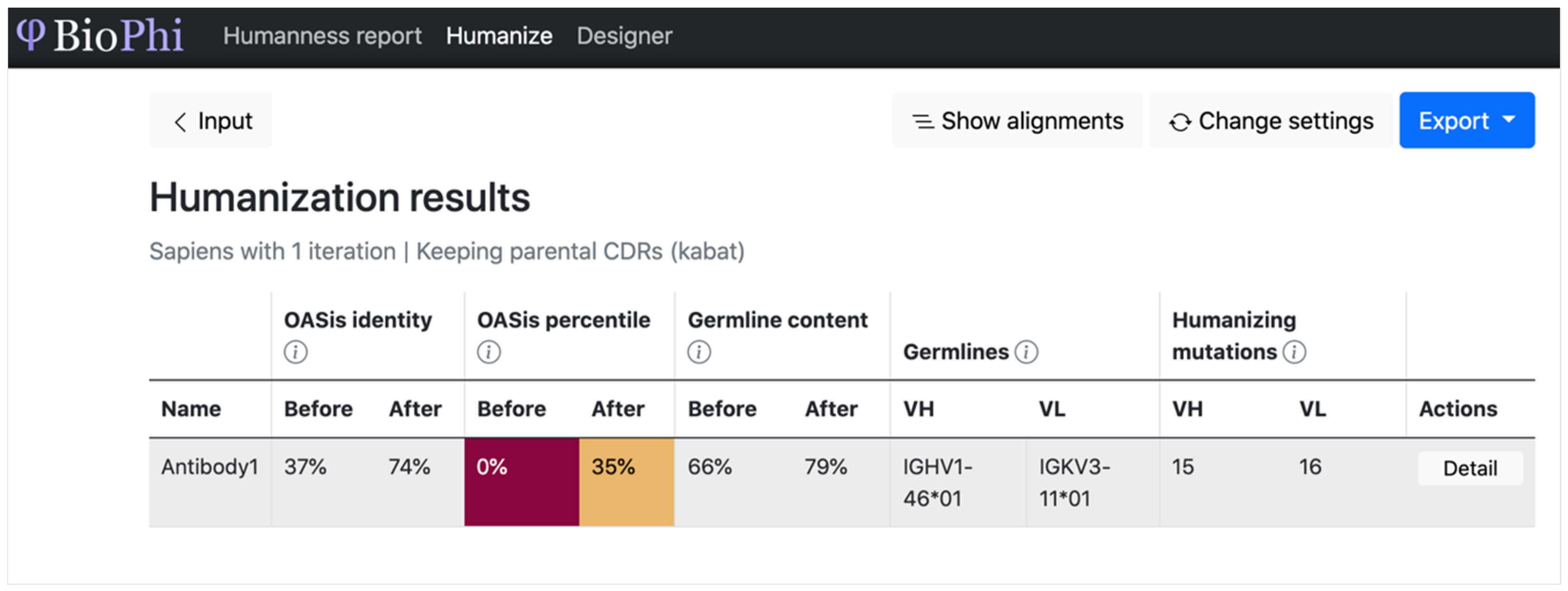
- Prihoda, D.; Maamary, J.; Waight, A.; Juan, V.; Fayadat-Dilman, L.; Svozil, D.; Bitton, D.A. BioPhi: A platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning. mAbs 2022, 14, 2020203. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.H.; Boyles, F.; Deane, C.M. Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci. A Publ. Protein Soc. 2022, 31, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Shuai, R.W.; Ruffolo, J.A.; Gray, J.J. Generative Language Modeling for Antibody Design. bioRxiv 2021. [Google Scholar] [CrossRef]
- Han, W.; Chen, N.; Xu, X.; Sahil, A.; Zhou, J.; Li, Z.; Zhong, H.; Gao, E.; Zhang, R.; Wang, Y.; et al. Predicting the antigenic evolution of SARS-COV-2 with deep learning. Nat. Commun. 2023, 14, 3478. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, I.; Chenthamarakshan, V.; Chen, P.-Y.; Das, P.; Dhurandhar, A.; Padhi, I.; Das, D. Reprogramming Pretrained Language Models for Antibody Sequence Infilling. arXiv 2022, arXiv:2210.07144. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Xu, J.; Mcpartlon, M.; Li, J. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell. 2021, 3, 601–609. [Google Scholar] [CrossRef]
- Vig, J. Visualizing Attention in Transformer-Based Language Representation Models. arXiv 2019, arXiv:1904.02679v2. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, X.; Wei, Y.; Huang, L.; Shi, H.; Liu, W.; Huang, T.S. CCNet: Criss-Cross Attention for Semantic Segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2020, 45, 6896–6908. [Google Scholar] [CrossRef] [PubMed]
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat. Methods 2020, 17, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Ruffolo, J.A.; Chu, L.-S.; Mahajan, S.P.; Gray, J.J. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Nat. Commun. 2023, 14, 2389. [Google Scholar] [CrossRef] [PubMed]
- Ferruz, N.; Höcker, B. Controllable protein design with language models. Nat. Mach. Intell. 2022, 4, 521–532. [Google Scholar] [CrossRef]
- Abanades, B.; Wong, W.K.; Boyles, F.; Georges, G.; Bujotzek, A.; Deane, C.M. ImmuneBuilder: Deep-Learning models for predicting the structures of immune proteins. Commun. Biol. 2023, 6, 575. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Buchanan, A.; Taddese, B.; Deane, C.M. DLAB: Deep learning methods for structure-based virtual screening of antibodies. Bioinformatics 2021, 38, 377–383. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Mahajan, S.; Peters, B.; Nielsen, M.; Marcatili, P. Antibody Specific B-Cell Epitope Predictions: Leveraging Information from Antibody-Antigen Protein Complexes. Front. Immunol. 2019, 10, 298. [Google Scholar] [CrossRef]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef]
- Imrie, F.; Bradley, A.R.; van der Schaar, M.; Deane, C.M. Protein Family-Specific Models Using Deep Neural Networks and Transfer Learning Improve Virtual Screening and Highlight the Need for More Data. J. Chem. Inf. Model. 2018, 58, 2319–2330. [Google Scholar] [CrossRef]
- Li, N.; Kaehler, O.; Pfeifer, N. A Comparison of Deep Learning Methods for Airborne Lidar Point Clouds Classification. IEEE J. Sel. Top. Appl. Earth Obs. Remote Sens. 2021, 14, 6467–6486. [Google Scholar] [CrossRef]
- Rosebrock, A. Are CNNs Invariant to Translation, Rotation, and Scaling? 2021. Available online: https://www.pyimagesearch.com/2021/05/14/are-cnns-invariant-to-translation-rotation-and-scaling/ (accessed on 5 February 2024).
- Balci, A.T.; Gumeli, C.; Hakouz, A.; Yuret, D.; Keskin, O.; Gursoy, A. DeepInterface: Protein-protein interface validation using 3D Convolutional Neural Networks. bioRxiv 2019. [Google Scholar] [CrossRef]
- Si, D.; Moritz, S.A.; Pfab, J.; Hou, J.; Cao, R.; Wang, L.; Wu, T.; Cheng, J. Deep Learning to Predict Protein Backbone Structure from High-Resolution Cryo-EM Density Maps. Sci. Rep. 2020, 10, 4282. [Google Scholar] [CrossRef]
- Bepler, T.; Zhong, E.D.; Kelley, K.; Brignole, E.; Berger, B.; Wallach, H. Explicitly disentangling image content from translation and rotation with spatial-VAE. arXiv 2019, arXiv:1909.11663. [Google Scholar]
- Zhou, Q.-Y.; Park, J.; Koltun, V. Open3D: A Modern Library for 3D Data Processing. arXiv 2018, arXiv:1801.09847. [Google Scholar] [CrossRef]
- Leem, J.; Dunbar, J.; Georges, G.; Shi, J.; Deane, C.M. ABodyBuilder: Automated antibody structure prediction with data-driven accuracy estimation. mAbs 2016, 8, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating Protein Docking in ZDOCK Using an Advanced 3D Convolution Library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef]
- Hie, B.L.; Shanker, V.R.; Xu, D.; Bruun, T.U.J.; Weidenbacher, P.A.; Tang, S.; Wu, W.; Pak, J.E.; Kim, P.S. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Outeiral, C.; Deane, C.M. Perfecting antibodies with language models. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, B.; Xu, F.; Li, C.; Xu, Z.; Su, X.; He, H.; Huang, Y.; Rossjohn, J.; Song, J.; et al. DeepAIR: A deep learning framework for effective integration of sequence and 3D structure to enable adaptive immune receptor analysis. Sci. Adv. 2023, 9, eabo5128. [Google Scholar] [CrossRef]
- Adolf-Bryfogle, J.; Kalyuzhniy, O.; Kubitz, M.; Weitzner, B.D.; Hu, X.; Adachi, Y.; Schief, W.R.; Dunbrack, R.L., Jr. RosettaAntibodyDesign (RAbD): A general framework for computational antibody design. PLoS Comput. Biol. 2018, 14, e1006112. [Google Scholar] [CrossRef] [PubMed]
- Martinkus, K.; Ludwiczak, J.; Cho, K.; Liang, W.-C.; Lafrance-Vanasse, J.; Hotzel, I.; Rajpal, A.; Wu, Y.; Bonneau, R.; Gligorijevic, V.; et al. AbDiffuser: Full-Atom Generation of In-Vitro Functioning Antibodies. arXiv 2023, arXiv:2308.05027. [Google Scholar]
- Alamdari, S.; Thakkar, N.; van den Berg, R.; Lu, A.X.; Fusi, N.; Amini, A.P.; Yang, K.K. Protein generation with evolutionary diffusion: Sequence is all you need. bioRxiv 2023. [Google Scholar] [CrossRef]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models. bioRxiv 2022. [Google Scholar] [CrossRef]
- Luo, S.; Su, Y.; Peng, X.; Wang, S.; Peng, J.; Ma, J. Antigen-Specific Antibody Design and Optimization with Diffusion-Based Generative Models for Protein Structures. bioRxiv 2022. [Google Scholar] [CrossRef]
- Chu, A.E.; Cheng, L.; El Nesr, G.; Xu, M.; Huang, P.S. An all-atom protein generative model. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, J.; Kim, P.M. Score-based generative modeling for de novo protein design. Nat. Comput. Sci. 2023, 3, 382–392. [Google Scholar] [CrossRef]
- Yim, J.; Trippe, B.L.; De Bortoli, V.; Mathieu, E.; Doucet, A.; Barzilay, R.; Jaakkola, T. SE(3) diffusion model with application to protein backbone generation. arXiv 2023, arXiv:2302.02277. [Google Scholar] [CrossRef]
- Ingraham, J.B.; Baranov, M.; Costello, Z.; Barber, K.W.; Wang, W.; Ismail, A.; Frappier, V.; Lord, D.M.; Ng-Thow-Hing, C.; Van Vlack, E.R.; et al. Illuminating protein space with a programmable generative model. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Ni, B.; Kaplan, D.L.; Buehler, M.J. Generative design of de novo proteins based on secondary-structure constraints using an attention-based diffusion model. Chem 2023, 9, 1828–1849. [Google Scholar] [CrossRef] [PubMed]
- Anand, N.; Achim, T. Protein Structure and Sequence Generation with Equivariant Denoising Diffusion Probabilistic Models. arXiv 2022, arXiv:2205.15019. [Google Scholar] [CrossRef]
- Lisanza, S.L.; JGershon, J.M.; Tipps, S.; Arnoldt, L.; Hendel, S.; Sims, J.N.; Li, X.; Baker, D. Joint Generation of Protein Sequence and Structure with RoseTTAFold Sequence Space Diffusion. bioRxiv 2023. [Google Scholar] [CrossRef]
- Nakata, S.; Mori, Y.; Tanaka, S. End-to-end protein–ligand complex structure generation with diffusion-based generative models. BMC Bioinform. 2023, 24, 233. [Google Scholar] [CrossRef]
- Bilbrey, J.; Ward, L.; Choudhury, S.; Kumar, N.; Sivaraman, G. Evening the Score: Targeting SARS-CoV-2 Protease Inhibition in Graph Generative Models for Therapeutic Candidates. arXiv 2021, arXiv:2105.10489. [Google Scholar]
- Ganea, O.-E.; Huang, X.; Bunne, C.; Bian, Y.; Barzilay, R.; Jaakkola, T.; Krause, A. Independent {SE}(3)-Equivariant Models for End-to-End Rigid Protein Docking. arXiv 2022, arXiv:2111.07786. [Google Scholar]
- Wang, X.; Zhu, H.; Jiang, Y.; Li, Y.; Tang, C.; Chen, X.; Li, Y.; Liu, Q.; Liu, Q. PRODeepSyn: Predicting anticancer synergistic drug combinations by embedding cell lines with protein–protein interaction network. Brief. Bioinform. 2022, 23, bbab587. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, Y.; Li, P.; Song, S.; Peng, J. Deep geometric representations for modeling effects of mutations on protein-protein binding affinity. PLoS Comput. Biol. 2021, 17, e1009284. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Gong, W.; Li, Z.; Yang, X.; Wang, J.; Wang, H. Predicting Protein–Protein Interactions via Gated Graph Attention Signed Network. Biomolecules 2021, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Mahbub, S.; Bayzid, M.S. EGRET: Edge aggregated graph attention networks and transfer learning improve protein–protein interaction site prediction. Brief. Bioinform. 2022, 23, bbab578. [Google Scholar] [CrossRef]
- Yuan, Q.; Chen, J.; Zhao, H.; Zhou, Y.; Yang, Y. Structure-aware protein-protein interaction site prediction using deep graph convolutional network. Bioinformatics 2021, 38, 125–132. [Google Scholar] [CrossRef]
- Réau, M.; Renaud, N.; Xue, L.C.; Bonvin, A.M.J.J. DeepRank-GNN: A Graph Neural Network Framework to Learn Patterns in Protein-Protein Interfaces. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kang, Y.; Leng, D.; Guo, J.; Pan, L. Sequence-based deep learning antibody design for in silico antibody affinity maturation. arXiv 2021, arXiv:2103.03724. [Google Scholar]
- Renz, P.; Van Rompaey, D.; Wegner, J.K.; Hochreiter, S.; Klambauer, G. On failure modes in molecule generation and optimization. Drug Discov. Today. Technol. 2019, 32–33, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Raybould, M.I.; Marks, C.; Krawczyk, K.; Taddese, B.; Nowak, J.; Lewis, A.P.; Bujotzek, A.; Shi, J.; Deane, C.M. Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. USA 2019, 116, 4025–4030. [Google Scholar] [CrossRef]
- Jin, W. Structured Refinement Network for Antibody Design. 2022. Available online: https://www.youtube.com/watch?v=uDTccbg_Ai4&list=PL27Hzl3ugX__okAYK-HmUJ8wHEVS1n_5u&index=1&t=1035s&ab_channel=ValenceDiscovery (accessed on 5 February 2024).
- Myung, Y.; Pires, D.E.V.; Ascher, D.B. CSM-AB: Graph-based antibody-antigen binding affinity prediction and docking scoring function. Bioinformatics 2021, 38, 1141–1143. [Google Scholar] [CrossRef]
- Julie Josse, N.P.; Scornet, E.; Varoquaux, G. On the consistency of supervised learning with missing values. arXiv 2020, arXiv:1902.06931. [Google Scholar]
- Peters, M.E.; Neumann, M.; Iyyer, M.; Gardner, M.; Clark, C.; Lee, K.; Zettlemoyer, L. Deep contextualized word representations. arXiv 2018, arXiv:1802.05365. [Google Scholar]
- Villegas-Morcillo, A.; Makrodimitris, S.; van Ham, R.C.; Gomez, A.M.; Sanchez, V.; Reinders, M.J. Unsupervised protein embeddings outperform hand-crafted sequence and structure features at predicting molecular function. Bioinformatics 2021, 37, 162–170. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Levinthal, C. Are there pathways for protein folding? J. Chim. Phys. 1968, 65, 44–45. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lima, W.C.; Gasteiger, E.; Marcatili, P.; Duek, P.; Bairoch, A.; Cosson, P. The ABCD database: A repository for chemically defined antibodies. Nucleic Acids Res. 2020, 48, D261–D264. [Google Scholar] [CrossRef] [PubMed]
- Raybould, M.I.J.; Kovaltsuk, A.; Marks, C.; Deane, C.M. CoV-AbDab: The coronavirus antibody database. Bioinformatics 2021, 37, 734–735. [Google Scholar] [CrossRef] [PubMed]
- Corrie, B.D.; Marthandan, N.; Zimonja, B.; Jaglale, J.; Zhou, Y.; Barr, E.; Knoetze, N.; Breden, F.M.; Christley, S.; Scott, J.K.; et al. iReceptor: A platform for querying and analyzing antibody/B-cell and T-cell receptor repertoire data across federated repositories. Immunol. Rev. 2018, 284, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Adam, D. The pandemic’s true death toll: Millions more than official counts. Nature 2022, 601, 312–315. [Google Scholar] [CrossRef]
- Shi, Z.; Li, X.; Wang, L.; Sun, Z.; Zhang, H.; Chen, X.; Cui, Q.; Qiao, H.; Lan, Z.; Zhang, X.; et al. Structural basis of nanobodies neutralizing SARS-CoV-2 variants. Structure 2022, 30, 707–720.e5. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Zhang, X.W.; Yap, Y.L. The 3D structure analysis of SARS-CoV S1 protein reveals a link to influenza virus neuraminidase and implications for drug and antibody discovery. Theochem 2004, 681, 137–141. [Google Scholar] [CrossRef]
- Chaouat, A.E.; Achdout, H.; Kol, I.; Berhani, O.; Roi, G.; Vitner, E.B.; Melamed, S.; Politi, B.; Zahavy, E.; Brizic, I.; et al. SARS-CoV-2 receptor binding domain fusion protein efficiently neutralizes virus infection. PLoS Pathog. 2021, 17, e1010175. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, Y.B.; Gonzalez, K.J.; Strauch, E.-M. Targeting Viral Surface Proteins through Structure-Based Design. Viruses 2021, 13, 1320. [Google Scholar] [CrossRef] [PubMed]
- Marcandalli, J.; Fiala, B.; Ols, S.; Perotti, M.; de van der Schueren, W.; Snijder, J.; Hodge, E.; Benhaim, M.; Ravichandran, R.; Carter, L.; et al. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 2019, 176, 1420–1431.e17. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Du, J.; Liu, J.; Wu, H.; Gui, F.; Zhang, N.; Deng, X.; Song, G.; Li, Y.; Lu, J.; et al. Screening of potent neutralizing antibodies against SARS-CoV-2 using convalescent patients-derived phage-display libraries. Cell Discov. 2021, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.Z.; Garg, P.; Wang, L.; Willis, J.R.; Kwan, E.; Hernandez, A.G.L.; Tuscano, E.; Sever, E.N.; Keane, E.; Soto, C.; et al. Rapid discovery of diverse neutralizing SARS-CoV-2 antibodies from large-scale synthetic phage libraries. mAbs 2022, 14, 2002236. [Google Scholar] [CrossRef] [PubMed]
- Shiakolas, A.R.; Kramer, K.J.; Johnson, N.V.; Wall, S.C.; Suryadevara, N.; Wrapp, D.; Periasamy, S.; Pilewski, K.A.; Raju, N.; Nargi, R.; et al. Efficient discovery of SARS-CoV-2-neutralizing antibodies via B cell receptor sequencing and ligand blocking. Nat. Biotechnol. 2022, 40, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Abubaker Bagabir, S.; Ibrahim, N.K.; Abubaker Bagabir, H.; Hashem Ateeq, R. COVID-19 and Artificial Intelligence: Genome sequencing, drug development and vaccine discovery. J. Infect. Public Health 2022, 15, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rincon, A.; Tonda, A.; Mendoza-Maldonado, L.; Mulders, D.G.; Molenkamp, R.; Perez-Romero, C.A.; Claassen, E.; Garssen, J.; Kraneveld, A.D. Classification and specific primer design for accurate detection of SARS-CoV-2 using deep learning. Sci. Rep. 2021, 11, 947. [Google Scholar] [CrossRef]
- Zeng, X.; Song, X.; Ma, T.; Pan, X.; Zhou, Y.; Hou, Y.; Zhang, Z.; Li, K.; Karypis, G.; Cheng, F. Repurpose Open Data to Discover Therapeutics for COVID-19 Using Deep Learning. J. Proteome Res. 2020, 19, 4624–4636. [Google Scholar] [CrossRef]
- Wang, B.; Jin, S.; Yan, Q.; Xu, H.; Luo, C.; Wei, L.; Zhao, W.; Hou, X.; Ma, W.; Xu, Z.; et al. AI-assisted CT imaging analysis for COVID-19 screening: Building and deploying a medical AI system. Appl. Soft Comput. 2021, 98, 106897. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, K.; Wang, R.; Nguyen, D.D.; Wei, G.-W. Review of COVID-19 Antibody Therapies. Annu. Rev. Biophys. 2021, 50, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Darmawan, J.T.; Leu, J.-S.; Avian, C.; Ratnasari, N.R.P. MITNet: A fusion transformer and convolutional neural network architecture approach for T-cell epitope prediction. Brief. Bioinform. 2023, 24, bbad202. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.N.H.; Jain, A.; Haq, E.; Mehbodniya, A.; Webber, J. Machine Learning Techniques for the Prediction of B-Cell and T-Cell Epitopes as Potential Vaccine Targets with a Specific Focus on SARS-CoV-2 Pathogen: A Review. Pathogens 2022, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jin, J.; Cui, Y.; Xiong, Z.; Nasiri, A.; Zhao, Y.; Hu, J. DeepSeqPanII: An interpretable recurrent neural network model with attention mechanism for peptide-HLA class II binding prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 19, 2188–2196. [Google Scholar] [CrossRef]
- Hess, M.; Keul, F.; Goesele, M.; Hamacher, K. Addressing inaccuracies in BLOSUM computation improves homology search performance. BMC Bioinform. 2016, 17, 189. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Peters, B.; Sette, A.; Justesen, S.; Buus, S.; Lund, O. Quantitative Predictions of Peptide Binding to Any HLA-DR Molecule of Known Sequence: NetMHCIIpan. PLoS Comput. Biol. 2008, 4, e1000107. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Kanyavuz, A.; Marey-Jarossay, A.; Lacroix-Desmazes, S.; Dimitrov, J.D. Breaking the law: Unconventional strategies for antibody diversification. Nature reviews. Immunology 2019, 19, 355–368. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Ong, E.; Wang, H.; Wong, M.U.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign-ML: Supervised machine learning reverse vaccinology model for improved prediction of bacterial protective antigens. Bioinformatics 2020, 36, 3185–3191. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.W.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. Chapter nineteen—Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. In Computer Methods, Part C; Johnson, M.L., Brand, L., Eds.; Academic Press: Hoboken, NJ, USA, 2011; Volume 487, pp. 545–574. [Google Scholar]
- Leaver-Fay, A.; Froning, K.J.; Atwell, S.; Aldaz, H.; Pustilnik, A.; Lu, F.; Huang, F.; Yuan, R.; Hassanali, S.; Chamberlain, A.K.; et al. Computationally Designed Bispecific Antibodies using Negative State Repertoires. Structure 2016, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198. [Google Scholar] [CrossRef]
- Miklos, A.E.; Kluwe, C.; Der, B.S.; Pai, S.; Sircar, A.; Hughes, R.A.; Berrondo, M.; Xu, J.; Codrea, V.; Buckley, P.E.; et al. Structure-based design of supercharged, highly thermoresistant antibodies. Chem. Biol. 2012, 19, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.N.; Jacobs, T.M.; Kuhlman, B. Boosting protein stability with the computational design of β-sheet surfaces. Protein Sci. 2016, 25, 702–710. [Google Scholar] [CrossRef]
- Harmalkar, A.; Rao, R.; Richard Xie, Y.; Honer, J.; Deisting, W.; Anlahr, J.; Hoenig, A.; Czwikla, J.; Sienz-Widmann, E.; Rau, D.; et al. Toward generalizable prediction of antibody thermostability using machine learning on sequence and structure features. mAbs 2023, 15, 2163584. [Google Scholar] [CrossRef]
- Liang, T.; Jiang, C.; Yuan, J.; Othman, Y.; Xie, X.Q.; Feng, Z. Differential performance of RoseTTAFold in antibody modeling. Brief. Bioinform. 2022, 23, bbac152. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; Kraml, J.; Georges, G.; Liedl, K.R. CDR-H3 loop ensemble in solution—Conformational selection upon antibody binding. mAbs 2019, 11, 1077–1088. [Google Scholar] [CrossRef]
- Gainza, P.; Sverrisson, F.; Monti, F.; Rodola, E.; Boscaini, D.; Bronstein, M.M.; Correia, B.E. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 2020, 17, 184–192. [Google Scholar] [CrossRef]
- Guo, L.; He, J.; Lin, P.; Huang, S.-Y.; Wang, J. TRScore: A three-dimensional RepVGG-based scoring method for ranking protein docking models. Bioinformatics 2022, 38, 2444–2451. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language Models are Few-Shot Learners. arXiv 2020, arXiv:2005.14165. [Google Scholar]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Temitope Sobodu. How to Deploy and Interpret AlphaFold2 with Minimal Compute. 2023. Available online: https://towardsdatascience.com/how-to-deploy-and-interpret-alphafold2-with-minimal-compute-9bf75942c6d7 (accessed on 5 February 2024).
- Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 2022, 31, e4379. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, F.J.; Graziadei, A.; Forbrig, C.; Bremenkamp, R.; Charles, K.; Lenz, S.; Elfmann, C.; Fischer, L.; Stülke, J.; Rappsilber, J. Protein complexes in cells by AI-assisted structural proteomics. Mol. Syst. Biol. 2023, 19, e11544. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2016.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Source | Description | Number of Entries |
|---|---|---|
| AbDb [45] | Expert-curated Ab structure database | ~2 k full structures |
| Absolut! [54] | In silico generated Ab–Ag bindings | 159 antigens times 6.9 million CDR-H3 murine sequences |
| AntiBodies Chemically Defined Database (ABCD) [145] | Manually curated depository of sequenced Abs | 23 k sequenced Abs against 4 k Ags |
| CoV-AbDab (in SAbDab) [146] | Coronavirus-binding Ab sequences and structures | 4 k homology models and 500 PDB structures |
| iReceptor [147] | Ab/B-cell and T-cell receptor repertoire data | >5 B |
| Observed Antibody Space (OAS) [84] | Paired and unpaired (VH/VL) Ab sequences | >1 B |
| SAbDab [44] | Ab structures available in PDB | >5 k |
| Data Source | Description | Number of Entries |
|---|---|---|
| IntAct [148] | Binary interactions from the literature and user submissions | >1 M |
| MINT (in IntAct) | Protein interaction information disseminated in the literature | >130 k |
| SKEMPI V2.0 | Structural Kinetic and Energetic database of Mutant Protein Interactions | 7 k |
| STRING [149] | Direct (physical) and indirect (functional) PPIs | >20 B |
| Model | Goal | Input Type | Output | Architecture | Metrics | Note |
|---|---|---|---|---|---|---|
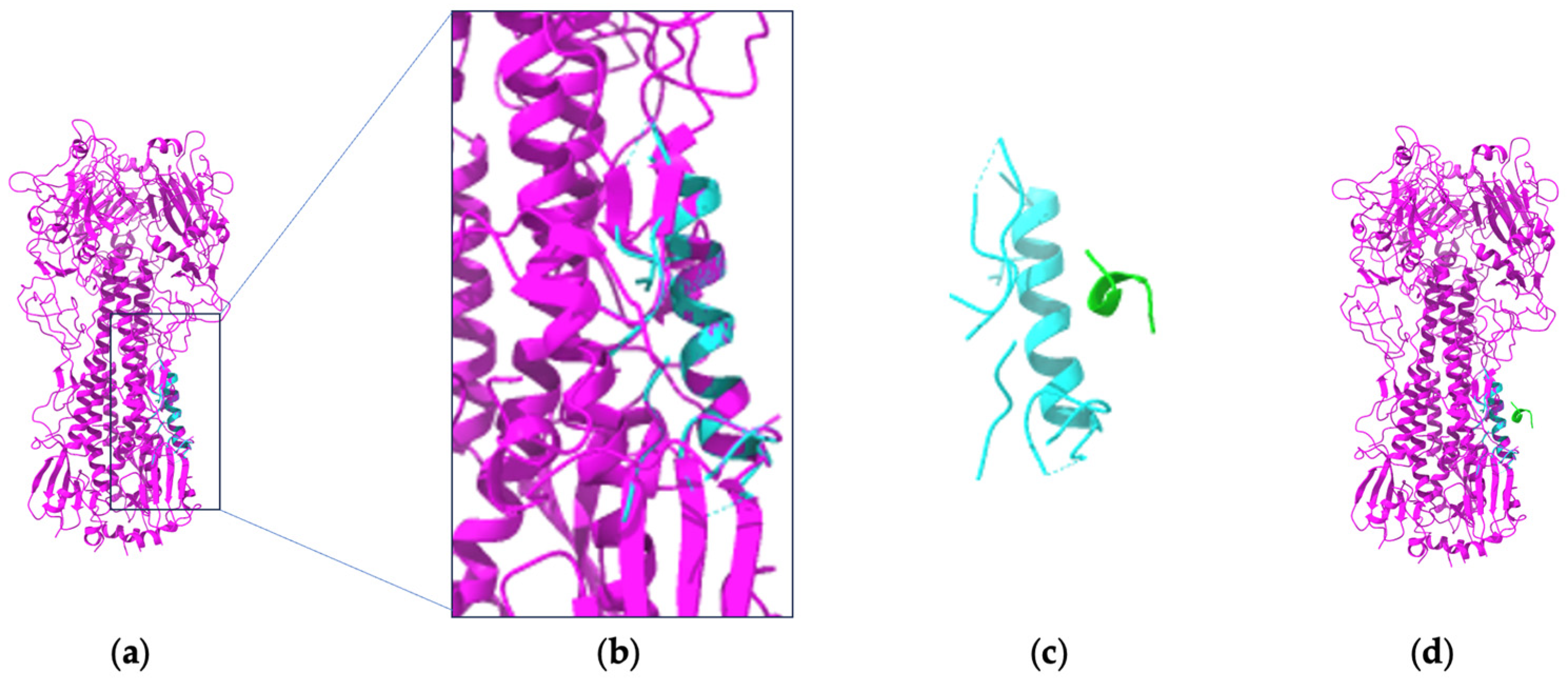
| Binding-ddg-predictor | Redesign the CDR to enhance Ab affinity (targeting multiple virus variants) | Sequence | Predicted binding affinity | Attention-based geometric neural network | kD (dissociation constant) | Through an iterative optimization procedure, this DL method found that the optimized Ab exhibited broader and much more potent neutralizing activity compared to the original Ab |
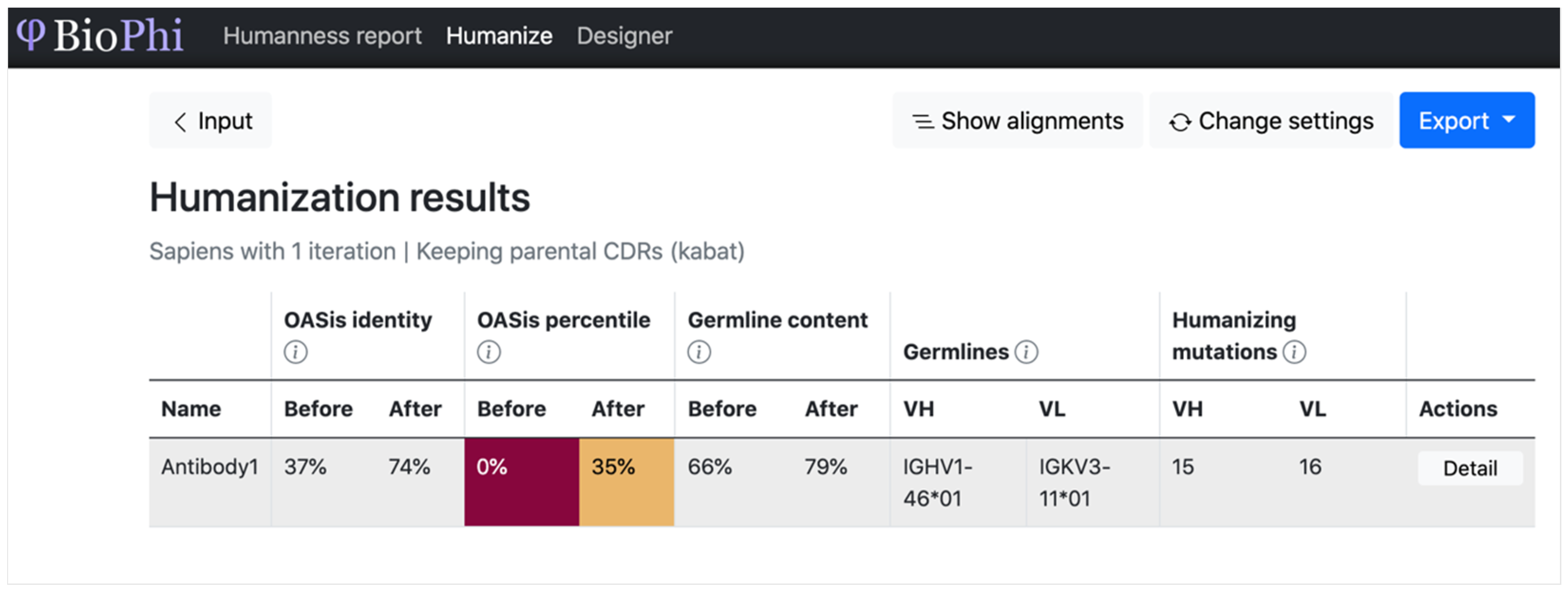
| BioPhi | Humanize the sequence and evaluate the humanness of the sequence | Sequence | Sequence and predicted humanization | Transformer | Accuracy (%), ROC, AUC, and R2 | Different methods were more successful in different cases, further encouraging the assembly of a diverse arsenal of humanization methods |
| DeepAb | Predict the Ab mutation effect on binding | Sequence | Structure and predicted affinity | RNN for sequence representation and ResNet to predict six distances and angles | Orientational coordinate distance and AUC | Provides an attention layer to interpret the features contributing to its predictions |
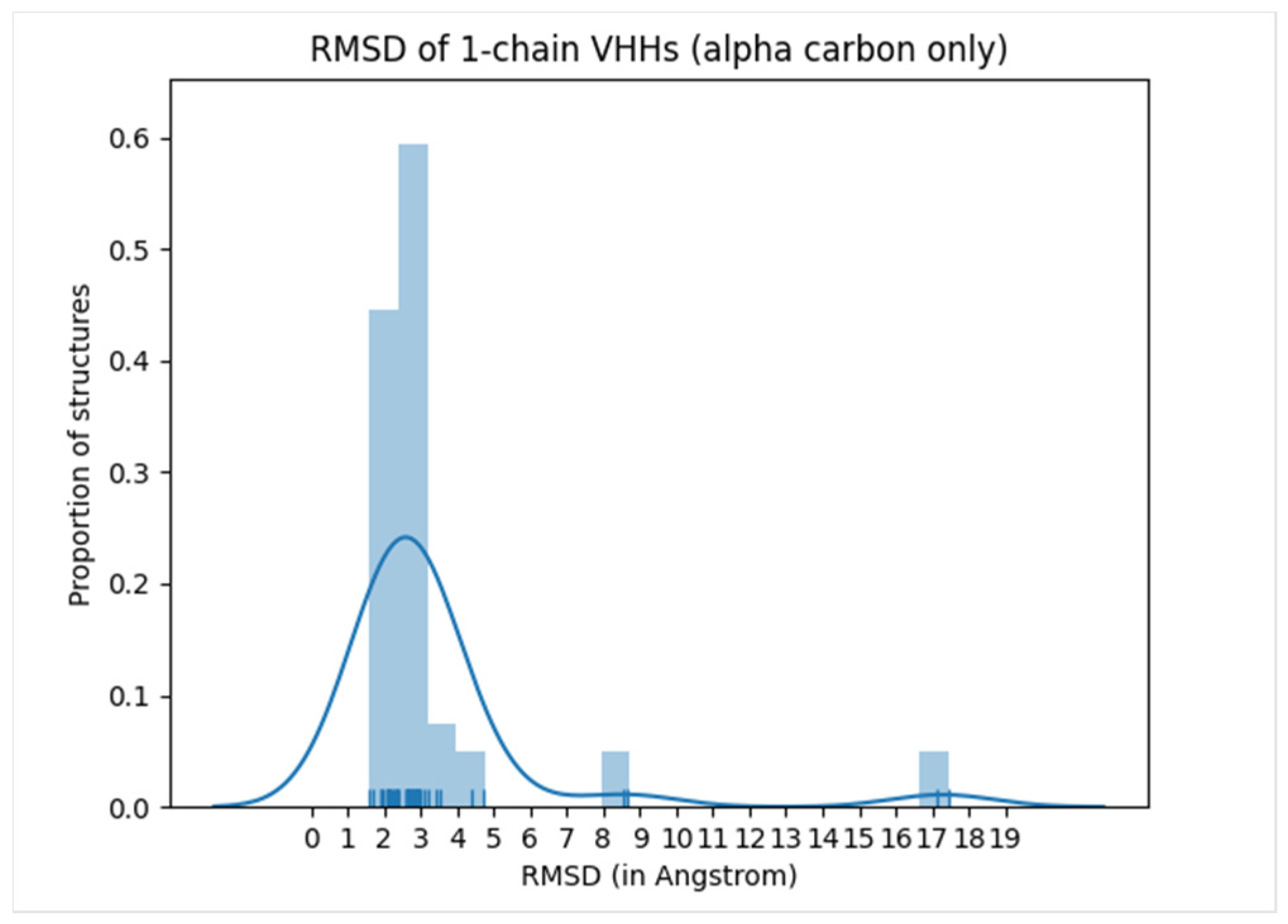
| IgFold | Predict Ab (Fv) structure | Sequence | Predicted Ab structures | Pre-trained language model followed by graph networks that directly predict backbone atom coordinates | Orientational coordinate distance and RMSD | Representations from IgFold may be useful as features for ML models |
| IG-VAE | Directly generate 3D coordinates of full-atom Abs | Known IG structures | Diversified IG structures | VAE | Distance matrix reconstruction and torsion angle inference | Intended for use with existing protein design suites such as Rosetta |
| iNNterfaceDesign | One-sided design of protein–protein interfaces | Both sequence and structure (features of protein receptors) | Redesigned protein interface sequence and structures | LSTM with attention | Recovery rates of the native sequence and hot spot | First neural network model for prediction of amino acid sequences for peptides involved into interchain interactions |
| RefineGNN | Co-design of the sequence and 3D structure of CDRs as graph | Both sequence and structure | Both sequence and structure | Autoregressive/generative graph neural network | Perplexity of sequences and the RMSD | Co-designs the sequence and 3D structure of CDRs as a graph |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.N.; McNaughton, A.D.; Kumar, N. Leveraging Artificial Intelligence to Expedite Antibody Design and Enhance Antibody–Antigen Interactions. Bioengineering 2024, 11, 185. https://doi.org/10.3390/bioengineering11020185
Kim DN, McNaughton AD, Kumar N. Leveraging Artificial Intelligence to Expedite Antibody Design and Enhance Antibody–Antigen Interactions. Bioengineering. 2024; 11(2):185. https://doi.org/10.3390/bioengineering11020185
Chicago/Turabian StyleKim, Doo Nam, Andrew D. McNaughton, and Neeraj Kumar. 2024. "Leveraging Artificial Intelligence to Expedite Antibody Design and Enhance Antibody–Antigen Interactions" Bioengineering 11, no. 2: 185. https://doi.org/10.3390/bioengineering11020185
APA StyleKim, D. N., McNaughton, A. D., & Kumar, N. (2024). Leveraging Artificial Intelligence to Expedite Antibody Design and Enhance Antibody–Antigen Interactions. Bioengineering, 11(2), 185. https://doi.org/10.3390/bioengineering11020185