
Low-Data Drug Design with Few-Shot Generative Domain Adaptation


Abstract
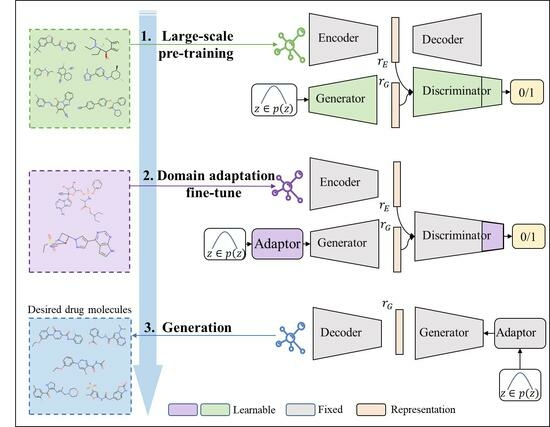
:
1. Introduction
2. Research Problem and Motivation
2.1. Research Problem
2.2. Limitation of Previous Methods
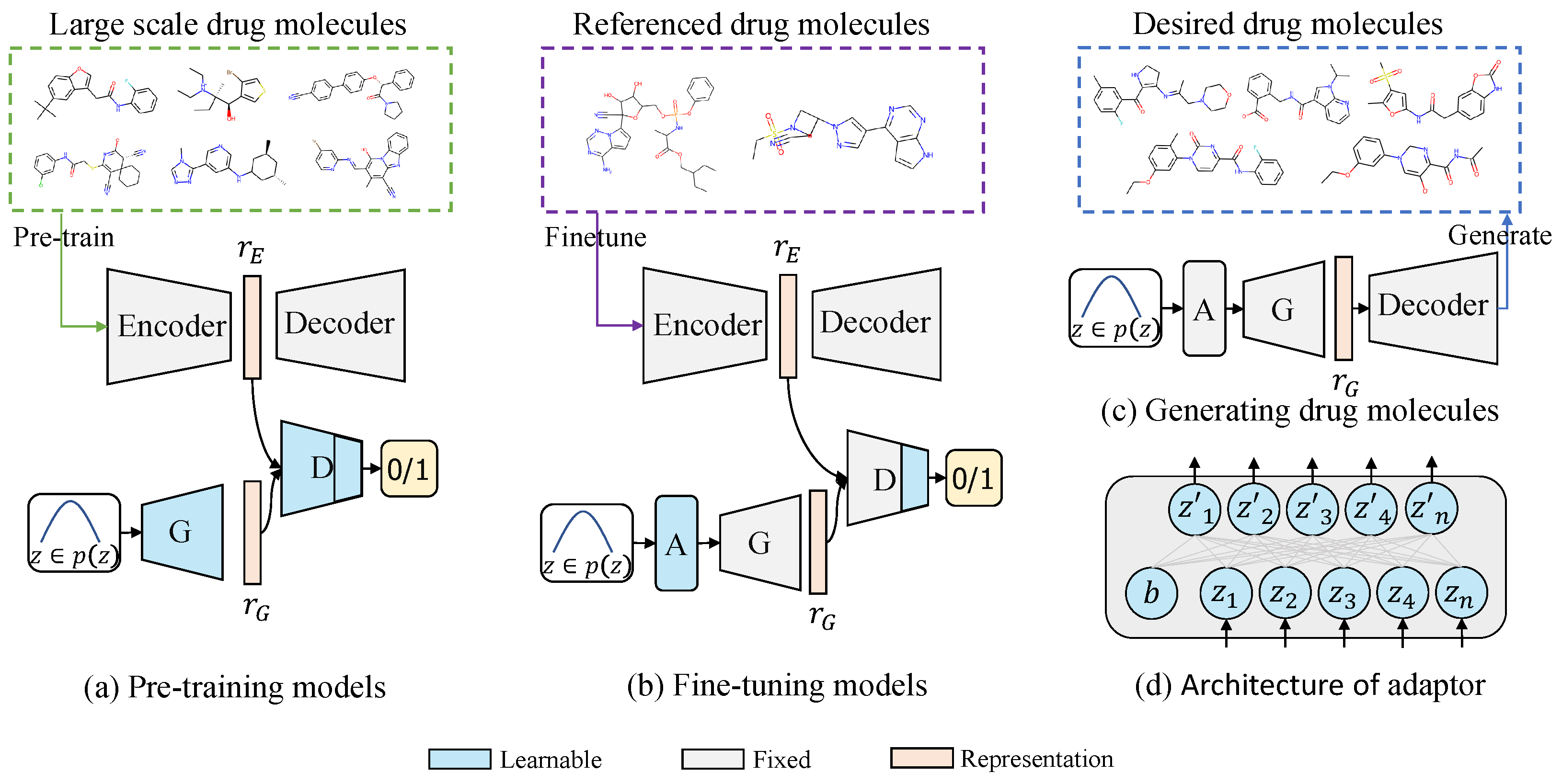
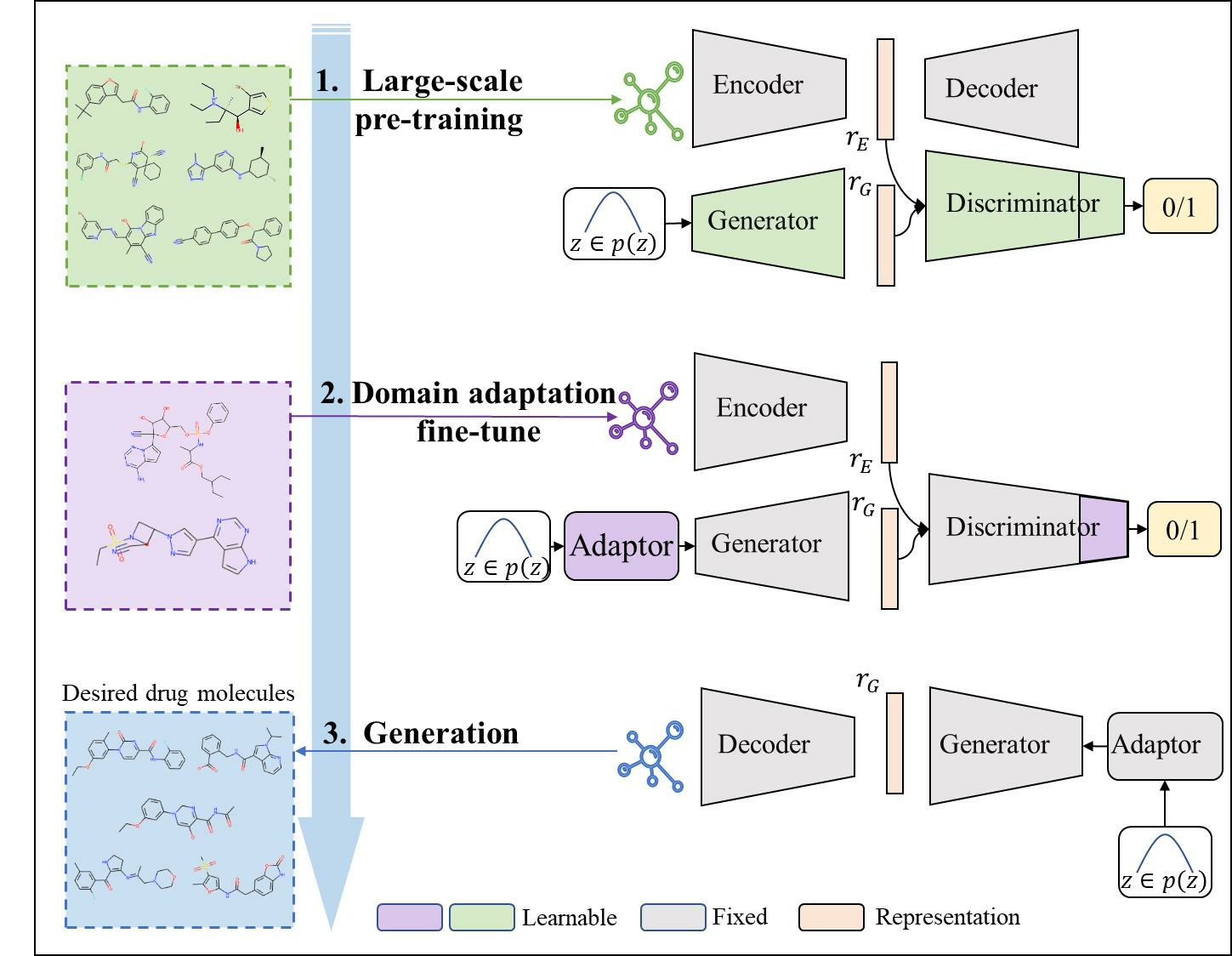
3. Our Method
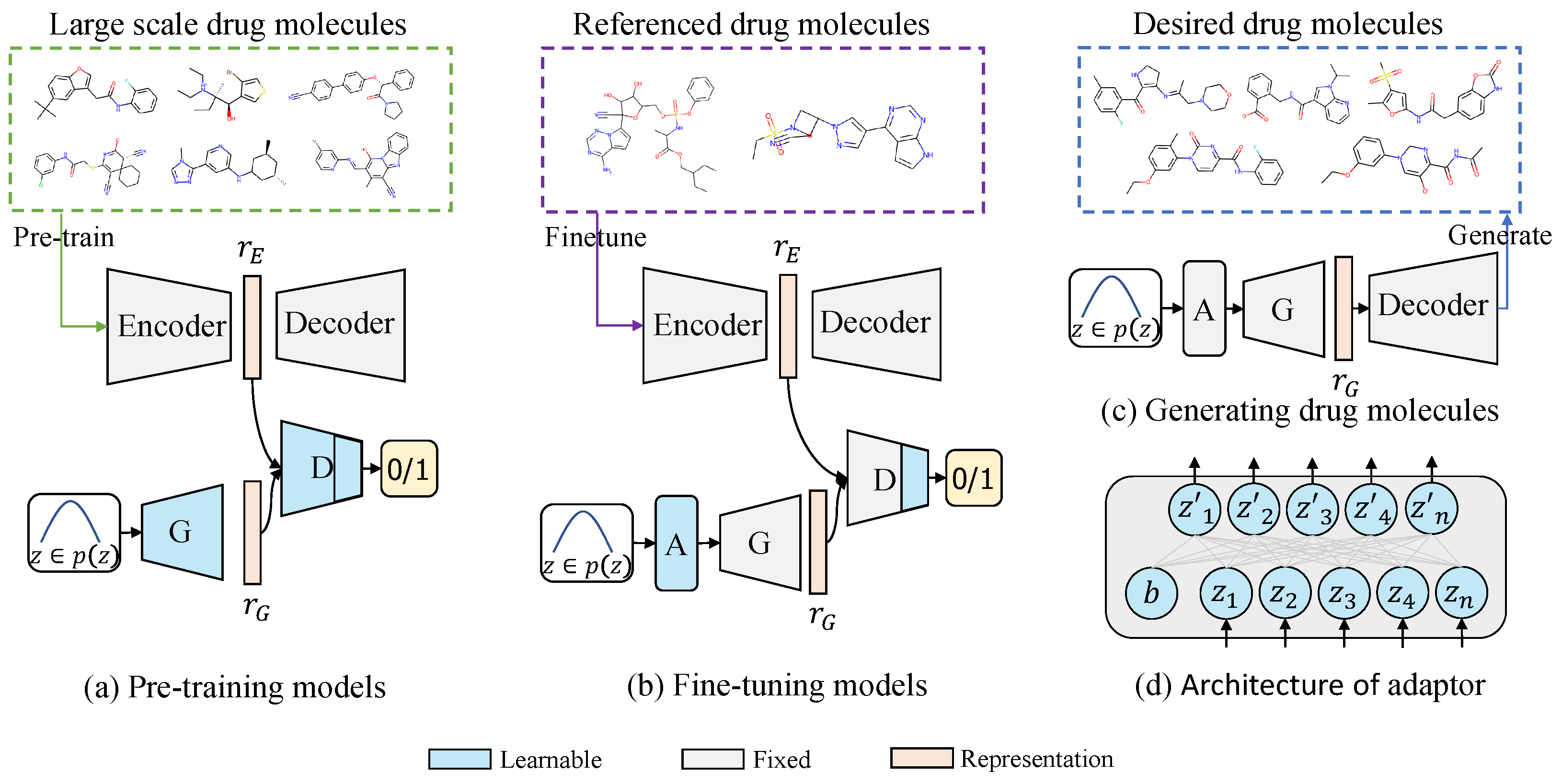
3.1. Large-Scale Pre-Training
| Algorithm 1: Large-scale pre-training |
 |
3.2. Generative Domain Adaptation
| Algorithm 2: Generative Domain Adaptation Fine Tuning |
 |
3.3. Constrained Molecule Generation
| Algorithm 3: Constrained Molecule Generation |
| Input: , trained adaptor parameters; , pre-trained generator parameters; , pre-trained decoder parameters; z, random noise. Output: , generated desired drug molecules; Hyperparameters:
|
4. Results
4.1. Data
4.2. Model and Training Configurations
4.3. Comparison to Previous Methods
4.4. Evaluation Metrics
4.5. Performance and Discussion
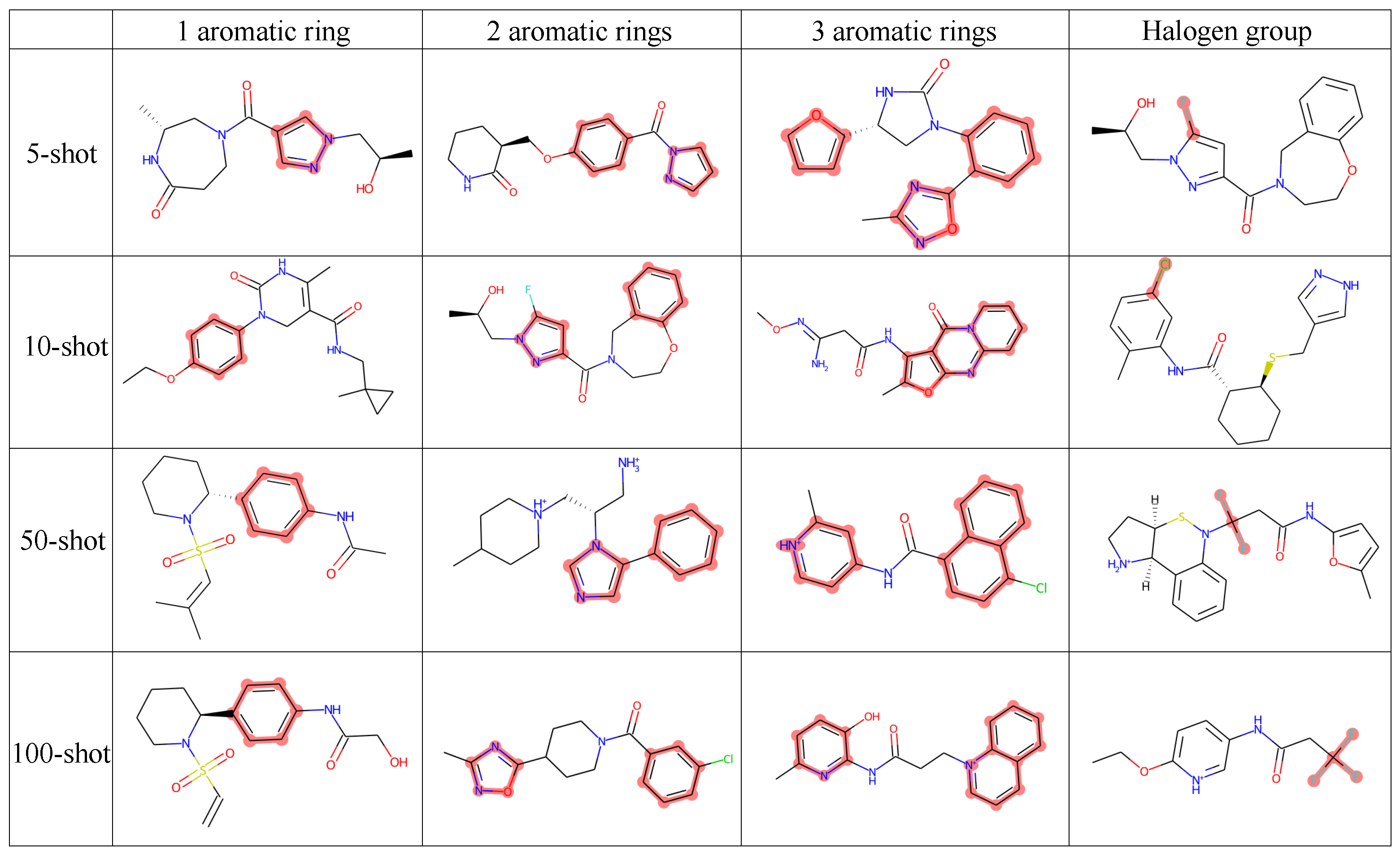
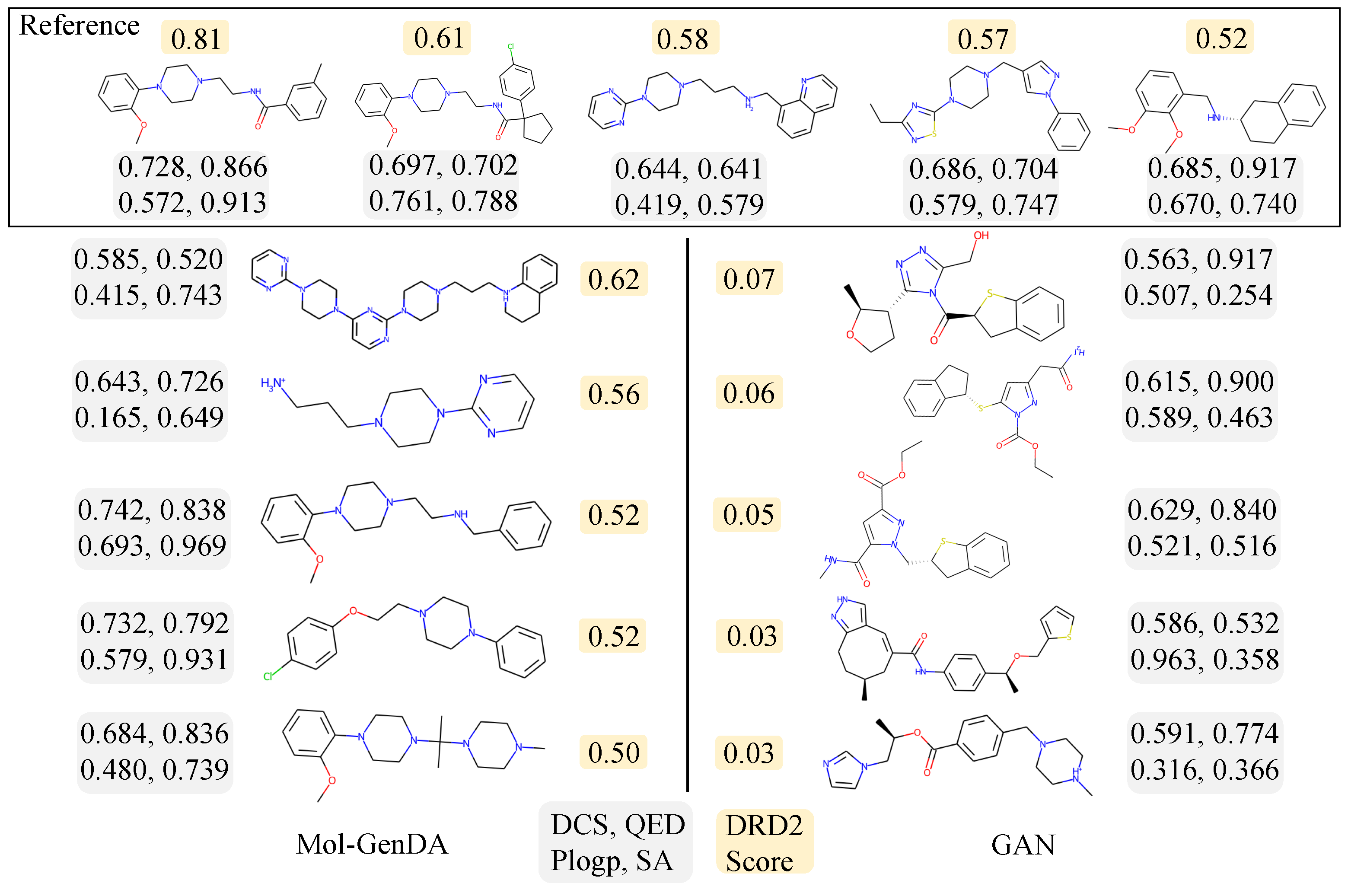
4.5.1. Structure-Constrained Generation
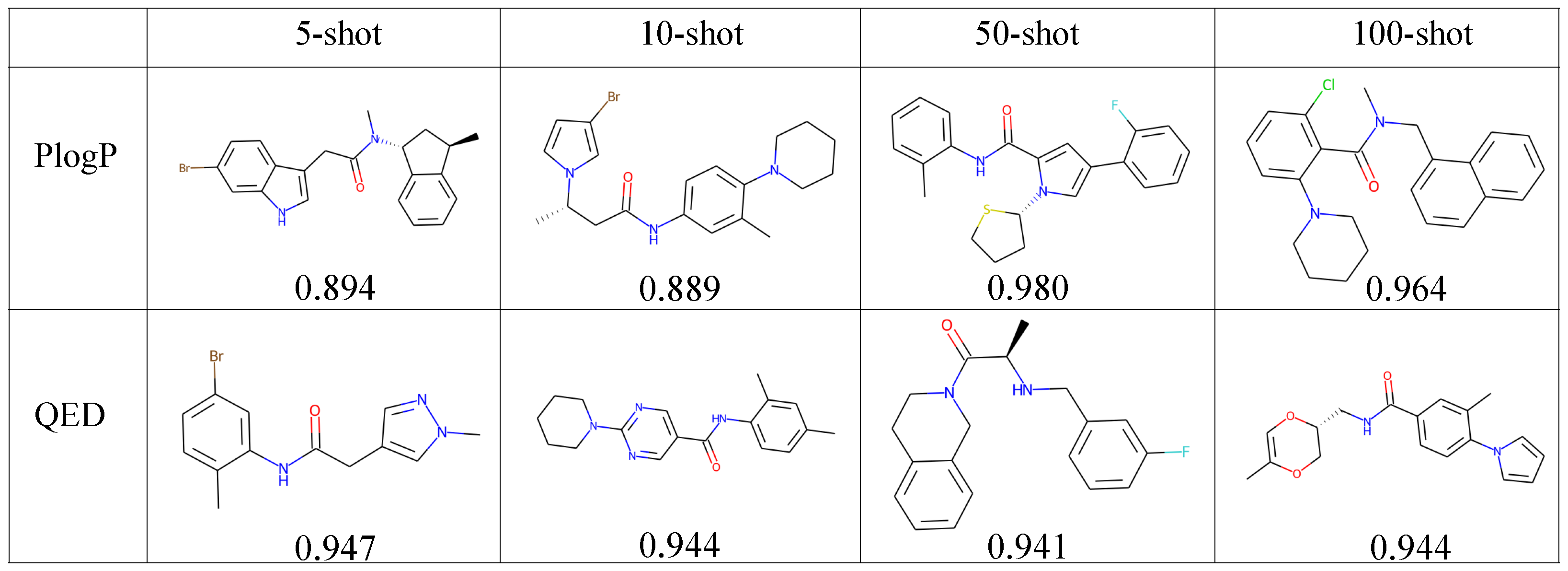
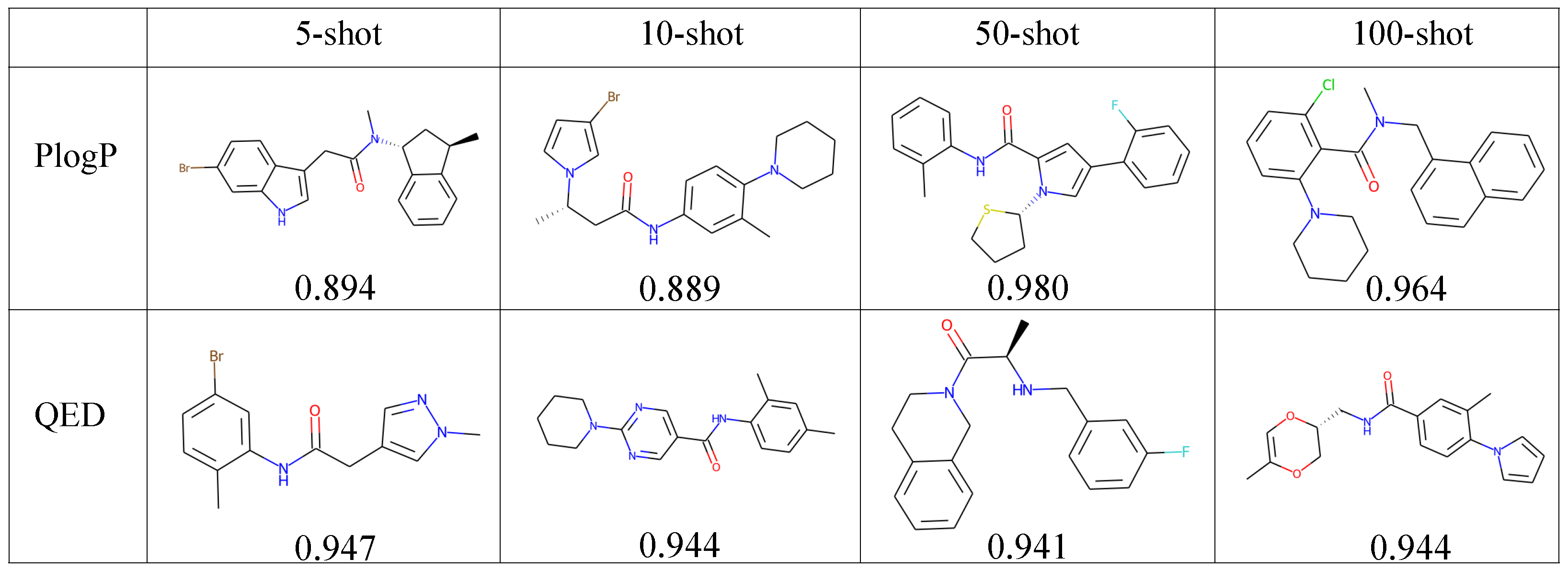
4.5.2. Property-Constrained Generation
4.5.3. Activity: Dopamine Receptor D2
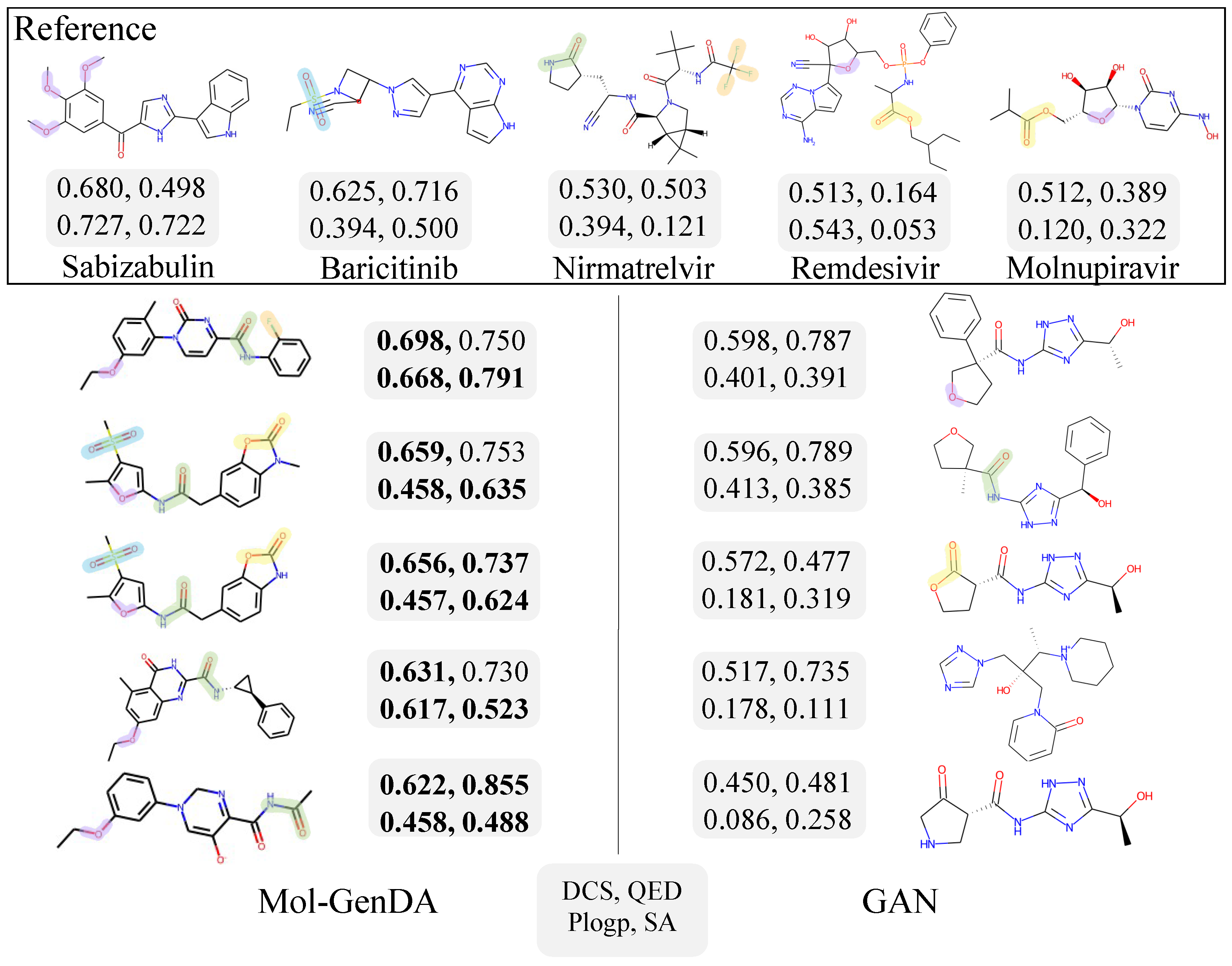
4.6. Case Study: Drug Generation for COVID-19
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salazar, D.E.; Gormley, G. Chapter 41—Modern Drug Discovery and Development. In Clinical and Translational Science, 2nd ed.; Robertson, D., Williams, G.H., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 719–743. [Google Scholar] [CrossRef]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, C.; Jin, W.; Jaakkola, T.; Barzilay, R.; Jensen, K.F. Generative models for molecular discovery: Recent advances and challenges. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1608. [Google Scholar] [CrossRef]
- Sharma, N.; Bora, K.S. Computer Aided Drug Design, 3D Printing, and Virtual Screening: Recent Advancement and Applications in the Pharma Field. ECS Trans. 2022, 107, 16423–16430. [Google Scholar] [CrossRef]
- Mullard, A. New drugs cost US $2.6 billion to develop. Nat. Rev. Drug Discov. 2014, 13, 877. [Google Scholar] [CrossRef]
- Reymond, J.L.; Awale, M. Exploring chemical space for drug discovery using the chemical universe database. ACS Chem. Neurosci. 2012, 3, 649–657. [Google Scholar] [CrossRef]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A., Jr.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef]
- Jing, X.; Xu, J. Fast and effective protein model refinement using deep graph neural networks. Nat. Comput. Sci. 2021, 1, 462–469. [Google Scholar] [CrossRef]
- Chen, Z.; Min, M.R.; Parthasarathy, S.; Ning, X. A deep generative model for molecule optimization via one fragment modification. Nat. Mach. Intell. 2021, 3, 1040–1049. [Google Scholar] [CrossRef]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- Walters, W.P.; Barzilay, R. Applications of deep learning in molecule generation and molecular property prediction. Accounts Chem. Res. 2020, 54, 263–270. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. In Proceedings of the 2nd International Conference on Learning Representations, ICLR 2014, Banff, AB, Canada, 14–16 April 2014. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. Adv. Neural Inf. Process. Syst. 2014, 27, 2672–2680. [Google Scholar]
- Rezende, D.; Mohamed, S. Variational inference with normalizing flows. In Proceedings of the International Conference on Machine Learning, Lille, France, 7–9 July 2015; pp. 1530–1538. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Krenn, M.; Häse, F.; Nigam, A.; Friederich, P.; Aspuru-Guzik, A. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 2020, 1, 045024. [Google Scholar] [CrossRef]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput.-Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef]
- Kusner, M.J.; Paige, B.; Hernández-Lobato, J.M. Grammar variational autoencoder. In Proceedings of the International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 1945–1954. [Google Scholar]
- Liu, Q.; Allamanis, M.; Brockschmidt, M.; Gaunt, A. Constrained graph variational autoencoders for molecule design. Adv. Neural Inf. Process. Syst. 2018, 31, 7795–7804. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proceedings of the International Conference on Machine Learning, Vienna, Austria, 25–31 July 2018; pp. 2323–2332. [Google Scholar]
- De Cao, N.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar]
- Pölsterl, S.; Wachinger, C. Adversarial learned molecular graph inference and generation. In Proceedings of the Machine Learning and Knowledge Discovery in Databases: European Conference, ECML PKDD 2020, Ghent, Belgium, 14–18 September 2020; Proceedings, Part II. Springer: Berlin/Heidelberg, Germany, 2021; pp. 173–189. [Google Scholar]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Wang, F. MoFlow: An invertible flow model for generating molecular graphs. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Virtual Event, 23–27 August 2020; pp. 617–626. [Google Scholar]
- Shi, C.; Xu, M.; Zhu, Z.; Zhang, W.; Zhang, M.; Tang, J. Graphaf: A flow-based autoregressive model for molecular graph generation. arXiv 2020, arXiv:2001.09382. [Google Scholar]
- Madhawa, K.; Ishiguro, K.; Nakago, K.; Abe, M. Graphnvp: An invertible flow model for generating molecular graphs. arXiv 2019, arXiv:1905.11600. [Google Scholar]
- Wang, Y.; Wu, C.; Herranz, L.; Van de Weijer, J.; Gonzalez-Garcia, A.; Raducanu, B. Transferring gans: Generating images from limited data. In Proceedings of the European Conference on Computer Vision (ECCV), Munich, Germany, 8–14 September 2018; pp. 218–234. [Google Scholar]
- Ojha, U.; Li, Y.; Lu, J.; Efros, A.A.; Lee, Y.J.; Shechtman, E.; Zhang, R. Few-shot image generation via cross-domain correspondence. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Nashville, TN, USA, 20–25 June 2021; pp. 10743–10752. [Google Scholar]
- Yang, C.; Shen, Y.; Zhang, Z.; Xu, Y.; Zhu, J.; Wu, Z.; Zhou, B. One-shot generative domain adaptation. arXiv 2021, arXiv:2111.09876. [Google Scholar]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminform. 2018, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, X. GF-VAE: A Flow-based Variational Autoencoder for Molecule Generation. In Proceedings of the CIKM’21: The 30th ACM International Conference on Information and Knowledge Management, Virtual Event, 1–5 November 2021; Demartini, G., Zuccon, G., Culpepper, J.S., Huang, Z., Tong, H., Eds.; ACM: New York, NY, USA, 2021; pp. 1181–1190. [Google Scholar] [CrossRef]
- Jabbar, A.; Li, X.; Omar, B. A survey on generative adversarial networks: Variants, applications, and training. ACM Comput. Surv. (CSUR) 2021, 54, 1–49. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Xu, M.; Cheng, J.; Liu, Y.; Huang, W. DeepGAN: Generating Molecule for Drug Discovery Based on Generative Adversarial Network. In Proceedings of the 2021 IEEE Symposium on Computers and Communications (ISCC), Rhodes Island, Greece, 5–8 September 2021; IEEE: New York, NY, USA, 2021; pp. 1–6. [Google Scholar]
- Maziarka, Ł.; Pocha, A.; Kaczmarczyk, J.; Rataj, K.; Danel, T.; Warchoł, M. Mol-CycleGAN: A generative model for molecular optimization. J. Cheminform. 2020, 12, 1–18. [Google Scholar]
- Li, Y.; Zhang, R.; Lu, J.; Shechtman, E. Few-shot Image Generation with Elastic Weight Consolidation. In Proceedings of the Advances in Neural Information Processing Systems 33: Annual Conference on Neural Information Processing Systems 2020, NeurIPS 2020, Virtual, 6–12 December 2020. [Google Scholar]
- Zhao, M.; Cong, Y.; Carin, L. On Leveraging Pretrained GANs for Generation with Limited Data. In Proceedings of the 37th International Conference on Machine Learning, ICML 2020, Virtual Event, 13–18 July 2020; Volume 119, pp. 11340–11351. [Google Scholar]
- Yang, C.; Lim, S.N. One-shot domain adaptation for face generation. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Seattle, WA, USA, 14–19 June 2020; pp. 5921–5930. [Google Scholar]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low data drug discovery with one-shot learning. ACS Cent. Sci. 2017, 3, 283–293. [Google Scholar] [CrossRef]
- Lv, Q.; Chen, G.; Yang, Z.; Zhong, W.; Chen, C.Y.C. Meta Learning With Graph Attention Networks for Low-Data Drug Discovery. IEEE Trans. Neural Netw. Learn. Syst. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wauchope, O.R.; Frank, A.T. Navigating chemical space by interfacing generative artificial intelligence and molecular docking. J. Chem. Inf. Model. 2021, 61, 5589–5600. [Google Scholar] [CrossRef]
- Gulrajani, I.; Ahmed, F.; Arjovsky, M.; Dumoulin, V.; Courville, A.C. Improved training of wasserstein gans. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar]
- Xu, Y.; Shen, Y.; Zhu, J.; Yang, C.; Zhou, B. Generative hierarchical features from synthesizing images. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Nashville, TN, USA, 20–25 June 2021; pp. 4432–4442. [Google Scholar]
- Dara, S.; Tumma, P. Feature extraction by using deep learning: A survey. In Proceedings of the 2018 Second International Conference on Electronics, Communication and Aerospace Technology (ICECA), Coimbatore, India, 29–31 March 2018; IEEE: New York, NY, USA, 2018; pp. 1795–1801. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019, 32. [Google Scholar]
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Greg Landrum 2013, 8, 31. [Google Scholar]
- Hsu, J. COVID-19: What now for remdesivir? BMJ 2020, 371. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.G.; Lee, T.C. Nirmatrelvir-ritonavir for COVID-19. CMAJ 2022, 194, E218. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.C.; Tse, C.L.; Burry, L.; Dresser, L.D. Baricitinib: A review of pharmacology, safety, and emerging clinical experience in COVID-19. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 843–856. [Google Scholar] [CrossRef]
- Markowski, M.C.; Tutrone, R.; Pieczonka, C.; Barnette, K.G.; Getzenberg, R.H.; Rodriguez, D.; Steiner, M.S.; Saltzstein, D.R.; Eisenberger, M.A.; Antonarakis, E.S. A Phase Ib/II Study of Sabizabulin, a Novel Oral Cytoskeleton Disruptor, in Men with Metastatic Castration-resistant Prostate Cancer with Progression on an Androgen Receptor–targeting Agent. Clin. Cancer Res. 2022, 13, 2789–2795. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, A.; Singh, R.; Misra, A. Molnupiravir in COVID-19: A systematic review of literature. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 102329. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task | Dataset | Num. | Diver. | Plogp | QED | 1 Ring | 2 Rings | 3 Rings | Halogen |
|---|---|---|---|---|---|---|---|---|---|
| Pre-training | ZINC | 2.5 × 10 | 0.915 | 0.561 | 0.731 | 75,580 | 98,222 | 47,603 | 87,556 |
| Structure- constrained | 1 ring | 5 | 0.902 | 0.519 | 0.799 | 5 | - | - | - |
| 10 | 0.905 | - | - | 10 | - | - | - | ||
| 50 | 0.906 | - | - | 50 | - | - | - | ||
| 100 | 0.909 | - | - | 100 | - | - | - | ||
| 2 rings | 5 | 0.893 | 0.668 | 0.839 | - | 5 | - | - | |
| 10 | 0.905 | - | - | - | 10 | - | - | ||
| 50 | 0.906 | - | - | - | 50 | - | - | ||
| 100 | 0.908 | - | - | - | 100 | - | - | ||
| 3 rings | 5 | 0.889 | - | - | - | - | 5 | - | |
| 10 | 0.902 | - | - | - | - | 10 | - | ||
| 50 | 0.907 | - | - | - | - | 50 | - | ||
| 100 | 0.908 | - | - | - | - | 100 | - | ||
| Halogen | 5 | 0.895 | - | - | 1 | - | 3 | 5 | |
| 10 | 0.900 | - | - | 2 | 3 | 3 | 10 | ||
| 50 | 0.908 | - | - | 11 | 28 | 6 | 50 | ||
| 100 | 0.911 | - | - | 25 | 52 | 15 | 100 | ||
| Property- constrained | QED | 5 | 0.864 | 0.604 | 0.947 | - | - | - | - |
| 10 | 0.888 | 0.599 | 0.947 | - | - | - | - | ||
| 50 | 0.891 | 0.602 | 0.947 | - | - | - | - | ||
| 100 | 0.893 | 0.602 | 0.947 | - | - | - | - | ||
| PlogP | 5 | 0.878 | 1.000 | 0.292 | - | - | - | - | |
| 10 | 0.891 | 1.000 | 0.288 | - | - | - | - | ||
| 50 | 0.901 | 1.000 | 0.357 | - | - | - | - | ||
| 100 | 0.905 | 1.000 | 0.390 | - | - | - | - |
| Inter- Polate | Random Sampling | GAN | Pre-Train GAN | Mol- GenDA | ||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 2 | ||||||
| 5-shot | QED | 0.681 | 0.486 | 0.519 | 0.497 | 0.729 | 0.749 | 0.771 |
| Diversity | 0.868 | 0.908 | 0.923 | 0.930 | 0.850 | 0.866 | 0.859 | |
| Uniqueness | 0.129 | 0.929 | 0.979 | 0.994 | 0.094 | 0.243 | 0.240 | |
| 10-shot | QED | 0.738 | 0.439 | 0.525 | 0.516 | 0.753 | 0.749 | 0.769 |
| Diversity | 0.886 | 0.902 | 0.920 | 0.927 | 0.832 | 0.866 | 0.865 | |
| Uniqueness | 0.395 | 0.929 | 0.979 | 0.994 | 0.900 | 0.243 | 0.246 | |
| 50-shot | QED | 0.721 | 0.439 | 0.506 | 0.501 | 0.736 | 0.749 | 0.749 |
| Diversity | 0.897 | 0.910 | 0.922 | 0.928 | 0.841 | 0.866 | 0.862 | |
| Uniqueness | 0.998 | 0.840 | 0.997 | 0.996 | 0.073 | 0.242 | 0.243 | |
| 100-shot | QED | 0.722 | 0.467 | 0.521 | 0.683 | 0.748 | 0.749 | 0.762 |
| Diversity | 0.897 | 0.914 | 0.921 | 0.927 | 0.852 | 0.866 | 0.866 | |
| Uniqueness | 0.988 | 0.897 | 0.999 | 0.997 | 0.066 | 0.243 | 0.263 | |
| Inter- Polate | Random Sampling | GAN | Pre-Train GAN | Mol- GenDA | ||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 2 | ||||||
| 5-shot | Plogp | 0.651 | 0.639 | 0.547 | 0.519 | 0.519 | 0.568 | 0.682 |
| Diversity | 0.880 | 0.908 | 0.923 | 0.930 | 0.825 | 0.866 | 0.936 | |
| Uniqueness | 0.234 | 0.929 | 0.979 | 0.994 | 0.069 | 0.243 | 0.252 | |
| 10-shot | Plogp | 0.660 | 0.669 | 0.667 | 0.609 | 0.562 | 0.568 | 0.679 |
| Diversity | 0.899 | 0.871 | 0.920 | 0.927 | 0.837 | 0.866 | 0.866 | |
| Uniqueness | 0.612 | 0.827 | 0.994 | 0.995 | 0.065 | 0.243 | 0.255 | |
| 50-shot | Plogp | 0.635 | 0.617 | 0.614 | 0.594 | 0.547 | 0.568 | 0.677 |
| Diversity | 0.893 | 0.910 | 0.922 | 0.928 | 0.849 | 0.866 | 0.865 | |
| Uniqueness | 0.982 | 0.840 | 0.997 | 0.996 | 0.068 | 0.243 | 0.232 | |
| 100-shot | Plogp | 0.659 | 0.653 | 0.607 | 0.590 | 0.484 | 0.568 | 0.672 |
| Diversity | 0.913 | 0.914 | 0.921 | 0.928 | 0.864 | 0.866 | 0.865 | |
| Uniqueness | 0.985 | 0.897 | 0.999 | 0.994 | 0.085 | 0.243 | 0.236 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Han, Y.; Gong, Z.; Xu, H. Low-Data Drug Design with Few-Shot Generative Domain Adaptation. Bioengineering 2023, 10, 1104. https://doi.org/10.3390/bioengineering10091104
Liu K, Han Y, Gong Z, Xu H. Low-Data Drug Design with Few-Shot Generative Domain Adaptation. Bioengineering. 2023; 10(9):1104. https://doi.org/10.3390/bioengineering10091104
Chicago/Turabian StyleLiu, Ke, Yuqiang Han, Zhichen Gong, and Hongxia Xu. 2023. "Low-Data Drug Design with Few-Shot Generative Domain Adaptation" Bioengineering 10, no. 9: 1104. https://doi.org/10.3390/bioengineering10091104
APA StyleLiu, K., Han, Y., Gong, Z., & Xu, H. (2023). Low-Data Drug Design with Few-Shot Generative Domain Adaptation. Bioengineering, 10(9), 1104. https://doi.org/10.3390/bioengineering10091104