3.1. Characterization of Synthesized Material
Element chemical analysis data of Mg and Al for all samples are included in
Table 1. Sodium and sulfur content are given for samples containing organic sulfates or sulfonates in the interlayer. The presence of sodium is not expected but it could be present in the samples if they have not been sufficiently washed after synthesis resulting in remaining sodium cations on the surface of the LDH crystallite. A similar effect could appear for alkyl sulfate or alkyl sulfonate species on the external surface of the LDHs. Hence, sulfur was analyzed to check for any deviations in sulfur content from the theoretical S:Al ratio of 1. Mg/Al and S/Al molar ratios for the samples, calculated from the element chemical analysis data, are also included in
Table 1.
The Mg/Al molar ratio was close to 3, with a maximum deviation of 10% in all cases, which is within the range usually accepted to yield well-defined hydrotalcite-like structures [
6]. In the case of sulfur-containing anions, the theoretical S/Al ratio is 1 if the monovalent organic anion is the only species in the interlayer to balance the positive charge of the Mg
2+/Al
3+ substituted main layer. A S/Al ratio of approximately 1 was calculated for the MgAl-8SO3 and MgAl-12SO3, despite having a slight sulfur defect in the former sample. On the contrary the S/Al molar ratio of the samples MgAl-18SO3 and MgAl-12SO4 is much higher than one. These two samples also contain appreciable amounts of sodium leading us to the assumption that there is a fraction of sodium sulfate or sulfonate (respectively, for samples MgAl-18SO3 and MgAl-12SO4) species adsorbed on the external surface of the crystallites, due to insufficient washing of the samples. The lack of match between the excess of sulfur and the amount of sodium can probably also be originated by residual amounts of sodium from the basic medium used to prepare the samples. Small amounts of sodium were also detected for some of the MgAl-COO samples.
Preliminary batch experiment tests on the organically modified LDHs showed that synthesized samples intercalated with organic anions having less than 10 carbon atoms in the alkyl chain are ineffective as retention materials. Consequently, the retention studies were limited to samples with more than ten carbon atoms in the alkyl chain of the organic interlayer anion. For systematic reasons, characterization was carried out on all synthesized samples.
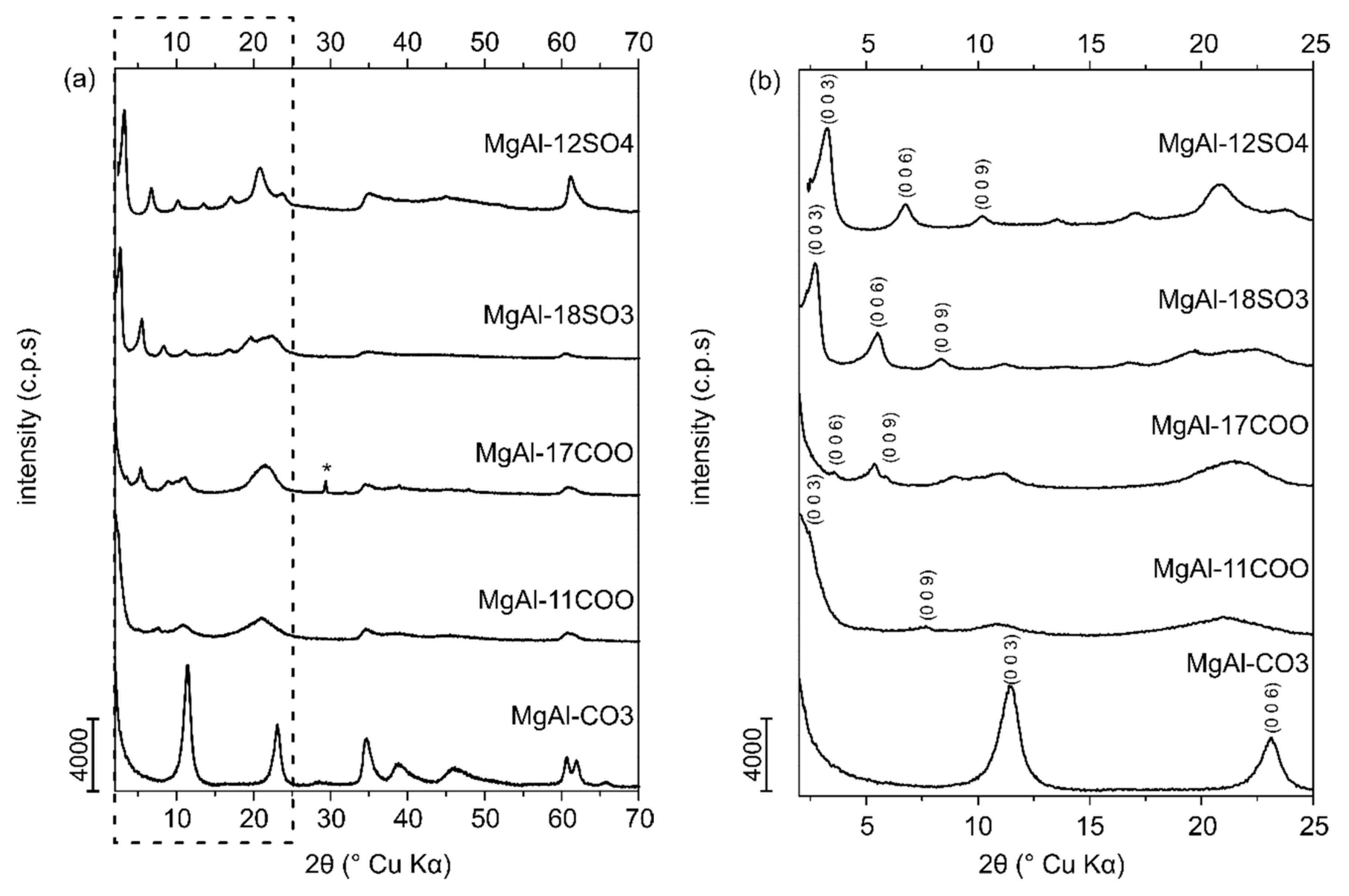
The PXRD patterns of all synthetized samples are included in the
Supplementary Materials Figures S1–S8; selected samples are shown in
Figure 2. The diagrams for the fully recorded diffraction angle range are shown in the left hand side panel (a), while on the right side the 2–25° (2θ) range is shown for a clear identification of the diffraction by basal planes (b). All studied samples show diffraction patterns corresponding to layered materials. The pattern of MgAl-CO3, used as the reference sample, is typical for that of LDHs with intercalated carbonate [
7,
15].
The first peak at 11.5° (2θ) (d = 7.7 Å) is due to the reflection of the (003) planes in the hydrotalcite-like structure with rhombohedral 3R stacking of the layers, for which the lattice parameter
c corresponds to three times the spacing value for this reflection,
c = 23.1 Å. The average distance between two neighbour cations in the brucite-like layers is given by lattice parameter
a and can be calculated as twice the spacing of the diffraction maximum of the (110) planes, which correspond to the first peak of the doublet close to 60° (2θ); the
a value for sample MgAl-CO3 is 3.1 Å. The second component of this doublet, a feature usually found in LDHs [
25], is hardly visible for the organically modified samples. Lattice parameters a, c’ and c of all synthesized samples are given in
Table 2.
The structural data of the carbonate-containing sample coincide with those reported in the literature [
15]. As expected, insertion of organic anions in the interlayer leads to swelling of the layers, with an increase in parameter
c. The largest swelling of the interlayer, by a factor of six compared to the reference sample MgAl-CO3 (7.7 Å), was detected in sample MgAl-17COO (47.7 Å). All values for the samples intercalated with organic anions agree with previously published data on LDHs with organic interlayer anions of similar length [
10,
25]. As expected, the height of the interlayer space increases with the length of the alkyl chain of the intercalated organic anion, confirming that these anions are not located parallel to the brucite-like layers but somewhat tilted. This free height was calculated assuming a width of 4.8 Å for the brucite-like layers [
6,
10,
11,
18] and adding the contour chain length, the distance between the ends of the organic molecule assuming it is streched out, of the organic molecule. Calculated contour chain lengths of all studied organic anions as well as their tilt angle can be found in
Table S3 (Supplementary Materials). As mentioned in literature [
10,
11,
18,
26] organic molecules can be present in the interlayer of an LDH in three different ways—simple intercalation, interdigitated intercalation or as a double interlayer. In addition to that it has been reported that the organic chains can be tilted with respect to the perpendicular of the layers.
For sample MgAl-5COO the organic anions seem to be minimal tilted with respect to the perpendicular of the main layer; there is only an increase of less than 1 Å in the free height of the interlayer space with respect to the reference sample, while the molecule contains a (C
11H
23) chain in addition to the functional group. This leads to the assumption that both organic molecules are interdigitatedly intercalated. However, when the length of the chain increases the formation of tilted bilayers becomes more likely, as it seems to be formed for samples MgAl-11COO and MgAl-17COO. A bilayer seems also to exist for samples MgAl-12SO3 and MgAl-12SO4, while perpendicular single bilayers are formed for samples MgAl-8SO3 and MgAl-18SO3 [
11,
12,
26]. In addition to the diffraction maxima typical of the hydrotalcite-like structure, sample MgAl-17COO showed weak extra peaks due to the presence of minor impurities of sodium nitrate, indicating an insufficient washing process for these samples; nevertheless, the presence of this impurity does not affect the capacity of these material for retention of chlorinated solvents.
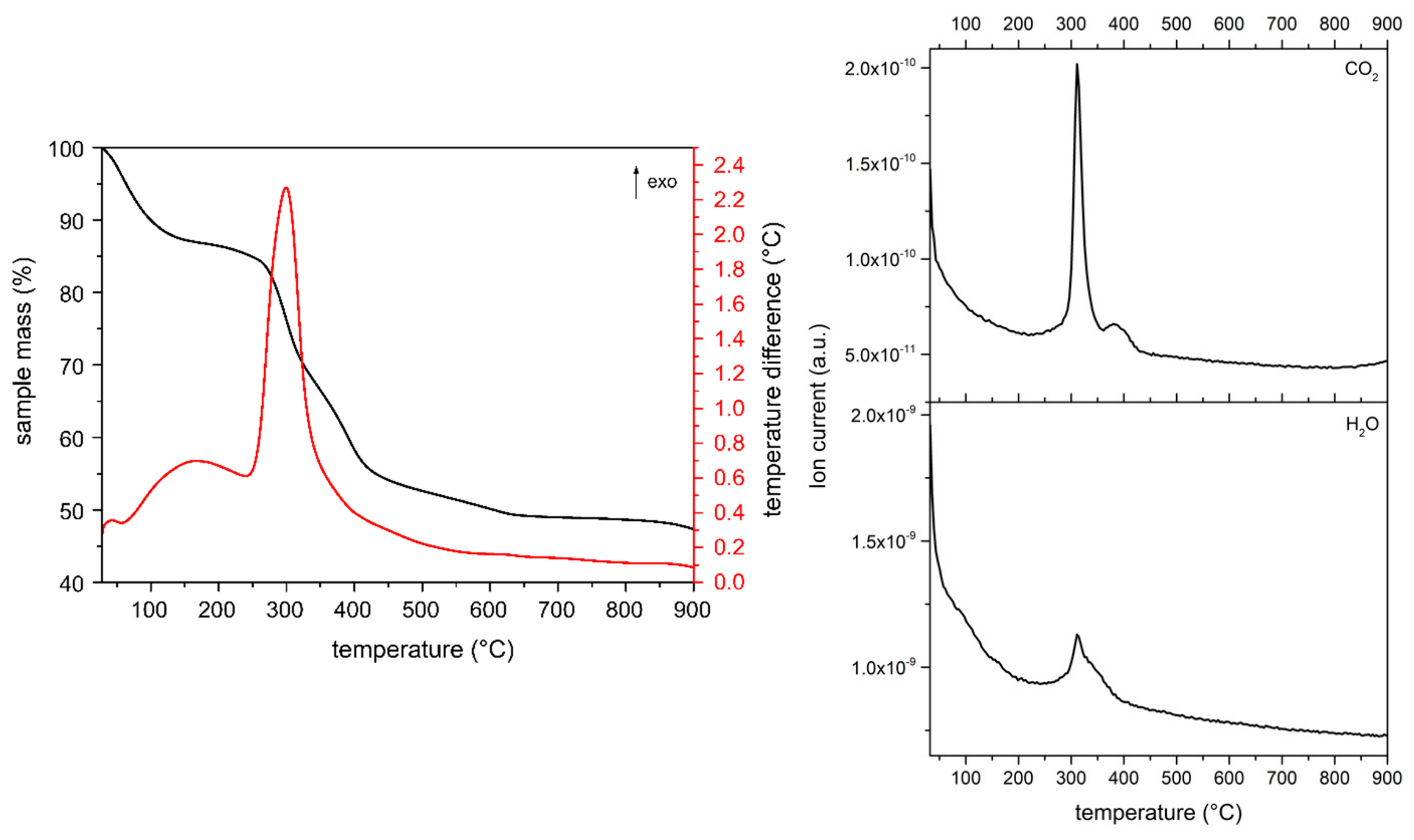
The thermal decomposition of the solids prepared was studied by TG-DTA; the nature of the gases evolved was determined by MS. The curves for sample MgAl-CO3 are included in the
Supplementary Materials section (Figure S9) and they show the expected mass losses [
27,
28]. A first mass loss up to ca. 200 °C, amounting in 15.49% of the initial sample mass, corresponds to removal of water from the interlayer or physisorbed on the external surface of the LDHs. This mass loss takes place in the same temperature range where a medium intensity endothermic peak is recorded in the DTA diagram. Quantitative analysis from the TG curves for all samples are summarized in
Table 3.
A very weak peak is recorded in the MS spectra for the mass-to-charge-ratio (m/z) of 44 corresponding to CO
2+. This derives from externally adsorbed carbonate species, formed by adsorption of atmospheric CO
2 onto the strongly basic surface of the solid. A second mass loss is recorded up to ca. 400 °C, which corresponds to the removal of water through condensation of hydroxyl groups and of CO
2, from decomposition of interlayer carbonate species. Both processes show two peaks in their MS spectra indicating a stepwise process with several reactions involved, as also concluded from the DTA curve. This agrees with results reported by Stanimirova et al. [
29]. The whole decomposition process ends around 600 °C.
For sample MgAl-5COO a first mass loss is recorded up to ca. 200 °C; but no signal at m/z = 18 (H
2O
+) was recorded in the MS data. Comparing the TG curves for samples MgAl-CO3 and MgAl-5COO (as well as all other MgAl-XCOO samples) in this temperature range, it can be observed that the TG curve slope for sample MgAl-CO3 is clearly larger than for the other samples, suggesting that in the case of sample MgAl-CO3 removal of water takes place rather suddenly in a narrower temperature range. If this is the case, the more gradual water elimination on the MgAl-XCOO samples would give rise to a much weaker and broader signal in the MS curve, being thus not clearly detected. For the MgAl-XCOO samples the mass loss percentage in this temperature range is lower than that for sample MgAl-CO3, as expected due to the larger contribution of the organic anion than carbonate to the mass of the solid; it is ca. 11% of the initial mass loss for all samples. Then, an important mass loss, amounting to up to 60% of the initial mass (sample MgAl-17COO,
Figure S12) is recorded in all cases; it should correspond to removal of the anion as water vapor and carbon dioxide through combustion of the organic anion, together with water released upon condensation of hydroxyl groups from the brucite-like layers. A very strong and sharp exothermic effect is recorded in all cases around 300 °C, probably due to the formation of CO
2. Compared with the MS data, it is evident that the maximum CO
2 evolution (signal at m/z = 44, CO
2+) is precisely recorded at this temperature; slightly above this temperature, a minor amount of CO
2 is also evolved, probably occluded within the channels and the pores of the solid. As expected, a signal at m/z = 18 (H
2O
+) is recorded in this temperature range, also very close to 300 °C, thus definitively confirming that the combustion process evolves CO
2 and H
2O.
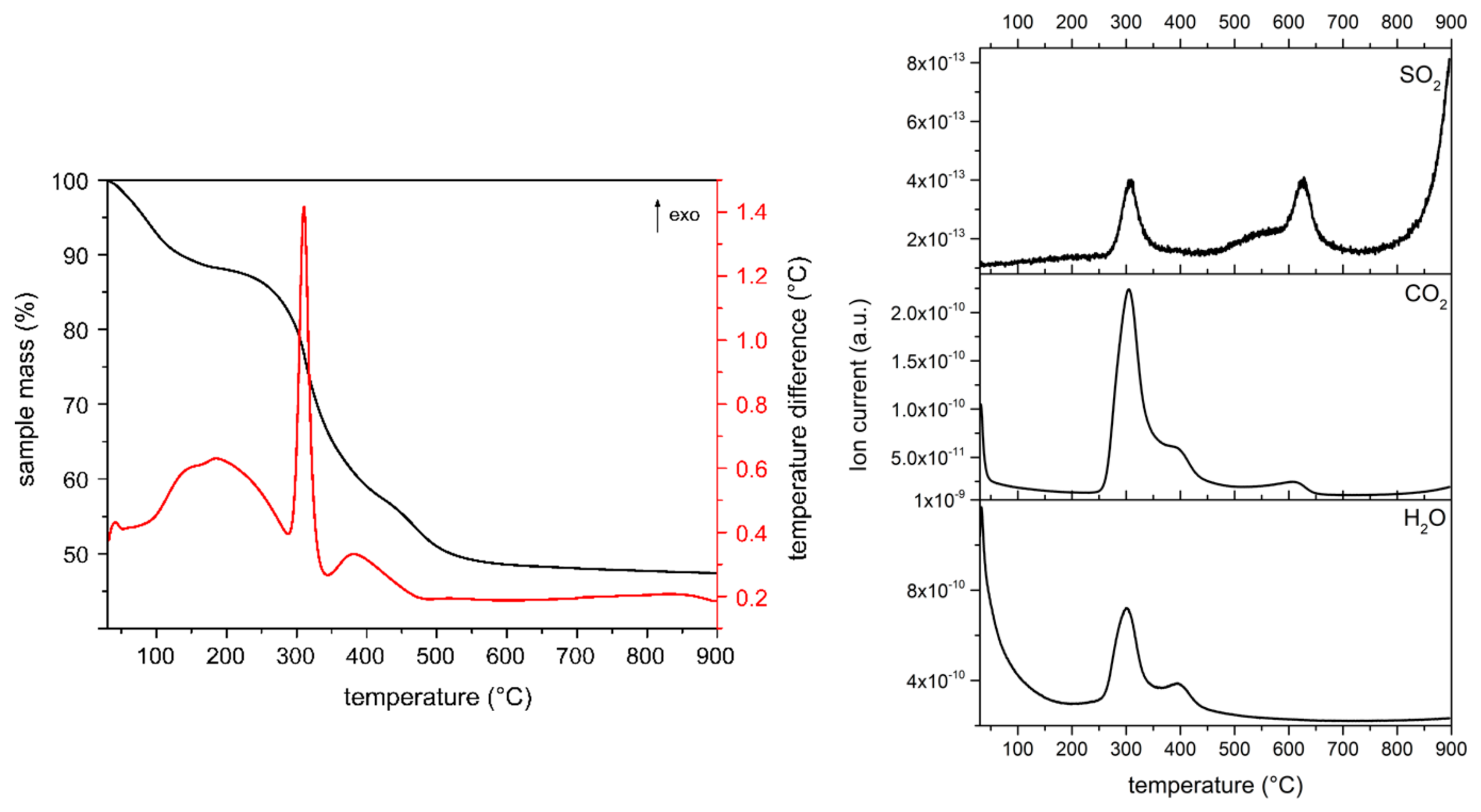
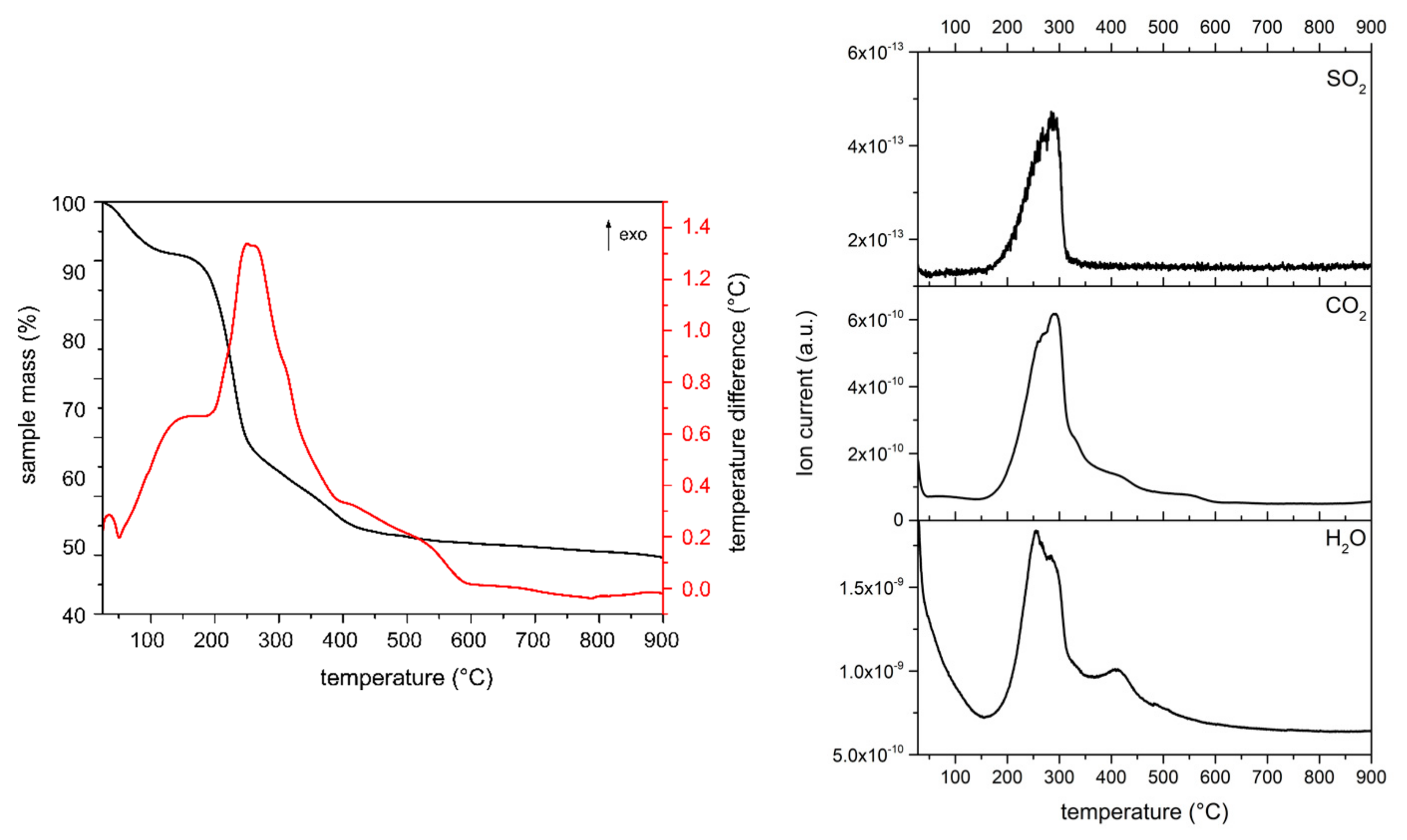
For the MgAl-XSO3 samples we have additionally monitored the presence of SO
2 during the mass spectrometry analysis of the released gases.
Figure 4 exemplarily includes that for sample MgAl-8SO3, while those for the other sulfonate samples are included in the
Supplementary Materials Section, Figures S13 and S14. A first mass loss occurs up to ca. 200 °C, probably due to removal of molecular water; although, as for the MgAl-XCOO samples, the signal at m/z = 18 (H
2O
+) is not recorded in the MS curves, probably because of the reason given above. Increasing temperature leads to an intense mass loss up to ca. 700 °C.
The second mass loss between 300–700 °C is due, according to the MS data, to removal of interlayer hydroxyl groups as water, combustion of the organic as water vapour and CO2. There is also a contribution due to the evolution of SO2. The relative intensity of the SO2 signal is extremely weak for sample MgAl-12SO3 but we have not found a reliable explanation for this result. However, it should be noticed that in this case the MS signal due to CO2 shows a shoulder at the high temperature side of the maximum, clearly noticeable for samples MgAl-8SO3 and MgAl-12SO3 and only recorded as an asymmetry for sample MgAl-18SO3; a second CO2 signal is recorded at 600–650 °C, a temperature range where a strong signal due to SO2 is also recorded, with small mass losses well. These results strongly suggest that decomposition of the alkyl sulfonates takes place in, at least, two steps, one around 300 °C, where most of the corresponding mass is lost and a second one at 600–650 °C, where residual species are probably removed. In all cases, the mass is constant from ca. 700 to 900 °C.
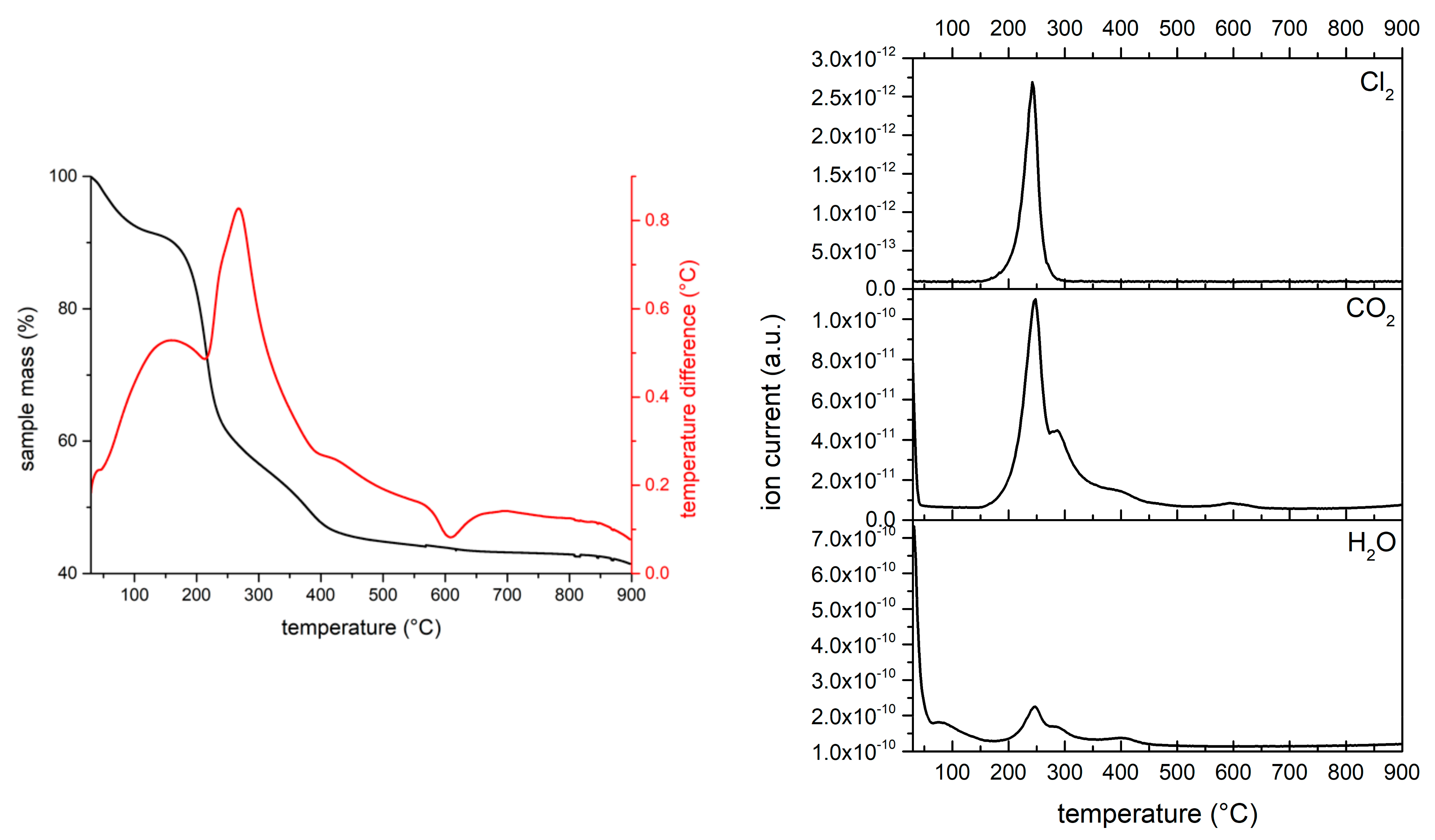
The thermal decomposition of sample MgAl-12SO4 is shown in
Figure 5. Results are rather different to those above reported for the MgAl-XSO3 samples—The first mass loss, probably due to water release up to 200 °C, represents a very small percentage of the original sample mass and a strong exothermic effect is recorded, as in the MgAl-XSO3 samples, around 300 °C, for which release of water, CO
2 and SO
2 has been observed. However, no high temperature mass loss, nor high temperature evolution of gases or water vapor is recorded, contrary to what has been observed for the similar sample MgAl-12SO3 but the release of water and CO
2 is observed at the high temperature side of the strongest MS signals. This means that, in this case, SO
2 is released rather easily together with fragments of the organic chain, but leaving behind some other organic fragments, so combustion would be responsible for the shoulders recorded in the MS signals at m/z = 18 (H
2O
+) and 44 (CO
2+).
The formula of the compounds prepared are given in
Table 4. The content of magnesium and aluminium have been calculated based on element chemical analysis (see raw data in
Table 1), the content of the anion has been determined assuming that it is the only responsible to balance the positive charge of the layers due to the presence of Al
3+ cations and the water content in the interlayer has been calculated from the TG curves.
Altogether, we can conclude that the samples prepared, whichever the interlayer anion, have a similar thermal stability behavior, releasing water at low temperature and decomposing upon burning at around 300 °C. For the sulfonate (MgAl-XSO3) samples, a second decomposition effect is recorded around 600–650 °C. It should be mentioned that intercalation of the organic anions will provide more hydrophobic properties to the interlayer region, hence we would expect a lower interlayer molecular content in these samples than in the carbonate-one. Looking at the formula of the compounds (
Table 4) it seems that the water content follows a rather random behavior. This might not be surprising if we accept that, although the more hydrophobic nature of the interlayer is expected to decrease the water content, as a result of the intercalation of these long-chain anions swelling the structure gives rise to more room in the interlayer, thus providing more space to accommodate water molecules. Probably both effects, together with minor differences in the drying processes, would account for the interlayer molecular water molecules number values calculated for each sample.
The Fourier transform infrared (FTIR) spectra of the studied materials show absorption bands due to the hydroxyl groups and water molecules, the interlayer anions and lattice vibrations within the layers. The spectrum for the reference sample is well known and throughly described by various authors [
30,
31,
32,
33,
34,
35]. The FTIR spectra of the reference sample and the most effective organo-LDHs are shown in
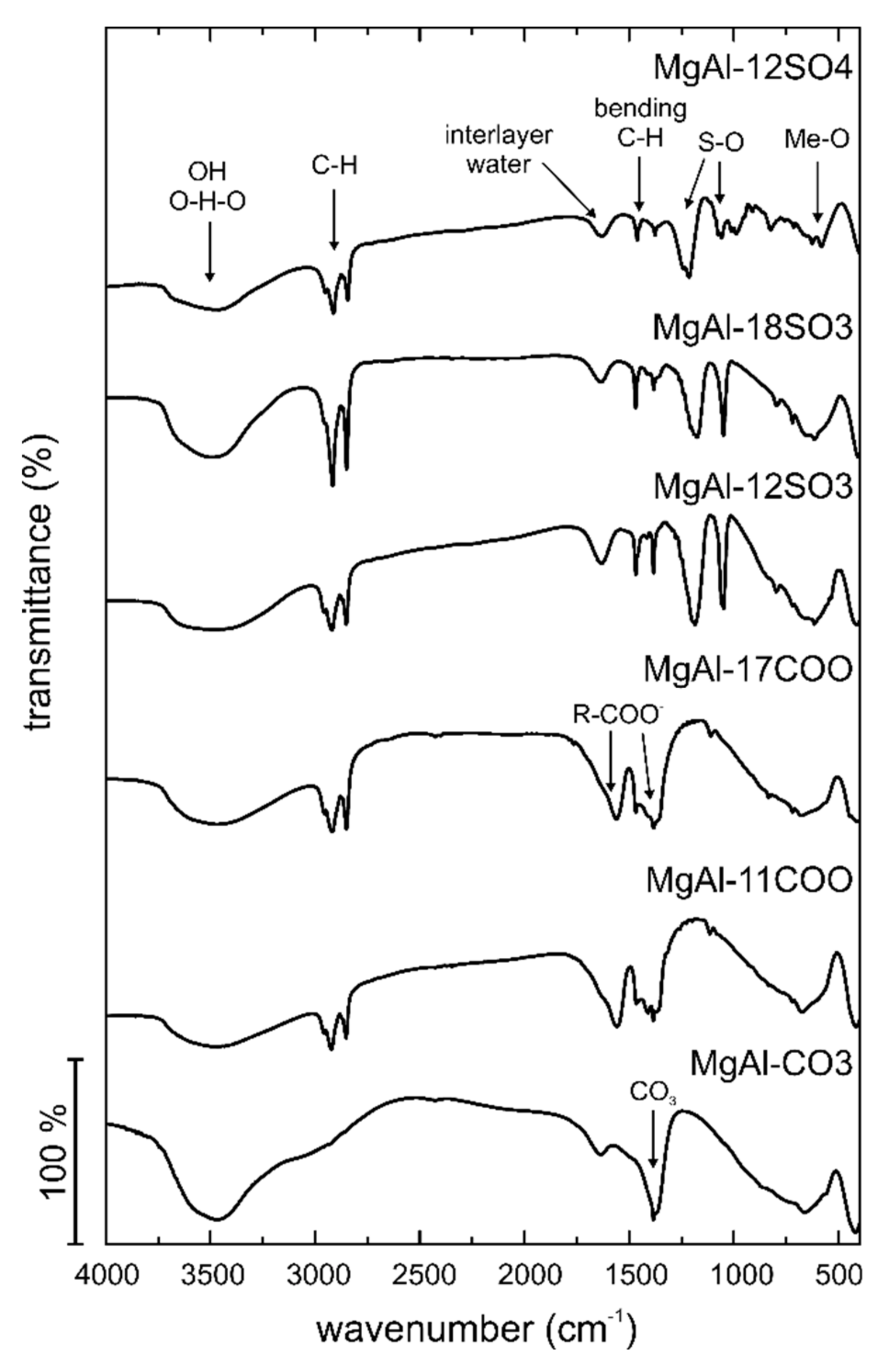
Figure 6 (see
supplementary data for spectra of all samples,
Figures S15–S23). All studied samples show absorption bands characteristic of LDHs, which can be assigned to vibrations of the multiatomic species existing in the main layer and in the interlayer region.
In all cases, a broad absorption band is recorded between ca. 3800–3000 cm−1, which corresponds to the stretching mode of hydroxyl groups, both from the brucite-like layers and from the interlayer water molecules. The medium intensity absorption band close to 1650–1600 cm−1 corresponds to the bending mode of water molecules. The bands below 700 cm−1 correspond to metal-hydroxyl vibrations. All other bands recorded are due to the interlayer anion.
The shoulder at ca. 2930 cm
−1 in the spectrum of sample MgAl-CO3 is due to the stretching mode of hydroxyl groups hydrogen-bonded to interlayer carbonate anions [
30,
33]. The sharp, very intense band at 1369 cm
−1 is due to mode ν
3 of interlayer carbonate anions. There is a sharp band overlapped to that one, recorded at 1384 cm
−1, which is due to the ν
3 mode of a nitrate impurity. Other carbonate bands are recorded at 865 cm
−1 (ν
2) and 663 cm
−1 (ν
4); it is worth noting that the band due to mode ν
1, expected around 1070 cm
−1, is not recorded. This band is forbidden in the IR spectrum for carbonate in a D3h geometry, indicating that no distortion of the carbonate structure exists upon its intercalation in the interlayer.
The spectra for the organically modified samples are rather different, although absorption bands corresponding to the brucite-like layers of the LDH are recorded at almost the same positions; the shift sometimes observed for the very broad absorption band in the 3500–3300 cm
−1 is due to the different hydration degree of the samples. In addition, the sharp absorption at 1384 cm
−1 of the nitrate impurity is recorded in most of the cases. Bands due to the organic components of the samples are usually sharp ones. Several bands due to the organic chain are also recorded below ca. 1500 cm
−1. The three sets of samples (carboxylate, sulfonate and sulfate) can be identified by the characteristic bands of these functional groups. Carboxylate is characterized by two bands at ca. 1570 and 1470 cm
−1, due to the antisymmetric and symmetric, respectively, vibration modes of the carboxylate group. The sulfonate group is mostly characterized by the very intense and sharp bands at 1180 cm
−1 and 1050 cm
−1 due to the antisymmetric and symmetric modes of the SO
2 group, and, finally, the sulfate group is characterized by the bands at 1221 cm
−1 and 1065 cm
−1 (
Supplementary Materials, Tables S4–S6).
The evaluation of the FTIR spectra confirms the presence of the organic anions in our samples and the lack (or very low intensity) of the carbonate-related bands confirms, together with the PXRD results discussed above, the insertion of the organic anions in the interlayer. The precise positions of the bands in all spectra recorded are given in
Tables S4–S6 (Supplementary Materials).
The particle size distribution was measured for selected samples and significant differences were found among the dried samples and those kept in a slurry after preparation. Nevertheless, some degree of aggregation was observed in all cases and, for that reason, the samples were treated with ultrasound for 5, 10 and 15 min. All the diagrams are included in the
Supplementary Materials Section, Figures S24–S37.
Within the dried carboxylate samples, the observed behavior was almost the same in all cases. The size extended in the 1–1000 μm range, with a maximum close to 300 μm; above this value, the curve decreased sharply but on the smaller particle size the decrease of the curve was more steadily and several shoulders seemed to exist. Application of ultrasounds up to 15 min gave raise to curves where the shoulders were more marked, such an effect being more evident as the length of the organic chain of the interlayer anion was increased. So, while for sample MgAl-5COO the intensity of the maximum somewhat decreased and weaker maxima at ca. 30 and 2 μm developed, some differences were found for sample MgAl-17COO, where the maximum for the untreated sample was recorded at ca. 100 μm, with a shoulder at ca. 300 μm; when the sample was submitted to ultrasound the maximum was now recorded at ca. 40 μm, with shoulders at 300 and 10 μm. A similar behavior was observed for the MgAl-XSO3 samples—no major differences were observed for sample MgAl-8SO3 as it was treated under ultrasound, with a maximum at 300 μm which intensity did not change upon ultrasound application, a treatment which gave rise to a weak but detectable, shoulder at ca. 70 μm. On the contrary, sample MgAl-18SO3 showed a maximum at ca. 300 μm which rapidly disappeared even after only 5 min ultrasound, the curve being now dominated by a main peak at ca. 30 μm, with minor shoulders at ca. 8 and 300 μm. According to the element chemical analysis results, see
Table 1, this sample contains an appreciable amount of sodium sulfonate adsorbed on its external surface and this might be the origin of its easy deaggregation. Sample MgAl-12SO4 also shows a peak at ca. 300 μm which does not shift after ultrasound treatment, while a new shoulder at around 80 μm develops.
Concerning the samples that were kept in a slurry after preparation, their particle size distribution curves (
Figures S33–S37 in the Supplementary Materials section) were rather different. First, the maxima in the curves for the original samples (not treated under ultrasound) were always at a lower value than those for the corresponding dried samples, confirming that drying enhances agglomeration. Within a given series, as the length of the organic chain was increased, deagglomeration was less marked. For sample MgAl-11COO the maximum was recorded at 5 μm, with a second, weaker one at 50 μm; upon ultrasound treatment three peaks were recorded at 350 μm, 70 μm and 10 μm, showing that agglomeration takes place. It is possible that ultrasound energy in some sort of way helps to agglomerate very small particles which are released during the treatment. On the contrary, sample MgAl-17COO showed an intense maximum at ca 50 μm, with a shoulder at 3 μm but upon ultrasound treatment only a shift towards lower values (25 μm) of the absolute maximum was observed. For the sulfonate samples also some differences could be observed as the length of the organic chain was changed. A bimodal particle size distribution with a double peak at 12 and 50 μm for the untreated sample MgAl-12SO3 was converted into a monomodal particle size distribution with a maximum around 10 μm. No change being further observed as the time of ultrasound treatment was extended. Sample MgAl-18SO3 showed a single maximum at 30 μm, with a medium intensity shoulder at 150 μm, being transformed to a single peak again at 30 μm and development of a weak peak at 900–1000 μm upon ultrasonic treatment. No change in the particle size distribution upon ultrasound treatment was observed for sample MgAl-12SO4, showing a monomodal particle size distribution with a maximum at 5 μm, probably due to the presence of sodium sulfate on its surface.
It can be concluded that drying of the samples gives rise to agglomeration of the particles and that such an agglomeration is less intense if the samples are kept in a slurry. This finding might be relevant concerning a probable usage of organically modified LDHs in remediation strategies adressing retention of chlorinated organic solvents. The samples seem to behave randomly concerning the change in the particle size distribution, as no clear relationship between the functional group (−COO, −SO
3, −O-SO
3) could be found, nor with the length of the organic chain. The d(0.9) values for all samples are included in
Table 5.
Adsorption-desorption studies of N
2 adsorption at −196 °C were conducted to determine the specific surface areas of the dried samples, as well as their surface texture. The isotherms are included in the
Supplementary Materials Section (Figures S38–S46) and the values determined for the BET specific surface areas are given in
Table 5. The reference sample MgAl-CO3 shows a value close to 100 m
2/g, typical of carbonate-containing hydrotalcites which had been submitted to treatments as that for our sample [
31]. The isotherm corresponds to type II in the International Union of Pure and Applied Chemistry (IUPAC) classification [
36], due to adsorption on non-porous or macroporous surfaces, with unrestricted monolayer-multilayer adsorption [
37]. A hysteresis loop is also recorded, its shape corresponds to type H3, typical for adsorbents formed by rigid aggregates of plate-like particles giving rise to slit-shaped pores [
36,
37]. The organically modified samples show clearly lower specific surface area values but no relationship seems to exist between the specific surface area and the chain of the organic anion; even the shape of the hysteresis loop does not follow any specific trend with the chain length. All four carboxylate samples show, as sample MgAl-CO3, type II isotherms, due to adsorption on non-porous or microporous solids. While for sample MgAl-11-COO the narrow hysteresis loop corresponds to type H3, due to adsorption on aggregates of plate-like particles forming slit-shaped pores, those for the other three samples of this series are markedly broader, approaching to type H2, typical for disordered materials with ill-defined distribution of shape and size of the pores. A similar behavior is observed for the sulfonate containing samples MgAl-XSO3. There, the isotherm for sample MgAl-8SO3 shows a type H3 hysteresis, while those for samples MgAl-7SO3 and MgAl-18SO3 are very narrow (actually, almost absent for the last sample), corresponding to type II. This last behavior is also observed for sample MgAl-12SO4. These findings suggest that the surface texture of the samples is probably a result of slight differences in the drying process, which can strength aggregation of the primary particles. Obviously, no measurements of this type were performed on samples, which were kept in slurry.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}