UV-Vis Spectroscopy and Chemometrics for the Monitoring of Organosolv Pretreatments



Abstract
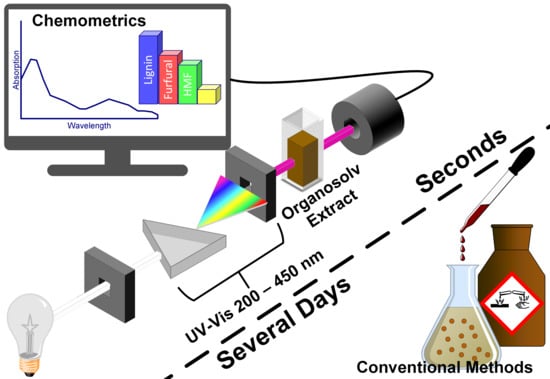
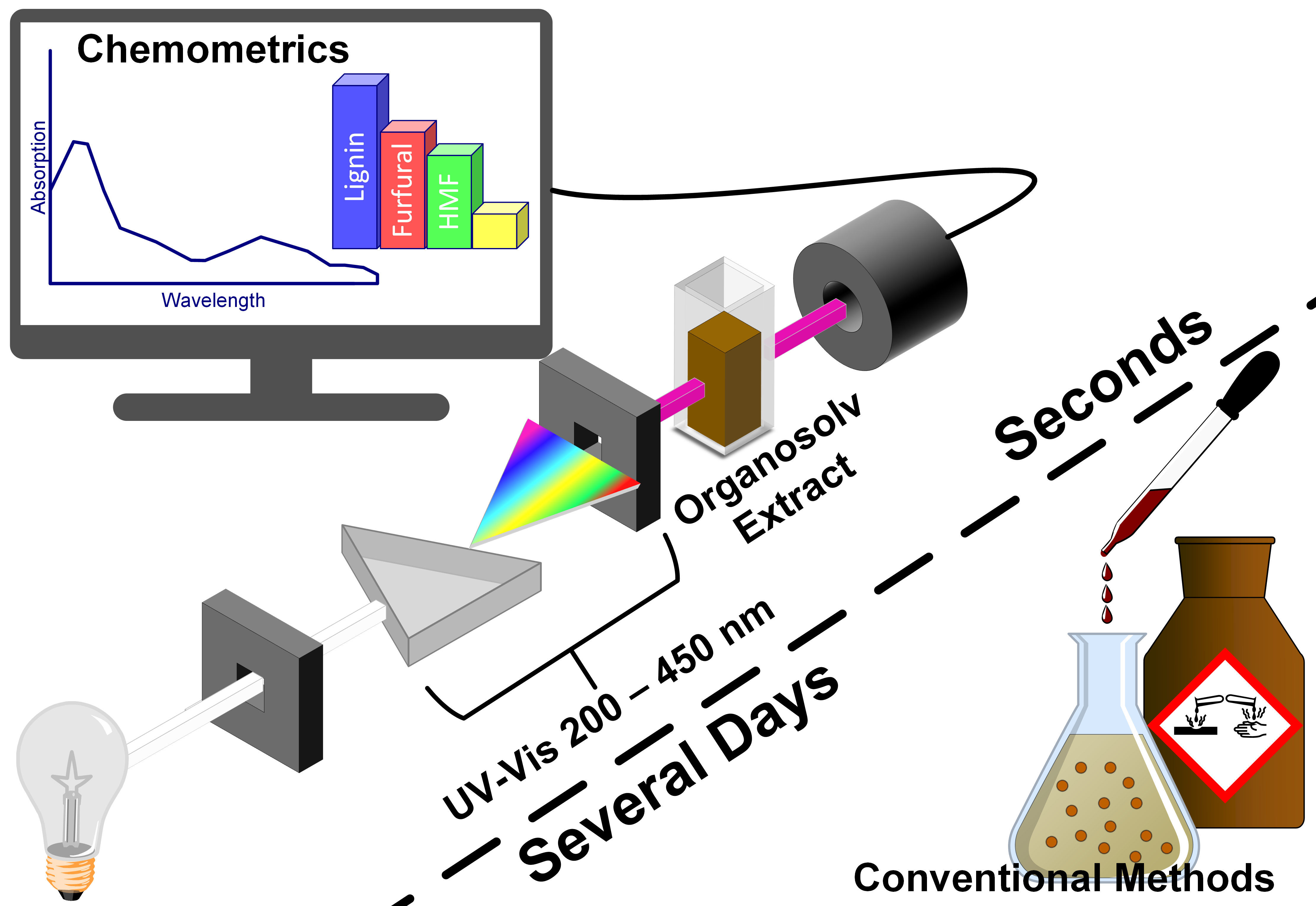
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Organosolv Pretreatment
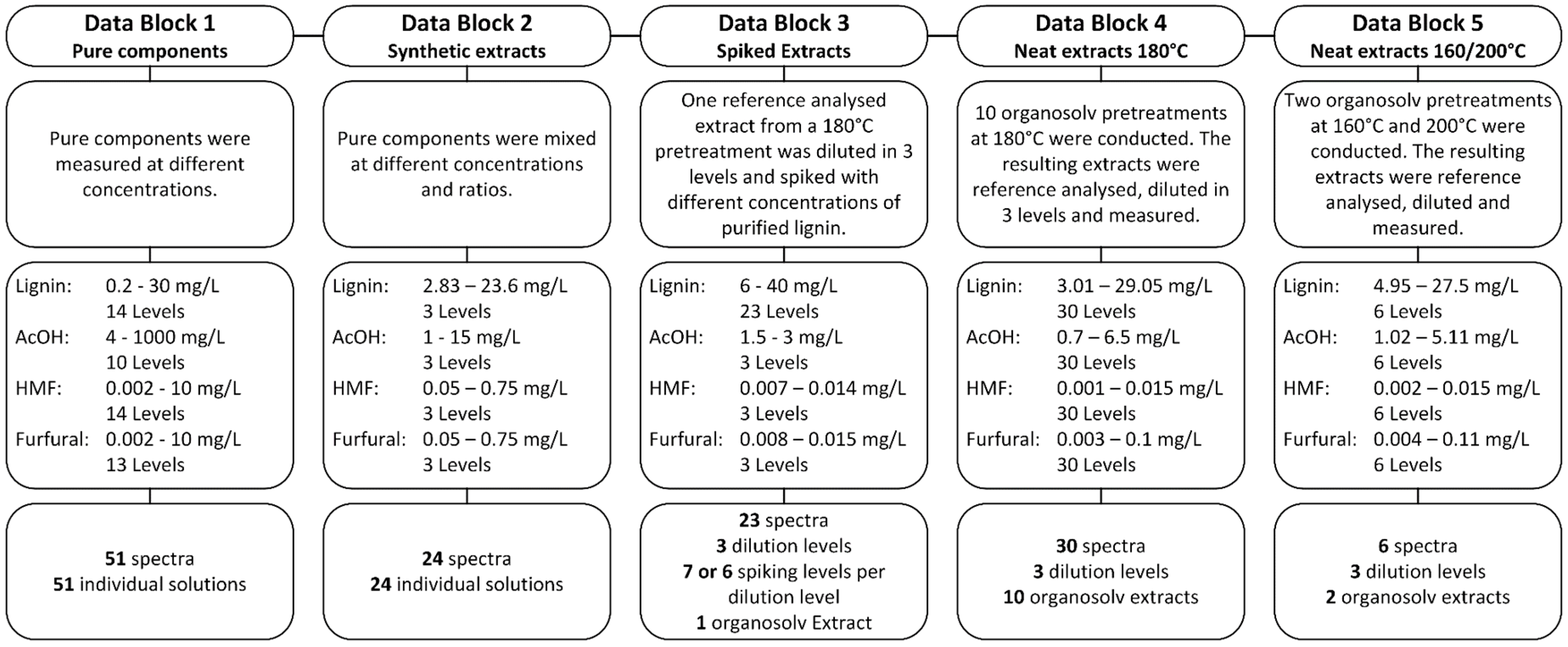
2.3. Reference Analysis and Synthetic Extract
2.4. UV-Vis Spectroscopy
2.5. Chemometrics
3. Results and Discussion
3.1. Single Component Spectra
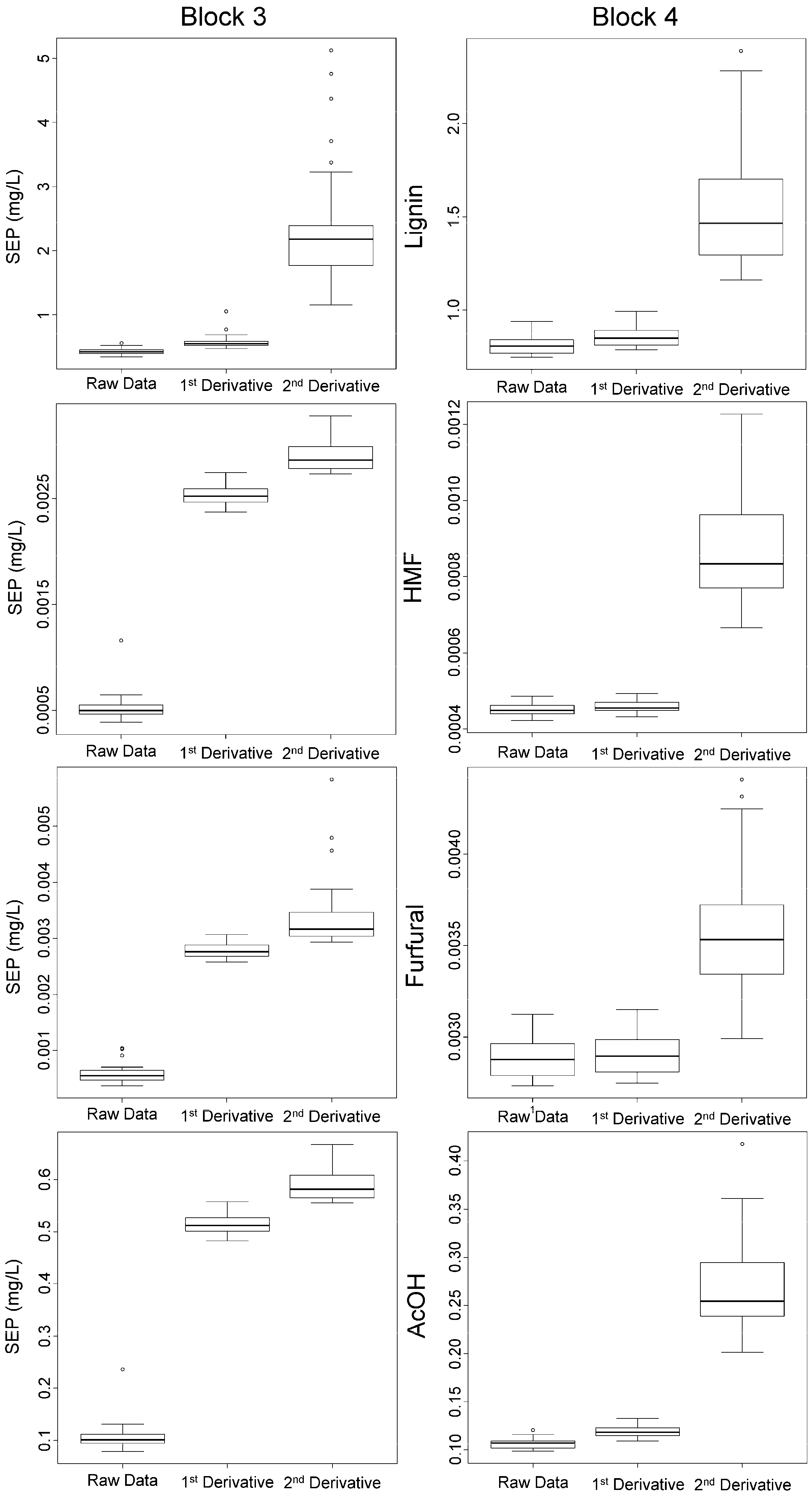
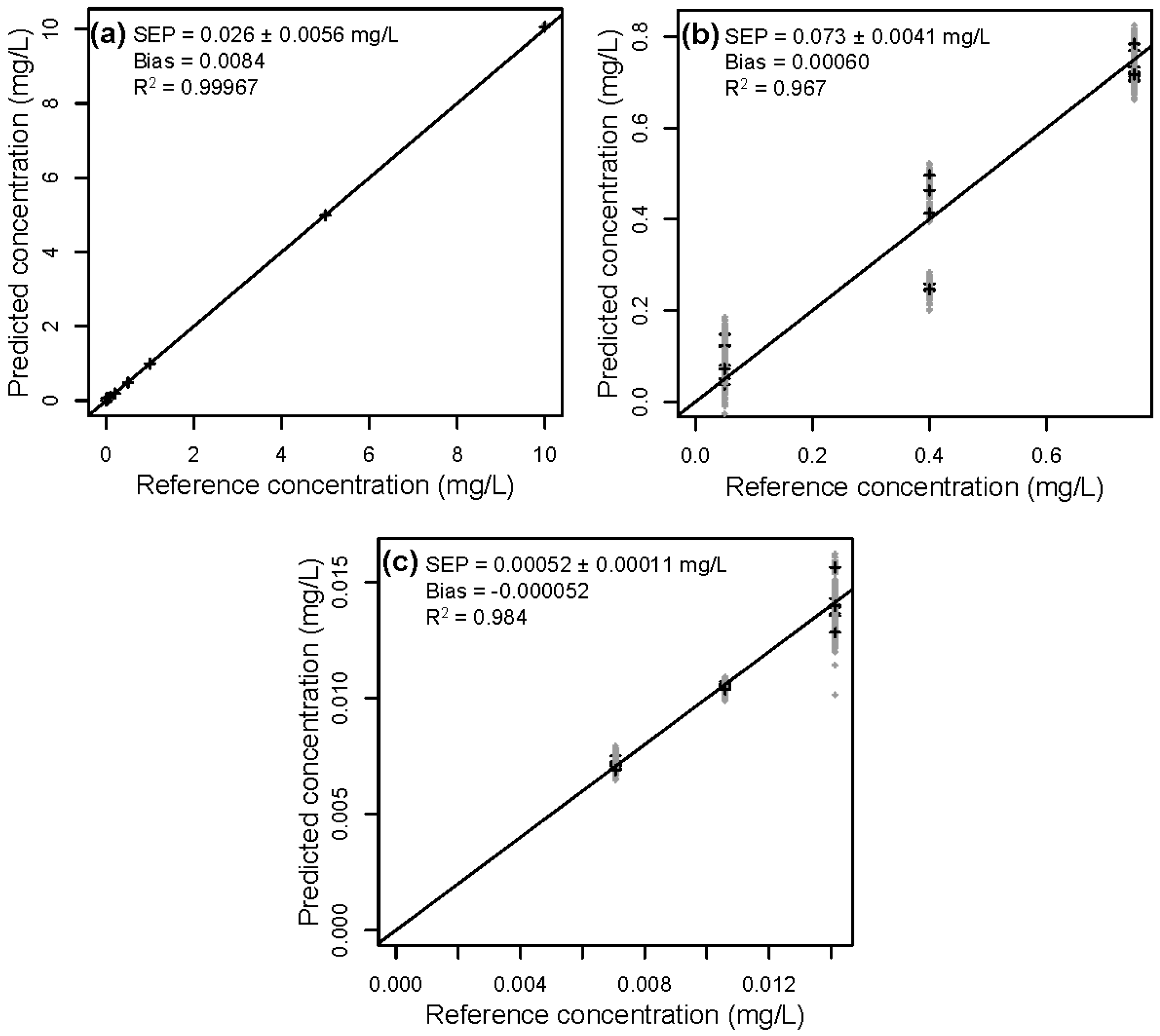
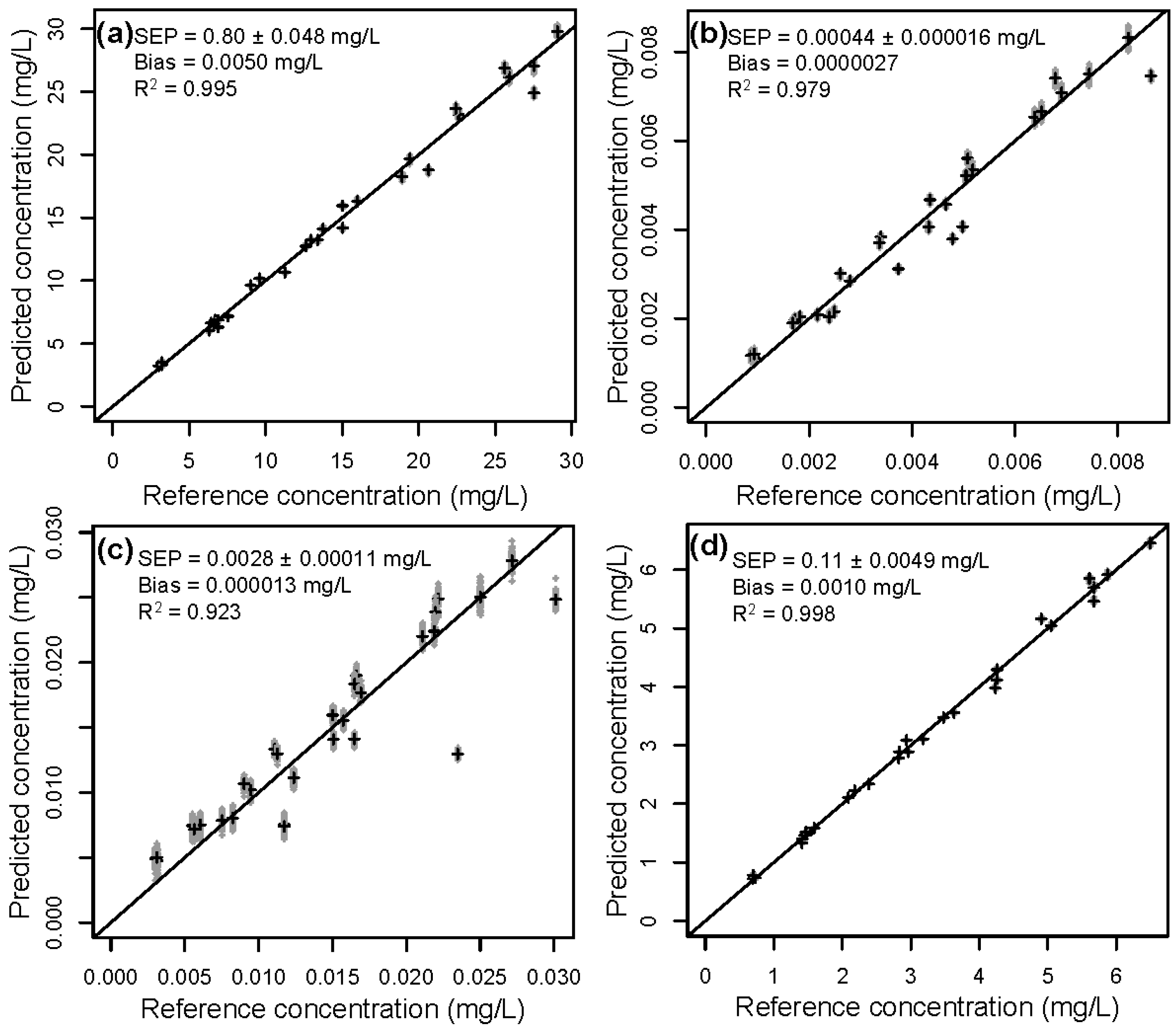
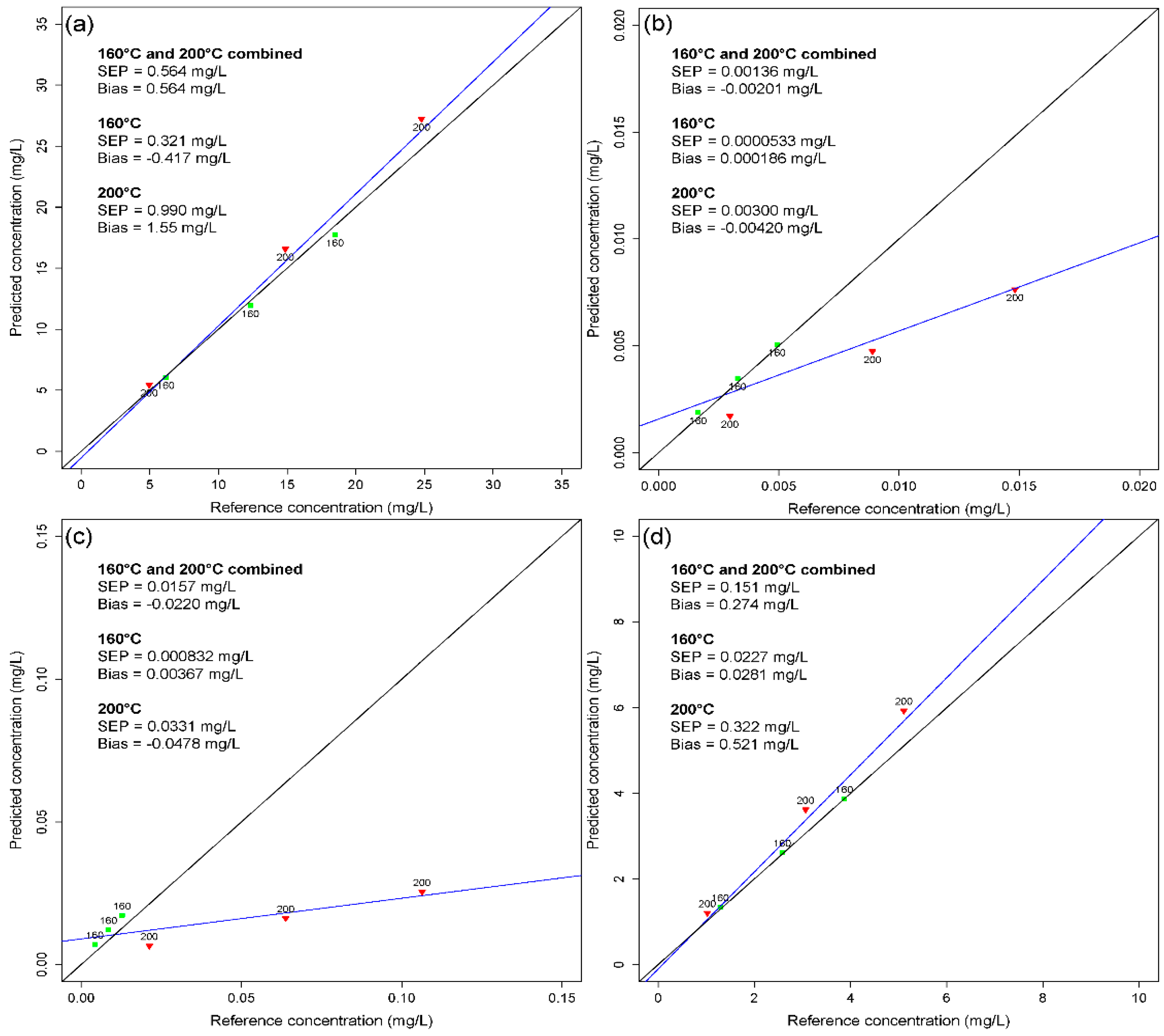
3.2. Validation of the Calibration
3.3. Model Application
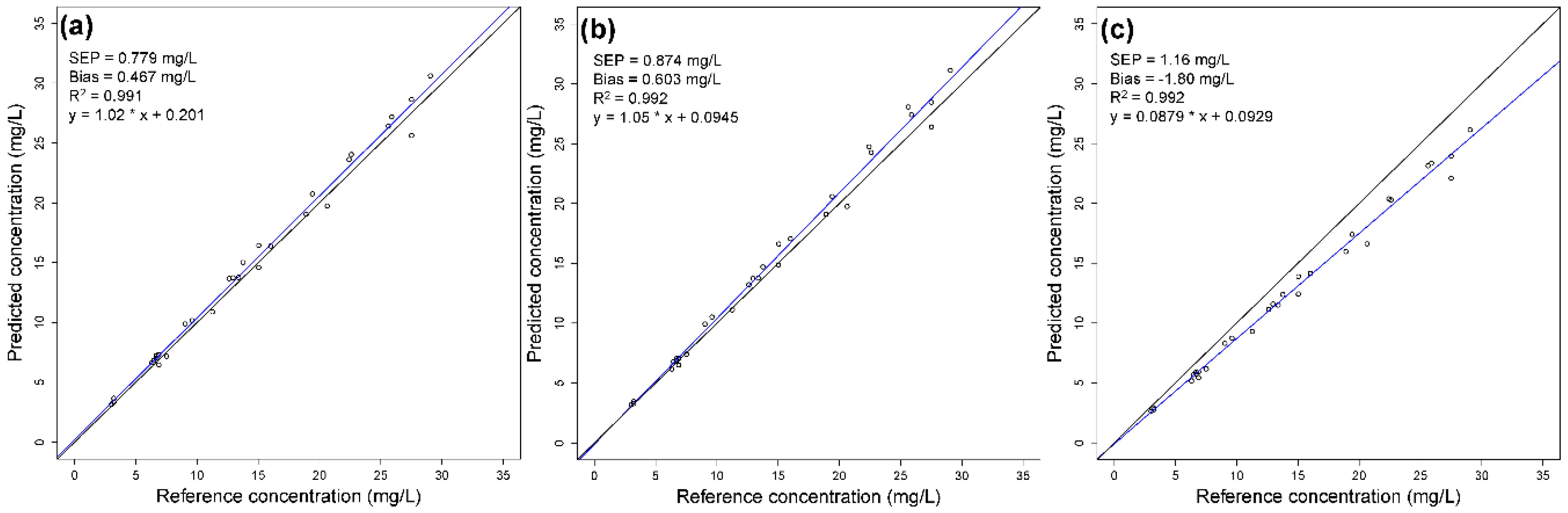
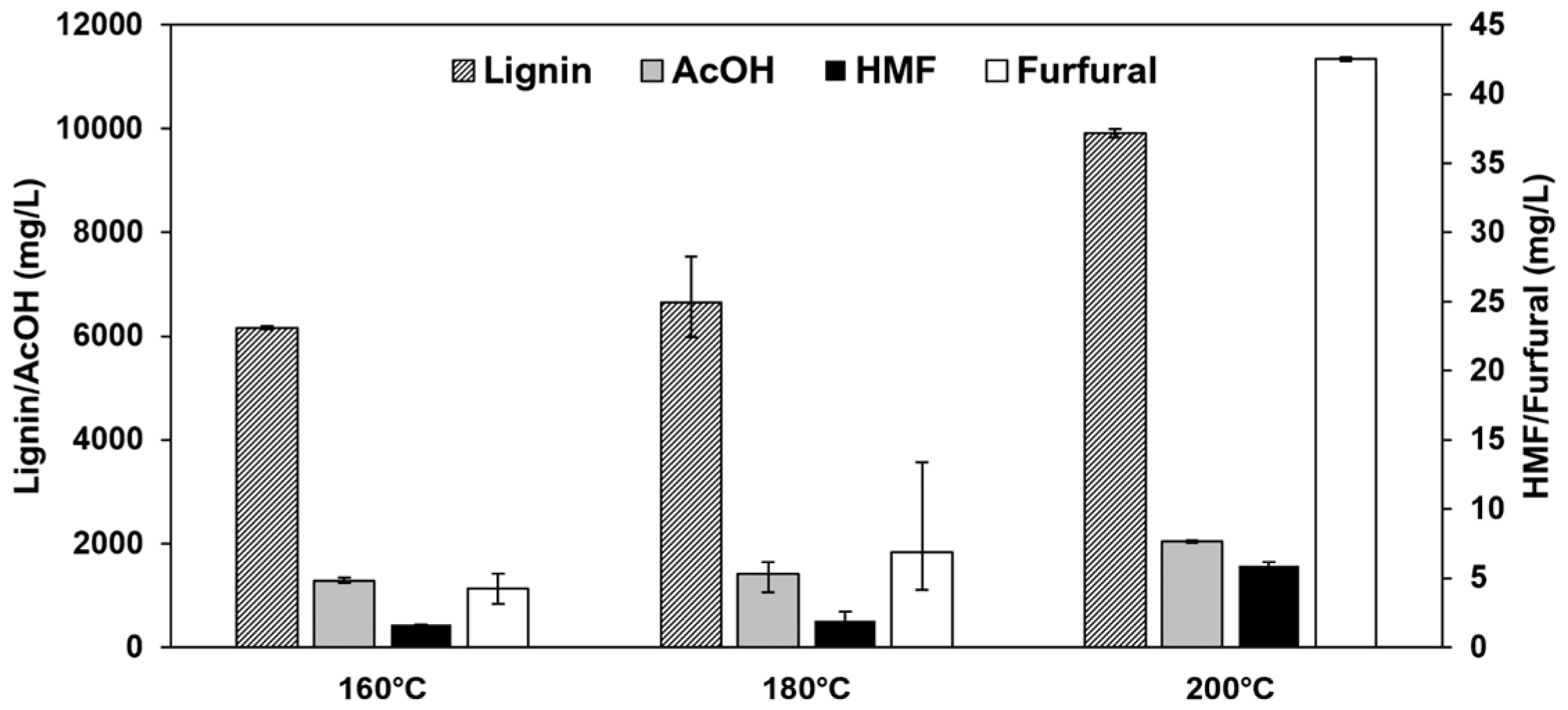
3.4. Influence of the Pretreatment Temperature
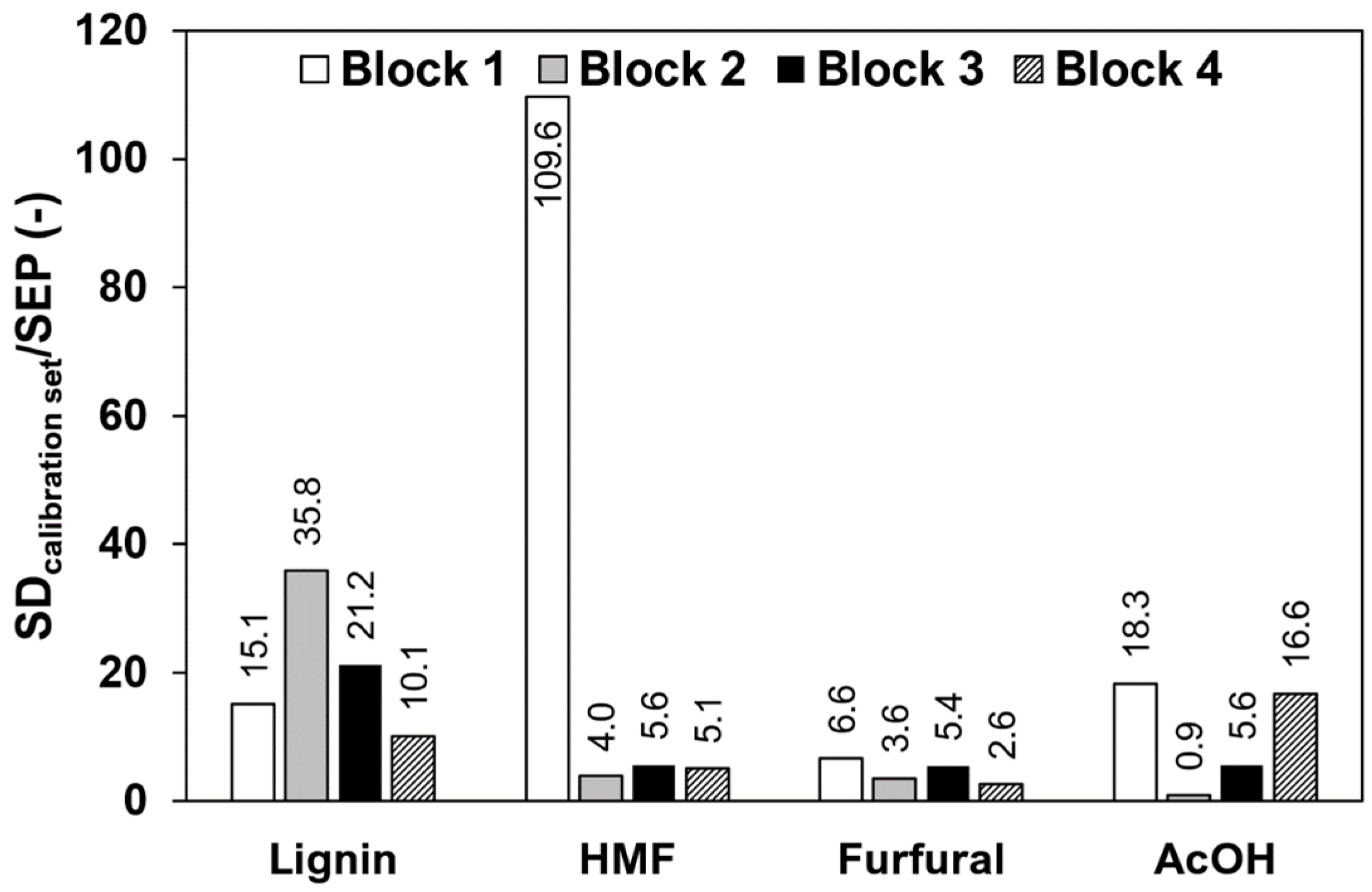
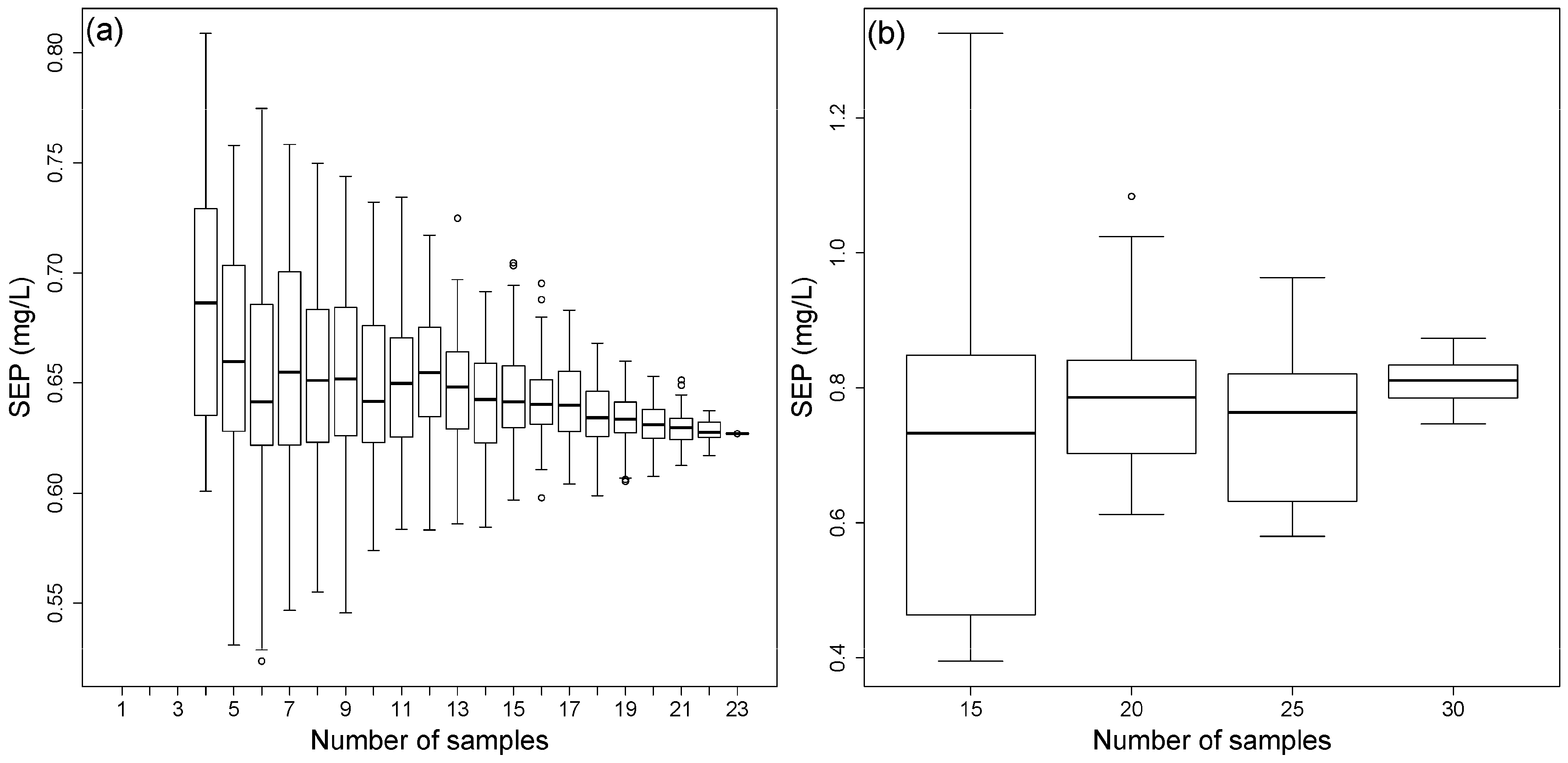
3.5. Reduction of the Calibration Effort
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kamm, B.; Kamm, M. Principles of biorefineries. Appl. Microbiol. Biotechnol. 2004, 64, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, Ó.J.; Cardona, C.A. Trends in biotechnological production of fuel ethanol from different feedstocks. Bioresour. Technol. 2008, 99, 5270–5295. [Google Scholar] [CrossRef] [PubMed]
- Tuck, C.O.; Perez, E.; Horvath, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of Biomass: Deriving More Value from Waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef] [PubMed]
- FitzPatrick, M.; Champagne, P.; Cunningham, M.F.; Whitney, R.A. A biorefinery processing perspective: Treatment of lignocellulosic materials for the production of value-added products. Bioresour. Technol. 2010, 101, 8915–8922. [Google Scholar] [CrossRef] [PubMed]
- Huijgen, W.J.J.; Smit, A.T.; de Wild, P.J.; den Uil, H. Fractionation of wheat straw by prehydrolysis, organosolv delignification and enzymatic hydrolysis for production of sugars and lignin. Bioresour. Technol. 2012, 114, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as Renewable Raw Material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Hames, B.R.; Thomas, S.R.; Sluiter, A.D.; Roth, C.J.; Templeton, D.W. Rapid biomass analysis. Appl. Biochem. Biotechnol. 2003, 105, 5–16. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.O.; Scarlata, C.; Sluiter, J.; Templeton, D. Determination of Structural Carbohydrates and Lignin in Biomass; Denver: Golden, CO, USA, 2008. [Google Scholar]
- Palmqvist, E.; Grage, H.; Meinander, N.Q.; Hahn-Hägerdal, B. Main and interaction effects of acetic acid, furfural, andp-hydroxybenzoic acid on growth and ethanol productivity of yeasts. Biotechnol. Bioeng. 1999, 63, 46–55. [Google Scholar] [CrossRef]
- Boyer, L.J.; Vega, J.L.; Klasson, K.T.; Clausen, E.C.; Gaddy, J.L. The effects of furfural on ethanol production by saccharomyces cereyisiae in batch culture. Biomass Bioenergy 1992, 3, 41–48. [Google Scholar] [CrossRef]
- Rasmussen, H.; Sørensen, H.R.; Meyer, A.S. Formation of degradation compounds from lignocellulosic biomass in the biorefinery: Sugar reaction mechanisms. Carbohydr. Res. 2014, 385, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C. Determination of Sugars, Byproducts, and Degradation Products in Liquid Fraction Process Samples; Denver: Golden, CO, USA, 2008. [Google Scholar]
- Lee, R.A.; Bédard, C.; Berberi, V.; Beauchet, R.; Lavoie, J.M. UV-Vis as quantification tool for solubilized lignin following a single-shot steam process. Bioresour. Technol. 2013, 144, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Dence, C.W. The Determination of Lignin. In Springer Series in Wood Science; Springer: Heidelberg/Berlin, Germany, 1992; pp. 33–61. ISBN 978-3-642-07768-5. [Google Scholar]
- Varmuza, K.; Filzmoser, P. Introduction to Multivariate Statistical Analysis in Chemometrics; CRC Press: Boca Raton, FL, USA, 2009; ISBN 978-1420059472. [Google Scholar]
- Filzmoser, P.; Liebmann, B.; Varmuza, K. Repeated double cross validation. J. Chemom. 2009, 23, 160–171. [Google Scholar] [CrossRef]
- R Development Core Team R. A Language and Environment for Statistical Computing; R Development Core Team, Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: www.r-project.org.
- Filzmoser, P.; Varmuza, K. “R” Package “chemometrics”. Available online: https://CRAN.R-project.org/package=chemometrics (accessed on 15 March 2017).
- Stevens, A.; Ramirez-Lopez, L. “R” Package ‘Prospectr’. Available online: https://CRAN.R-project.org/package=prospectr (accessed on 3 April 2018).
- Mevik, B.-H.; Wehrens, R.; Liland, K.H. “R” Package “pls”. Available online: https://CRAN.R-project.org/package=pls (accessed on 23 April 2017).
- Martinez, A.; Rodriguez, M.E.; York, S.W.; Preston, J.F.; Ingram, L.O. Use of UV absorbance to monitor furans in dilute acid hydrolysates of biomass. Biotechnol. Prog. 2000, 16, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, B.; Friedl, A.; Varmuza, K. Applicability of near-infrared spectroscopy for process monitoring in bioethanol production. Biochem. Eng. J. 2010, 52, 187–193. [Google Scholar] [CrossRef]
- Hatfield, R.; Fukushima, R.S. Can lignin be accurately measured? Crop Sci. 2005, 45, 832–839. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning. In Springer Series in Statistics; Springer: New York, NY, USA, 2009; Volume 99, ISBN 978-0-387-84857-0. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Block No. | Component | Standard Deviation Calibration Set | Raw Data | 1st Derivative | 2nd Derivative | Δ284/320 nm |
|---|---|---|---|---|---|---|
| 1 | Lignin | 9.35 | 0.62 ± 0.075 | 0.56 ± 0.14 | 1.7 ± 0.30 | na 1 |
| HMF | 2.85 | 0.026 ± 0.0056 | 0.020 ± 0.0043 | 0.53 ± 0.43 | na 1 | |
| Furfural | 2.96 | 0.45 ± 0.36 | 0.46 ± 0.37 | 0.69 ± 0.54 | na 1 | |
| AcOH | 308 | 17 ± 7.0 | 43 ± 28 | 201 ± 51 | na 1 | |
| 2 | Lignin | 8.28 | 0.23 ± 0.018 | 0.26 ± 0.017 | 0.62 ± 0.075 | 6.5 ± 0.30 |
| HMF | 0.290 | 0.073 ± 0.0041 | 0.076 ± 0.0066 | 0.21 ± 0.030 | 0.30 ± 0.022 | |
| Furfural | 0.290 | 0.081 ± 0.0083 | 0.085 ± 0.0085 | 0.24 ± 0.033 | 0.31 ± 0.032 | |
| AcOH | 5.84 | 6.5 ± 0.38 | 6.6 ± 0.40 | 7.0 ± 0.36 | 6.5 ± 0.39 | |
| 3 | Lignin | 8.85 | 0.42 ± 0.041 | 0.57 ± 0.091 | 2.40 ± 0.81 | 5.2 ± 0.31 |
| HMF | 0.00291 | 0.00052 ± 0.00011 | 0.0025 ± 0.000090 | 0.0028 ± 0.00014 | 0.0026 ± 0.00015 | |
| Furfural | 0.00316 | 0.00058 ± 0.00014 | 0.0027 ± 0.00014 | 0.0033 ± 0.00052 | 0.0034 ± 0.00023 | |
| AcOH | 0.591 | 0.11 ± 0.022 | 0.50 ± 0.018 | 0.58 ± 0.028 | 0.53 ± 0.031 | |
| 4 | Lignin | 8.08 | 0.80 ± 0.048 | 0.85 ± 0.056 | 1.6 ± 0.32 | 7.3 ± 0.27 |
| HMF | 0.00223 | 0.00044 ± 0.000016 | 0.00045 ± 0.000015 | 0.00088 ± 0.00016 | 0.0021 ± 0.000073 | |
| Furfural | 0.00750 | 0.0028 ± 0.00011 | 0.0029 ± 0.00011 | 0.0035 ± 0.00031 | 0.0073 ± 0.00030 | |
| AcOH | 1.75 | 0.11 ± 0.0049 | 0.12 ± 0.0061 | 0.27 ± 0.045 | 1.6 ± 0.060 |
| Block No. | Lignin | AcOH | HMF | Furfural |
|---|---|---|---|---|
| 1 | 2 | 1 | 3 | 1 |
| 2 | 2 | 1 | 3 | 3 |
| 3 | 2 | 4 | 4 | 4 |
| 4 | 1 | 1 | 1 | 1 |
| Lignin | HMF | Furfural | AcOH | |||||
|---|---|---|---|---|---|---|---|---|
| Calibration Block | SEP (mg/L) | Bias (mg/L) | SEP (mg/L) | Bias (mg/L) | SEP (mg/L) | Bias (mg/L) | SEP (mg/L) | Bias (mg/L) |
| 1 | 0.779 | 0.467 | 0.767 | 1.35 | 0.134 | −0.197 | 1108 | 1954 |
| 2 | 0.874 | 0.603 | 0.0942 | −0.0378 | 0.305 | 0.628 | 1.81 | 4.89 |
| 3 | 1.16 | −1.80 | 0.00690 | 0.0128 | 0.00386 | 0.00420 | 0.160 | 0.336 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beisl, S.; Binder, M.; Varmuza, K.; Miltner, A.; Friedl, A. UV-Vis Spectroscopy and Chemometrics for the Monitoring of Organosolv Pretreatments. ChemEngineering 2018, 2, 45. https://doi.org/10.3390/chemengineering2040045
Beisl S, Binder M, Varmuza K, Miltner A, Friedl A. UV-Vis Spectroscopy and Chemometrics for the Monitoring of Organosolv Pretreatments. ChemEngineering. 2018; 2(4):45. https://doi.org/10.3390/chemengineering2040045
Chicago/Turabian StyleBeisl, Stefan, Mathias Binder, Kurt Varmuza, Angela Miltner, and Anton Friedl. 2018. "UV-Vis Spectroscopy and Chemometrics for the Monitoring of Organosolv Pretreatments" ChemEngineering 2, no. 4: 45. https://doi.org/10.3390/chemengineering2040045
APA StyleBeisl, S., Binder, M., Varmuza, K., Miltner, A., & Friedl, A. (2018). UV-Vis Spectroscopy and Chemometrics for the Monitoring of Organosolv Pretreatments. ChemEngineering, 2(4), 45. https://doi.org/10.3390/chemengineering2040045