Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders



Abstract
1. Introduction
2. Source and Characteristics of PM
Particle Size-Dependent Translocation and Toxicity in the Airway Mucosa
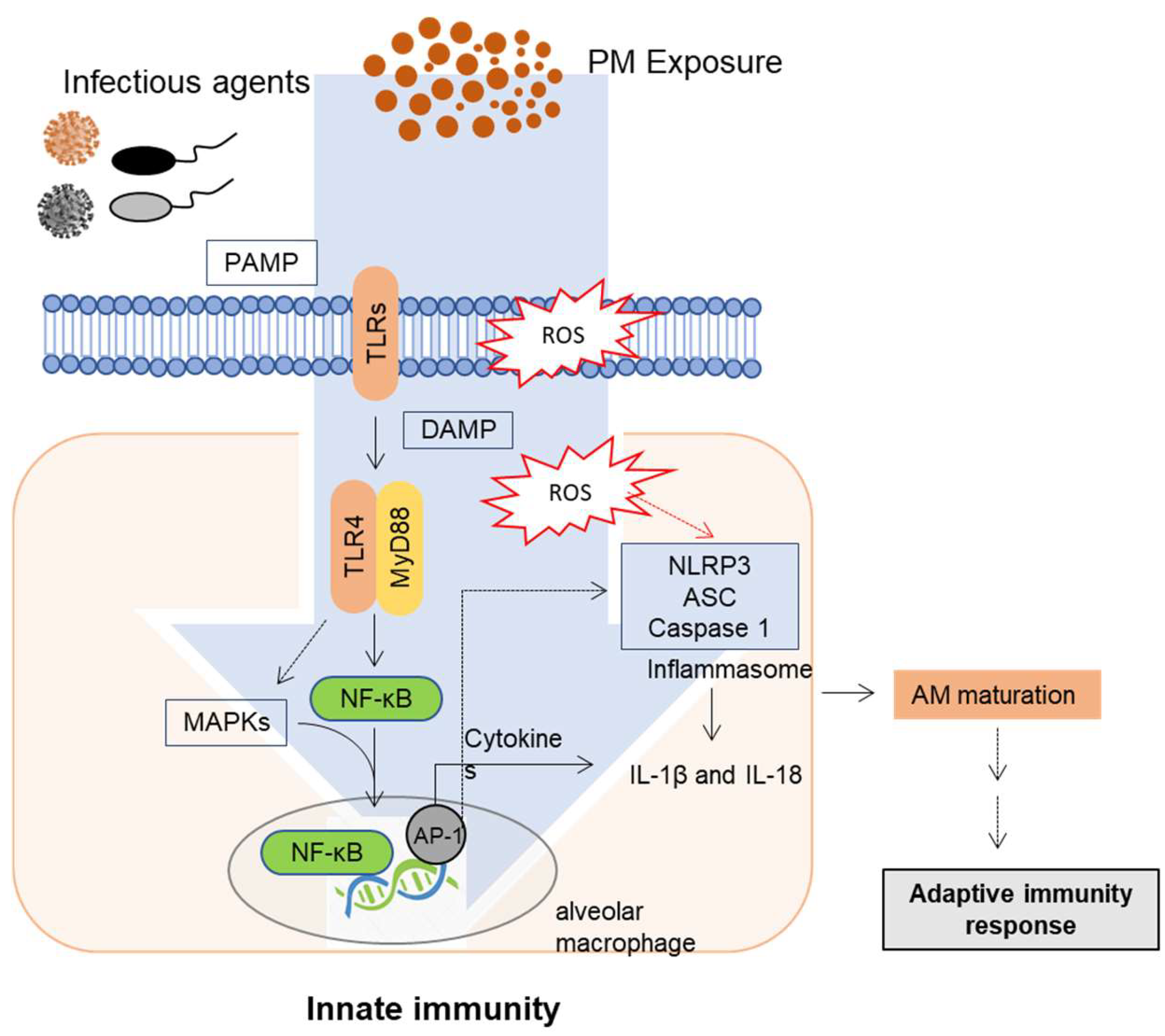
3. Mucosal Exposure and Innate Immune Responses to PM
Innate Immune Regulation and Signaling in the Lung Barrier
4. Adaptive Immune Response to PM Exposure
4.1. Influence of PM Exposure on T Cell Population
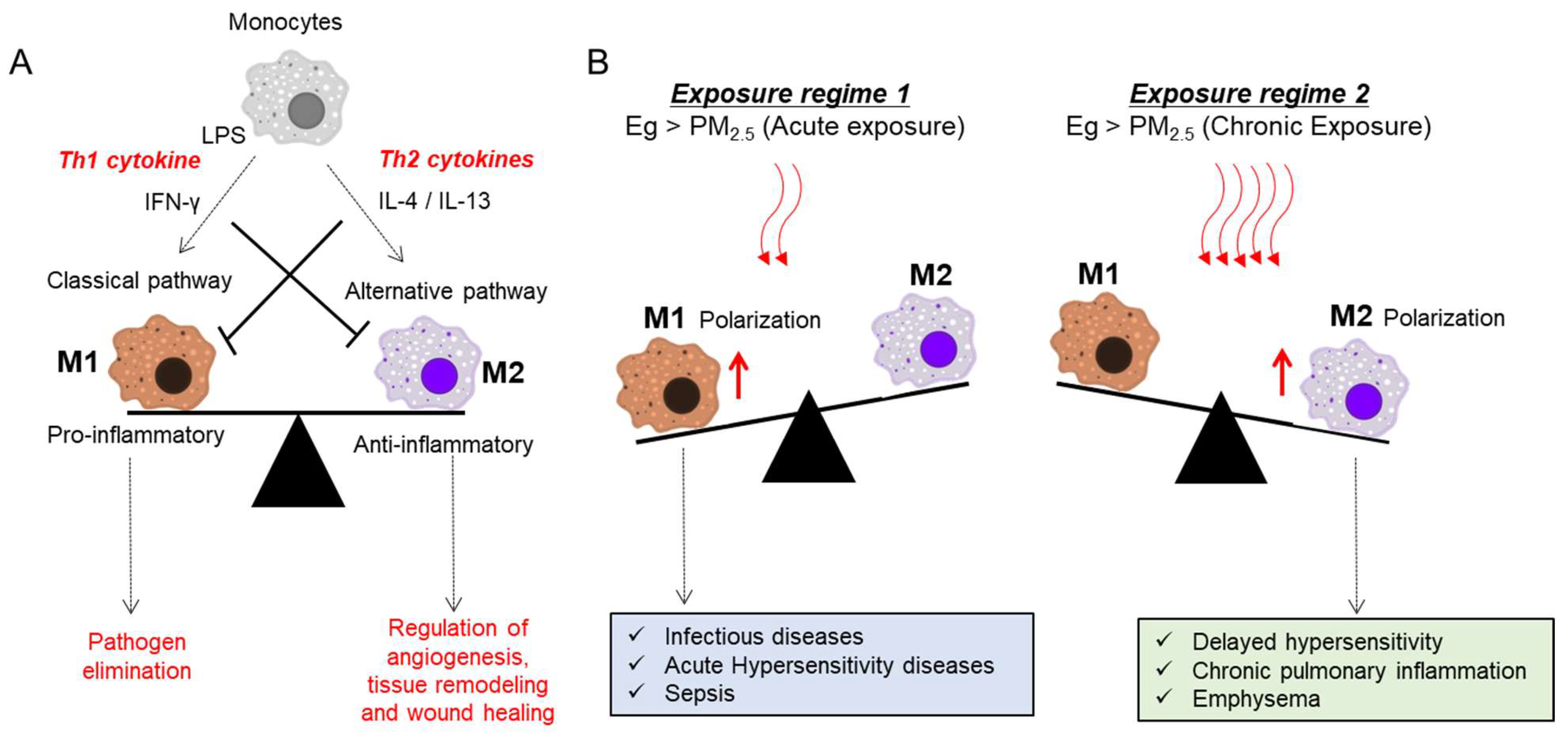
4.1.1. AMs as Pivotal Mediators of T Cell Skewing
4.1.2. Adjuvant-Like Actions of PM in T Cell Regulation
4.2. PM-Induced Suppression of T Cell Immune Responses
4.3. Controversy Regarding the Effect of PM on Adaptive Immunity
5. Effects of PM on Host Resistance to Infection
5.1. Roles of Airway Macrophages in Response to Infection and PM Exposure
5.2. PM Exposure in Bacterial Infection
5.3. PM Exposure in Viral Infection
6. PM-Linked Hypersensitivity
6.1. Effect on Acute and Chronic Hypersensitivity Diseases
6.2. PM Enhances Allergen Sensitization
6.3. Effect of Prenatal and Early-Life Exposure on Hypersensitivity Diseases
6.4. Molecular Etiologies of PM-Linked Hypersensitivity
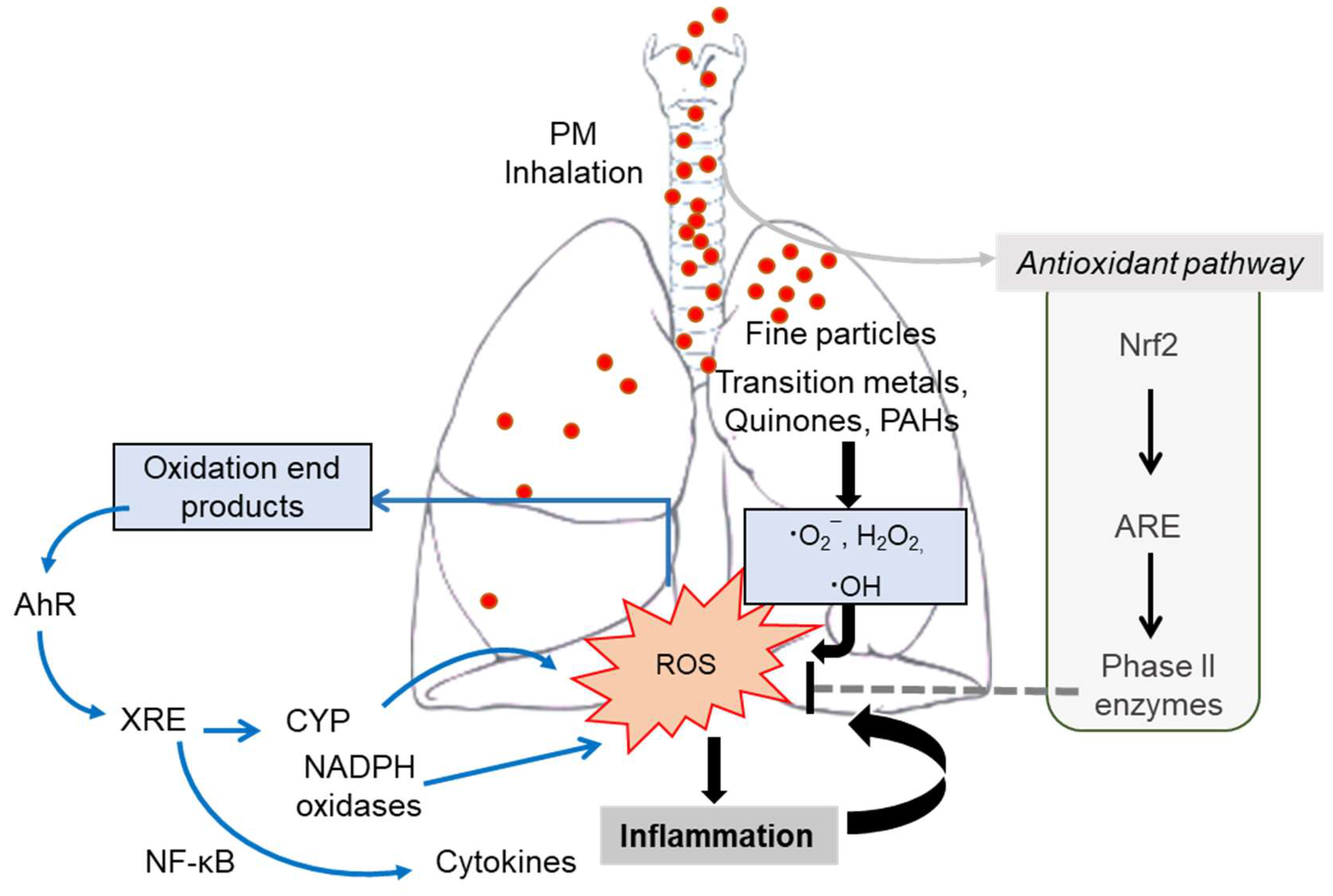
6.4.1. PM-Derived Oxidative Stress
6.4.2. Inflammatory Insults
6.4.3. Crosstalk with Allergens
6.4.4. Epigenetics and miRNA Regulation during PM-Linked Hypersensitivity
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mishra, R.; Krishnamoorthy, P.; Gangamma, S.; Raut, A.A.; Kumar, H. Particulate matter (PM10) enhances RNA virus infection through modulation of innate immune responses. Environ. Pollut. 2020, 266, 115148. [Google Scholar] [CrossRef]
- Namork, E.; Johansen, B.V.; Løvik, M. Detection of allergens adsorbed to ambient air particles collected in four European cities. Toxicol. Lett. 2006, 165, 71–78. [Google Scholar] [CrossRef]
- Vincent, R.; Goegan, P.; Johnson, G.; Brook, J.R.; Kumarathasan, P.; Bouthillier, L.; Burnett, R.T. Regulation of Promoter-CAT Stress Genes in HepG2 Cells by Suspensions of Particles from Ambient Air. Fundam. Appl. Toxicol. 1997, 39, 18–32. [Google Scholar] [CrossRef]
- Pope, C.A., 3rd; Burnett, R.T.; Thun, M.J.; Calle, E.E.; Krewski, D.; Ito, K.; Thurston, G.D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. J. Am. Med. Assoc. 2002, 287, 1132–1141. [Google Scholar] [CrossRef]
- Loomis, D.A.N.A.; Huang, W.; Chen, G. The International Agency for Research on Cancer (IARC) evaluation of the carcinogenicity of outdoor air pollution: Focus on China. Chin. J. Cancer 2014, 33, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K. The carcinogenicity of outdoor air pollution. Lancet Oncol. 2013, 14, 1262–1263. [Google Scholar] [CrossRef]
- Hamra, G.B.; Guha, N.; Cohen, A.; Laden, F.; Raaschou-Nielsen, O.; Samet, J.M.; Vineis, P.; Forastiere, F.; Saldiva, P.; Yorifuji, T.; et al. Outdoor Particulate Matter Exposure and Lung Cancer: A Systematic Review and Meta-Analysis. Environ. Health Perspect. 2014, 122, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, L.-W.; Huang, J.-J.; Song, F.-J.; Zhang, L.-P.; Qian, Z.-M.; Trevathan, E.; Mao, H.-J.; Han, B.; Vaughn, M.; et al. Long-term exposure to urban air pollution and lung cancer mortality: A 12-year cohort study in Northern China. Sci. Total. Environ. 2016, 571, 855–861. [Google Scholar] [CrossRef]
- Wen, C.P.; Gao, W. PM 2.5: An important cause for chronic obstructive pulmonary disease? Lancet Planet. Health 2018, 2, e105–e106. [Google Scholar] [CrossRef]
- Khafaie, M.A.; Salvi, S.S.; Yajnik, C.S.; Ojha, A.; Khafaie, B.; Gore, S.D. Air pollution and respiratory health among diabetic and non-diabetic subjects in Pune, India—Results from the Wellcome Trust Genetic Study. Environ. Sci. Pollut. Res. 2017, 24, 15538–15546. [Google Scholar] [CrossRef]
- Ling, S.H.; van Eeden, S.F. Particulate matter air pollution exposure: Role in the development and exacerbation of chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2009, 4, 233–43. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.M.; Kim, H.R.; Park, Y.J.; Lee, S.Y.; Chung, K.H. Organic extracts of urban air pollution particulate matter (PM2.5)-induced genotoxicity and oxidative stress in human lung bronchial epithelial cells (BEAS-2B cells). Mutat. Res. Toxicol. Environ. Mutagen. 2011, 723, 142–151. [Google Scholar] [CrossRef]
- Harrison, C.M.; Pompilius, M.; Pinkerton, K.E.; Ballinger, S.W. Mitochondrial Oxidative Stress Significantly Influences Atherogenic Risk and Cytokine-Induced Oxidant Production. Environ. Health Perspect. 2011, 119, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Weinmayr, G.; Pedersen, M.; Stafoggia, M.; Andersen, Z.J.; Galassi, C.; Munkenast, J.; Jaensch, A.; Oftedal, B.; Krog, N.H.; Aamodt, G.; et al. Particulate matter air pollution components and incidence of cancers of the stomach and the upper aerodigestive tract in the European Study of Cohorts of Air Pollution Effects (ESCAPE). Environ. Int. 2018, 120, 163–171. [Google Scholar] [CrossRef]
- Crinnion, W. Particulate Matter Is a Surprisingly Common Contributor to Disease. Integr. Med. 2017, 16, 8–12. [Google Scholar]
- Zhang, P.; Dong, G.; Sun, B.; Zhang, L.; Chen, X.; Ma, N.; Yu, F.; Guo, H.; Huang, H.; Lee, Y.L.; et al. Long-Term Exposure to Ambient Air Pollution and Mortality Due to Cardiovascular Disease and Cerebrovascular Disease in Shenyang, China. PLoS ONE 2011, 6, e20827. [Google Scholar] [CrossRef] [PubMed]
- Katanoda, K.; Sobue, T.; Satoh, H.; Tajima, K.; Suzuki, T.; Nakatsuka, H.; Takezaki, T.; Nakayama, T.; Nitta, H.; Tanabe, K.; et al. An Association Between Long-Term Exposure to Ambient Air Pollution and Mortality from Lung Cancer and Respiratory Diseases in Japan. J. Epidemiology 2011, 21, 132–143. [Google Scholar] [CrossRef]
- Riddervold, I.S.; Bønløkke, J.H.; Olin, A.-C.; Grønborg, T.K.; Schlünssen, V.; Skogstrand, K.; Hougaard, D.M.; Massling, A.; Sigsgaard, T. Effects of wood smoke particles from wood-burning stoves on the respiratory health of atopic humans. Part. Fibre Toxicol. 2012, 9, 12. [Google Scholar] [CrossRef]
- Jamriska, M.; Thomas, S.; Morawska, L.; Clark, B.A. Relation between indoor and outdoor exposure to fine particles near a busy arterial road. Indoor Air 1999, 9, 75–84. [Google Scholar] [CrossRef]
- Erlandsson, L.; Lindgren, R.; Nääv, Åsa; Krais, A.M.; Strandberg, B.; Lundh, T.; Boman, C.; Isaxon, C.; Hansson, S.R.; Malmqvist, E. Exposure to wood smoke particles leads to inflammation, disrupted proliferation and damage to cellular structures in a human first trimester trophoblast cell line. Environ. Pollut. 2020, 264, 114790. [Google Scholar] [CrossRef]
- Rokoff, L.B.; Koutrakis, P.; Garshick, E.; Karagas, M.R.; Oken, E.; Gold, D.R.; Fleisch, A.F. Wood Stove Pollution in the Developed World: A Case to Raise Awareness Among Pediatricians. Curr. Probl. Pediatr. Adolesc. Health Care 2017, 47, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Soukup, J.M.; Case, M.; Dailey, L.; Richards, J.; Berntsen, J.; Devlin, R.B.; Stone, S.; Rappold, A. Exposure to wood smoke particles produces inflammation in healthy volunteers. Occup. Environ. Med. 2011, 69, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Levi, M.; Garcia-Aymerich, J.; Villar, J.; Ramírez-Sarmiento, A.; Antó, J.M.; Gea, J. Wood smoke exposure and risk of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Rajpandey, M. Domestic smoke pollution and chronic bronchitis in a rural community of the Hill Region of Nepal. Thorax 1984, 39, 337–339. [Google Scholar] [CrossRef]
- Ezzati, M.; Kammen, D.M. Quantifying the effects of exposure to indoor air pollution from biomass combustion on acute respiratory infections in developing countries. Environ. Health Perspect. 2001, 109, 481–488. [Google Scholar] [CrossRef]
- Smith, K.R.; Samet, J.M.; Romieu, I.; Bruce, N. Indoor air pollution in developing countries and acute lower respiratory infections in children. Thorax 2000, 55, 518–532. [Google Scholar] [CrossRef]
- Thompson, J.E. Airborne Particulate Matter: Human Exposure and Health Effects. J. Occup. Environ. Med. 2018, 60, 392–423. [Google Scholar] [CrossRef]
- Wei, T.; Tang, M. Biological effects of airborne fine particulate matter (PM 2.5) exposure on pulmonary immune system. Environ. Toxicol. Pharmacol. 2018, 60, 195–201. [Google Scholar] [CrossRef]
- Mukherjee, A.; Agrawal, M. A Global Perspective of Fine Particulate Matter Pollution and Its Health Effects. Rev. Environ. Contam. Toxicol. 2018, 244, 5–51. [Google Scholar] [CrossRef]
- Alfaro-Moreno, E.; Nawrot, T.S.; Nemmar, A.; Nemery, B. Particulate matter in the environment: Pulmonary and cardiovascular effects. Curr. Opin. Pulm. Med. 2007, 13, 98–106. [Google Scholar] [CrossRef]
- Gu, X.-Y.; Chu, X.; Zeng, X.-L.; Bao, H.-R.; Liu, X.-J. Effects of PM2.5 exposure on the Notch signaling pathway and immune imbalance in chronic obstructive pulmonary disease. Environ. Pollut. 2017, 226, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Glencross, D.A.; Ho, T.-R.; Camiña, N.; Hawrylowicz, C.M.; Pfeffer, P.E. Air pollution and its effects on the immune system. Free Radic. Biol. Med. 2020, 151, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Miyata, R.; Van Eeden, S.F. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol. Appl. Pharmacol. 2011, 257, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Deng, L.; Miao, Y.; Guo, X.; Li, Y. Particle deposition in the human lung: Health implications of particulate matter from different sources. Environ. Res. 2019, 169, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Idani, E.; Geravandi, S.; Akhzari, M.; Goudarzi, G.; Alavi, N.; Yari, A.R.; Mehrpour, M.; Khavasi, M.; Bahmaei, J.; Bostan, H.; et al. Characteristics, sources, and health risks of atmospheric PM10-bound heavy metals in a populated middle eastern city. Toxin Rev. 2020, 39, 266–274. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, J.; Yang, X.; Zhang, Y.; Chen, Z. The Role and Potential Pathogenic Mechanism of Particulate Matter in Childhood Asthma: A Review and Perspective. J. Immunol. Res. 2020, 2020, 1–8. [Google Scholar] [CrossRef]
- Cheung, K.; Daher, N.; Kam, W.; Shafer, M.M.; Ning, Z.; Schauer, J.J.; Sioutas, C. Spatial and temporal variation of chemical composition and mass closure of ambient coarse particulate matter (PM10–2.5) in the Los Angeles area. Atmos. Environ. 2011, 45, 2651–2662. [Google Scholar] [CrossRef]
- Kim, K.-H.; Kabir, E.; Kabir, S. A review on the human health impact of airborne particulate matter. Environ. Int. 2015, 74, 136–143. [Google Scholar] [CrossRef]
- Bernstein, J.A.; Alexis, N.; Barnes, C.; Bernstein, I.L.; Nel, A.; Peden, D.; Diaz-Sanchez, D.; Tarlo, S.M.; Williams, P.B. Health effects of air pollution. J. Allergy Clin. Immunol. 2004, 114, 1116–1123. [Google Scholar] [CrossRef]
- Naeher, L.P.; Brauer, M.; Lipsett, M.; Zelikoff, J.T.; Simpson, C.D.; Koenig, J.Q.; Smith, K.R. Woodsmoke Health Effects: A Review. Inhal. Toxicol. 2007, 19, 67–106. [Google Scholar] [CrossRef]
- Larson, T.V.; Koenig, J.Q. Wood Smoke: Emissions and Noncancer Respiratory Effects. Annu. Rev. Public Health 1994, 15, 133–156. [Google Scholar] [CrossRef] [PubMed]
- Dastoorpoor, M.; Sekhavatpour, Z.; Masoumi, K.; Mohammadi, M.J.; Aghababaeian, H.; Khanjani, N.; Hashemzadeh, B.; Vahedian, M. Air pollution and hospital admissions for cardiovascular diseases in Ahvaz, Iran. Sci. Total. Environ. 2019, 652, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Aghababaeian, H.; Dastoorpoor, M.; Ghasemi, A.; Kiarsi, M.; Khanjani, N.; Ahvazi, L.A. Cardiovascular and respiratory emergency dispatch due to short-term exposure to ambient PM10 in Dezful, Iran. J. Cardiovasc. Thorac. Res. 2019, 11, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.-H.; Kao, S.-W.; Tantoh, D.M.; Ko, P.-C.; Lan, S.-J.; Liaw, Y.-P. Association between fine particulate matter and oral cancer among Taiwanese men. J. Investig. Med. 2018, 67, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Consonni, D.; Carugno, M.; De Matteis, S.; Nordio, F.; Randi, G.; Bazzano, M.; Caporaso, N.E.; Tucker, M.A.; Bertazzi, P.A.; Pesatori, A.C.; et al. Outdoor particulate matter (PM10) exposure and lung cancer risk in the EAGLE study. PLoS ONE 2018, 13, e0203539. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, C.E.; Sarkar, S.; Cantarella, P.; Osornio-Vargas, A.; Quintana-Belmares, R.; Meng, Q.; Kirn, T.J.; Ohman Strickland, P.; Chow, J.C.; Watson, J.G.; et al. Air Pollution Particulate Matter Alters Antimycobacterial Respiratory Epithelium Innate Immunity. Infect. Immun. 2015, 83, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Rivas-Santiago, C.; Ibironke, O.A.; Carranza, C.; Meng, Q.; Osornio-Vargas, A.R.; Zhang, J.; Torres, M.; Chow, J.C.; Watson, J.G.; et al. Season and size of urban particulate matter differentially affect cytotoxicity and human immune responses to Mycobacterium tuberculosis. PLoS ONE 2019, 14, e0219122. [Google Scholar] [CrossRef]
- Saygin, M.; Gonca, T.; Ozturk, O.; Has, M.; Caliskan, S.; Has, Z.G. To Investigate the Effects of Air Pollution (PM10 and SO2) on the Respiratory Diseases Asthma and Chronic Obstructive Pulmonary Disease. Turk. Thorac. J. 2017, 18, 33–39. [Google Scholar] [CrossRef]
- Tecer, L.H.; Alagha, O.; Karaca, F.; Tuncel, G.; Eldes, N. Particulate Matter (PM2.5, PM10-2.5, and PM10) and Children’s Hospital Admissions for Asthma and Respiratory Diseases: A Bidirectional Case-Crossover Study. J. Toxicol. Environ. Health Part A 2008, 71, 512–520. [Google Scholar] [CrossRef]
- Dellinger, B.; Pryor, W.A.; Cueto, R.; Squadrito, G.L.; Hegde, V.; Deutsch, W.A. Role of Free Radicals in the Toxicity of Airborne Fine Particulate Matter. Chem. Res. Toxicol. 2001, 14, 1371–1377. [Google Scholar] [CrossRef]
- Zhao, J.; Xie, Y.; Qian, C.; Li, L.; Jiang, R.; Kan, H.; Chen, R.; Song, W. Imbalance of Th1 and Th2 cells in cardiac injury induced by ambient fine particles. Toxicol. Lett. 2012, 208, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Hoet, P.H.M.; Vanquickenborne, B.; Dinsdale, D.; Thomeer, M.; Hoylaerts, M.F.; Vanbilloen, H.; Mortelmans, L.; Nemery, B. Passage of Inhaled Particles into the Blood Circulation in Humans. Circulation 2002, 105, 411–414. [Google Scholar] [CrossRef]
- Nemmar, A.; Vanbilloen, H.; Hoylaerts, M.F.; Hoet, P.H.M.; Verbruggen, A.; Nemery, B. Passage of Intratracheally Instilled Ultrafine Particles from the Lung into the Systemic Circulation in Hamster. Am. J. Respir. Crit. Care Med. 2001, 164, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, S.; Karg, E.; Roth, C.; Schulz, H.; Ziesenis, A.; Heinzmann, U.; Schramel, P.; Heyder, J. Pulmonary and systemic distribution of inhaled ultrafine silver particles in rats. Environ. Health Perspect. 2001, 109, 547–551. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Li, G.; Zhang, Y.; Li, J.; Haosheng, C. Fluorescent reconstitution on deposition of PM2.5 in lung and extrapulmonary organs. Proc. Natl. Acad. Sci. USA 2019, 116 (Suppl. 4), 2488–2493. [Google Scholar] [CrossRef]
- Kwon, J.; Hwang, S.; Jin, H.; Kim, D.; Minai-Tehrani, A.; Yoon, H.; Choi, M.; Yoon, T.; Han, D.; Kang, Y.; et al. Body Distribution of Inhaled Fluorescent Magnetic Nanoparticles in the Mice. J. Occup. Health 2008, 50, 1–6. [Google Scholar] [CrossRef]
- Sarlo, K.; Blackburn, K.L.; Clark, E.D.; Grothaus, J.; Chaney, J.; Neu, S.; Flood, J.; Abbott, D.; Bohne, C.; Casey, K.; et al. Tissue distribution of 20nm, 100nm and 1000nm fluorescent polystyrene latex nanospheres following acute systemic or acute and repeat airway exposure in the rat. Toxicology 2009, 263, 117–126. [Google Scholar] [CrossRef]
- Pillay, V.; Murugan, K.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; Du Toit, L.C. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int. J. Nanomed. 2015, 10, 2191–2206. [Google Scholar] [CrossRef]
- Balakrishna, S.; Saravia, J.; Thevenot, P.; Ahlert, T.; Lomnicki, S.M.; Dellinger, B.; Cormier, S. Environmentally persistent free radicals induce airway hyperresponsiveness in neonatal rat lungs. Part. Fibre Toxicol. 2011, 8, 11. [Google Scholar] [CrossRef]
- Thevenot, P.T.; Saravia, J.; Jin, N.; Giaimo, J.D.; Chustz, R.E.; Mahne, S.; Kelley, M.A.; Hebert, V.Y.; Dellinger, B.; Dugas, T.R.; et al. Radical-Containing Ultrafine Particulate Matter Initiates Epithelial-to-Mesenchymal Transitions in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2013, 48, 188–197. [Google Scholar] [CrossRef]
- Tang, Q.; Huang, K.; Liu, J.; Wu, S.; Shen, D.; Dai, P.; Li, C. Fine particulate matter from pig house induced immune response by activating TLR4/MAPK/NF-κB pathway and NLRP3 inflammasome in alveolar macrophages. Chemosphere 2019, 236, 124373. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ichinose, T.; Song, Y.; Yoshida, Y.; Bekki, K.; Arashidani, K.; Yoshida, S.; Nishikawa, M.; Takano, H.; Shibamoto, T.; et al. Desert dust induces TLR signaling to trigger Th2-dominant lung allergic inflammation via a MyD88-dependent signaling pathway. Toxicol. Appl. Pharmacol. 2016, 296, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Shoenfelt, J.; Mitkus, R.J.; Zeisler, R.; Spatz, R.O.; Powell, J.; Fenton, M.J.; Squibb, K.A.; Medvedev, A.E. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J. Leukoc. Biol. 2009, 86, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Hirota, J.A.; Hirota, S.A.; Warner, S.M.; Stefanowicz, D.; Shaheen, F.; Beck, P.L.; Macdonald, J.A.; Hackett, T.-L.; Sin, D.D.; Van Eeden, S.; et al. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J. Allergy Clin. Immunol. 2012, 129, 1116–1125.e6. [Google Scholar] [CrossRef]
- Rylance, J.; Fullerton, D.G.; Scriven, J.; Aljurayyan, A.N.; Mzinza, D.; Barrett, S.; Wright, A.K.A.; Wootton, D.G.; Glennie, S.J.; Baple, K.; et al. Household Air Pollution Causes Dose-Dependent Inflammation and Altered Phagocytosis in Human Macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 52, 584–593. [Google Scholar] [CrossRef]
- Bauer, R.N.; Diaz-Sanchez, D.; Jaspers, I. Effects of air pollutants on innate immunity: The role of Toll-like receptors and nucleotide-binding oligomerization domain–like receptors. J. Allergy Clin. Immunol. 2012, 129, 14–24. [Google Scholar] [CrossRef]
- Becker, S.; Fenton, M.J.; Soukup, J.M. Involvement of Microbial Components and Toll-like Receptors 2 And 4 in Cytokine Responses to Air Pollution Particles. Am. J. Respir. Cell Mol. Biol. 2002, 27, 611–618. [Google Scholar] [CrossRef]
- Hirota, J.A.; Marchant, D.J.; Singhera, G.K.; Moheimani, F.; Dorscheid, D.R.; Carlsten, C.; Sin, D.; Knight, D. Urban particulate matter increases human airway epithelial cell IL-1β secretion following scratch wounding and H1N1 influenza A exposurein vitro. Exp. Lung Res. 2015, 41, 353–362. [Google Scholar] [CrossRef]
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted Disruption of the MyD88 Gene Results in Loss of IL-1- and IL-18-Mediated Function. Immunology 1998, 9, 143–150. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, C.A., Jr. MyD88 Is an Adaptor Protein in the hToll/IL-1 Receptor Family Signaling Pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Mukaida, N.; Mahe, Y.; Matsushima, K. Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J. Biol. Chem. 1990, 265, 21128–21133. [Google Scholar] [CrossRef]
- Yang, Z.; Kong, B.; Mosser, D.M.; Zhang, X. TLRs, macrophages, and NK cells: Our understandings of their functions in uterus and ovary. Int. Immunopharmacol. 2011, 11, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.-Y.; Yoon, S.R.; Kim, T.-D.; Choi, I.; Jung, H. Toll-Like Receptors in Natural Killer Cells and Their Application for Immunotherapy. J. Immunol. Res. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.-U.; Kamada, N.; Muñoz-Planillo, R.; Kim, Y.-G.; Kim, D.; Koizumi, Y.; Hasegawa, M.; Himpsl, S.D.; Browne, H.P.; Lawley, T.D.; et al. Distinct Commensals Induce Interleukin-1β via NLRP3 Inflammasome in Inflammatory Monocytes to Promote Intestinal Inflammation in Response to Injury. Immunology 2015, 42, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Reibman, J.; Hsu, Y.; Chen, L.-C.; Bleck, B.; Gordon, T. Airway Epithelial Cells Release MIP-3α/CCL20 in Response to Cytokines and Ambient Particulate Matter. Am. J. Respir. Cell Mol. Biol. 2003, 28, 648–654. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.W.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef]
- Matthews, N.C.; Faith, A.; Pfeffer, P.E.; Lu, H.; Kelly, F.; Hawrylowicz, C.M.; Lee, T.H. Urban Particulate Matter Suppresses Priming of Th1 Cells by GM-CSF-Activated Human Dendritic Cells. Am. J. Respir. Cell Mol. Biol. 2013, 50, 281–291. [Google Scholar] [CrossRef]
- Fujii, T.; Hayashi, S.; Hogg, J.C.; Vincent, R.; Van Eeden, S.F. Particulate Matter Induces Cytokine Expression in Human Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2001, 25, 265–271. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef]
- Trinchieri, G.; Pflanz, S.; Kastelein, R. The IL-12 Family of Heterodimeric Cytokines. Immunology 2003, 19, 641–644. [Google Scholar] [CrossRef]
- Kidd, P. Th1/Th2 balance: The hypothesis, its limitations, and implications for health and disease. Altern. Med. Rev. J. Clin. Ther. 2003, 8, 223–246. [Google Scholar]
- Zhao, H.; Ma, J.K.; Barger, M.W.; Mercer, R.R.; Millecchia, L.; Schwegler-Berry, D.; Castranova, V.; Ma, J.Y. Reactive Oxygen Species- and Nitric Oxide-Mediated Lung Inflammation and Mitochondrial Dysfunction in Wild-Type and iNOS-Deficient Mice Exposed to Diesel Exhaust Particles. J. Toxicol. Environ. Health Part A 2009, 72, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Roh, J.; Kim, Y.; Park, K.; Kim, D.-S.; Yu, S.-D. PM 2.5 collected in a residential area induced Th1-type inflammatory responses with oxidative stress in mice. Environ. Res. 2011, 111, 348–355. [Google Scholar] [CrossRef]
- Huang, K.-L.; Liu, S.-Y.; Chou, C.C.K.; Lee, Y.-H.; Cheng, T.-J. The effect of size-segregated ambient particulate matter on Th1/Th2-like immune responses in mice. PLoS ONE 2017, 12, e0173158. [Google Scholar] [CrossRef]
- Van Eeden, S.F.; Tan, W.C.; Suwa, T.; Mukae, H.; Terashima, T.; Fujii, T.; Qui, D.; Vincent, R.; Hogg, J.C. Cytokines Involved in the Systemic Inflammatory Response Induced by Exposure to Particulate Matter Air Pollutants (PM10). Am. J. Respir. Crit. Care Med. 2001, 164, 826–830. [Google Scholar] [CrossRef]
- Bach, N.; Bølling, A.K.; Brinchmann, B.C.; Totlandsdal, A.I.; Skuland, T.; Holme, J.; Låg, M.; Schwarze, P.E.; Øvrevik, J. Cytokine responses induced by diesel exhaust particles are suppressed by PAR-2 silencing and antioxidant treatment, and driven by polar and non-polar soluble constituents. Toxicol. Lett. 2015, 238, 72–82. [Google Scholar] [CrossRef]
- Hamilton, R.F.; Holian, A.; Morandi, M.T. A comparison of asbestos and urban particulate matter in the in vitro modification of human alveolar macrophage anti-gen-presenting cell function. Exp. Lung Res. 2004, 30, 147–162. [Google Scholar] [CrossRef]
- Kang, C.-M.; Jang, A.-S.; Ahn, M.-H.; Shin, J.-A.; Kim, J.-H.; Choi, Y.-S.; Rhim, T.-Y.; Park, C.-S. Interleukin-25 and Interleukin-13 Production by Alveolar Macrophages in Response to Particles. Am. J. Respir. Cell Mol. Biol. 2005, 33, 290–296. [Google Scholar] [CrossRef]
- Zhu, Z.; Homer, R.J.; Wang, Z.; Chen, Q.; Geba, G.P.; Wang, J.; Zhang, Y.; Elias, J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Investig. 1999, 103, 779–788. [Google Scholar] [CrossRef]
- Gour, N.; Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015, 75, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Wynn, T. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.. IL-13 effector functions. Annu. Rev. Immunol. 2003, 21, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 Induces IL-4, IL-5, and IL-13 and Th2-Associated Pathologies In Vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef]
- Corrigan, C.J.; Wang, W.; Meng, Q.; Fang, C.; Eid, G.; Caballero, M.R.; Lv, Z.; An, Y.; Wang, Y.-H.; Liu, Y.-J.; et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J. Allergy Clin. Immunol. 2011, 128, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.-Q.; Dong, W.-Y.; Xu, J.; Hong, Z.-C.; Zhao, R.-W.; Deng, C.; Zhuang, G.; Zhang, R. T-Helper Type 1-T-Helper Type 2 Shift and Nasal Remodeling after Fine Particulate Matter Exposure in a Rat Model of Allergic Rhinitis. Am. J. Rhinol. Allergy 2017, 31, 148–155. [Google Scholar] [CrossRef]
- Zhang, X.; Zhong, W.; Meng, Q.; Lin, Q.; Fang, C.; Huang, X.; Li, C.; Huang, Y.; Tan, J. Ambient PM2.5 exposure exacerbates severity of allergic asthma in previously sensitized mice. J. Asthma 2015, 52, 785–794. [Google Scholar]
- He, M.; Ichinose, T.; Kobayashi, M.; Arashidani, K.; Yoshida, S.; Nishikawa, M.; Takano, H.; Sun, G.; Shibamoto, T. Differences in allergic inflammatory responses between urban PM2.5 and fine particle derived from desert-dust in murine lungs. Toxicol. Appl. Pharmacol. 2016, 297, 41–55. [Google Scholar] [CrossRef]
- Galli, S.J.; Borregaard, N.; Wynn, T. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef]
- Alexis, N.E.; Lay, J.C.; Zeman, K.; Bennett, W.E.; Peden, D.B.; Soukup, J.M.; Devlin, R.B.; Becker, S. Biological material on inhaled coarse fraction particulate matter activates airway phagocytes in vivo in healthy volunteers. J. Allergy Clin. Immunol. 2006, 117, 1396–1403. [Google Scholar] [CrossRef]
- Williams, M.A.; Rangasamy, T.; Bauer, S.M.; Killedar, S.; Karp, M.; Kensler, T.W.; Yamamoto, M.; Breysse, P.; Biswal, S.; Georas, S.N. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J. Immunol. 2008, 181, 4545–4559. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.M.S.S.; Soukup, J. Coarse(PM 2.5–10), Fine(PM 2.5), and Ultrafine Air Pollution Particles Induce/Increase Immune Costimulatory Receptors on Human Blood-Derived Monocytes but not on Alveolar Macrophages. J. Toxicol. Environ. Health Part A 2003, 66, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Porter, M.; Karp, M.; Killedar, S.; Bauer, S.M.; Guo, J.; Williams, D.; Breysse, P.; Georas, S.N.; Williams, M.A. Diesel-Enriched Particulate Matter Functionally Activates Human Dendritic Cells. Am. J. Respir. Cell Mol. Biol. 2007, 37, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Siebenlist, U. Roles of the NF- B Pathway in Lymphocyte Development and Function. Cold Spring Harb. Perspect. Biol. 2009, 2, a000182. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ning, Z.; Majumdar, R.; Cui, J.; Takabe, W.; Jen, N.; Sioutas, C.; Hsiai, T.K. Ultrafine particles from diesel vehicle emissions at different driving cycles induce differential vascular pro-inflammatory responses: Implication of chemical components and NF-κB signaling. Part. Fibre Toxicol. 2010, 7, 6–12. [Google Scholar] [CrossRef]
- Nadra, I.; Boccaccini, A.R.; Philippidis, P.; Whelan, L.C.; McCarthy, G.M.; Haskard, D.O.; Landis, R. Effect of particle size on hydroxyapatite crystal-induced tumor necrosis factor alpha secretion by macrophages. Atherosclerosis 2008, 196, 98–105. [Google Scholar] [CrossRef]
- Ding, M.; Kisin, E.; Zhao, J.; Bowman, L.; Lu, Y.; Jiang, B.; Leonard, S.; Vallyathan, V.; Castranova, V.; Murray, A. Size-dependent effects of tungsten carbide–cobalt particles on oxygen radical production and activation of cell signaling pathways in murine epidermal cells. Toxicol. Appl. Pharmacol. 2009, 241, 260–268. [Google Scholar] [CrossRef]
- Castañeda, A.R.; Vogel, C.F.A.; Bein, K.J.; Hughes, H.; Smiley-Jewell, S.; Pinkerton, K.E. Ambient particulate matter enhances the pulmonary allergic immune response to house dust mite in a BALB/c mouse model by augmenting Th2- and Th17-immune responses. Physiol. Rep. 2018, 6, e13827. [Google Scholar] [CrossRef]
- Jiang, A.; Bloom, O.; Ono, S.; Cui, W.; Unternaehrer, J.; Jiang, S.; Whitney, J.A.; Connolly, J.; Banchereau, J.; Mellman, I. Disruption of E-Cadherin-Mediated Adhesion Induces a Functionally Distinct Pathway of Dendritic Cell Maturation. Immunology 2007, 27, 610–624. [Google Scholar] [CrossRef]
- Saravia, J.; You, D.; Thevenot, P.; Lee, G.I.; Shrestha, B.; Lomnicki, S.; Cormier, S. Early-life exposure to combustion-derived particulate matter causes pulmonary immunosuppression. Mucosal Immunol. 2014, 7, 694–704. [Google Scholar] [CrossRef]
- Jaligama, S.; Saravia, J.; You, D.; Yadav, N.; Lee, G.I.; Shrestha, B.; Cormier, S. Regulatory T cells and IL10 suppress pulmonary host defense during early-life exposure to radical containing combustion derived ultrafine particulate matter. Respir. Res. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, K.A.; Secrest, J.R.; Lau, C.; Pulczinski, J.; Zamora, M.L.; Leal, J.; Langley, R.; Myatt, L.G.; Raju, M.; Chang, R.C.-A.; et al. In utero ultrafine particulate matter exposure causes offspring pulmonary immunosuppression. Proc. Natl. Acad. Sci. USA 2019, 116, 3443–3448. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G.; Kinney, P.L.; Chillrud, S.N.; Jack, D. A Systematic Review of Innate Immunomodulatory Effects of Household Air Pollution Secondary to the Burning of Biomass Fuels. Ann. Glob. Health 2015, 81, 368–374. [Google Scholar] [CrossRef]
- Zhu, L.; Zhao, Q.; Yang, T.; Ding, W.; Zhao, Y. Cellular Metabolism and Macrophage Functional Polarization. Int. Rev. Immunol. 2014, 34, 82–100. [Google Scholar] [CrossRef]
- Shen, Y.; Song, J.; Wang, Y.; Chen, Z.; Zhang, L.; Yu, J.; Zhu, D.; Zhong, M. M2 macrophages promote pulmonary endothelial cells regeneration in sepsis-induced acute lung injury. Ann. Transl. Med. 2019, 7, 142. [Google Scholar] [CrossRef]
- Vlahos, R.; Bozinovski, S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front. Immunol. 2014, 5, 435. [Google Scholar] [CrossRef]
- Ma, Q.-Y.; Huang, D.; Zhang, H.-J.; Wang, S.; Chen, X.-F. Exposure to particulate matter 2.5 (PM2.5) induced macrophage-dependent inflammation, characterized by increased Th1/Th17 cytokine secretion and cytotoxicity. Int. Immunopharmacol. 2017, 50, 139–145. [Google Scholar] [CrossRef]
- Shi, Q.; Zhao, L.; Xu, C.; Zhang, L.; Zhao, H. High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation. Molecules 2019, 24, 1766. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, Y.; Wang, Q.; Chen, H.; Zhou, X. Fine particulate matter exposure promotes M2 macrophage polarization through inhibiting histone deacetylase 2 in the pathogenesis of chronic obstructive pulmonary disease. Ann. Transl. Med. 2020, 8, 1303. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef]
- Chandra, V.; Mahajan, S.; Saini, A.; Dkhar, H.K.; Nanduri, R.; Raj, E.B.; Kumar, A.; Gupta, P. Human IL10 Gene Repression by Rev-erbα Ameliorates Mycobacterium tuberculosis Clearance. J. Biol. Chem. 2013, 288, 10692–10702. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Luo, Q.; Guo, Y.; Chen, J.; Xiong, G.; Peng, Y.; Ye, J.; Li, J. Mycobacterium tuberculosis-Induced Polarization of Human Macrophage Orchestrates the Formation and Development of Tuberculous Granulomas In Vitro. PLoS ONE 2015, 10, e0129744. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.S.; Schoenbach, V.J.; Richardson, D.B.; Gammon, M.D. Particulate air pollution and susceptibility to the development of pulmonary tuberculosis disease in North Carolina: An ecological study. Int. J. Environ. Health Res. 2014, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cui, L.; Hou, L.; Yu, C.; Tao, N.; Liu, J.; Li, Y.; Zhou, C.; Yang, G.; Li, H. Ambient Air Pollution Exposures and Newly Diagnosed Pulmonary Tuberculosis in Jinan, China: A Time Series Study. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Ibironke, O.; Carranza, C.; Sarkar, S.; Torres, M.; Choi, H.T.; Nwoko, J.; Black, K.; Quintana-Belmares, R.O.; Osornio-Vargas, A.R.; Strickland, P.O.; et al. Urban Air Pollution Particulates Suppress Human T-Cell Responses to Mycobacterium Tuberculosis. Int. J. Environ. Res. Public Health 2019, 16, 4112. [Google Scholar] [CrossRef]
- Becker, J.M.S.S. Exposure to urban air particulates alters the macrophage-mediated inflammatory response to respiratory viral infection. J. Toxicol. Environ. Health Part A 1999, 57, 445–457. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Tanwar, V.; Adelstein, J.M.; Wold, L.E. Double Trouble: Combined Cardiovascular Effects of Particulate Matter Exposure and COVID-19. Cardiovasc. Res. 2020, 117, 85–95. [Google Scholar] [CrossRef]
- Setti, L.; Passarini, F.; De Gennaro, G.; Barbieri, P.; Perrone, M.G.; Borelli, M.; Palmisani, J.; Di Gilio, A.; Torboli, V.; Fontana, F.; et al. SARS-Cov-2RNA found on particulate matter of Bergamo in Northern Italy: First evidence. Environ. Res. 2020, 188, 109754. [Google Scholar] [CrossRef]
- Kaan, P.M.; Hegele, R.G. Interaction between respiratory syncytial virus and particulate matter in guinea pig alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2003, 28, 697–704. [Google Scholar] [CrossRef]
- Comunian, S.; Dongo, D.; Milani, C.; Palestini, P. Air Pollution and COVID-19: The Role of Particulate Matter in the Spread and Increase of COVID-19′s Morbidity and Mortality. Int. J. Environ. Res. Public Health 2020, 17, 4487. [Google Scholar] [CrossRef] [PubMed]
- Mantecca, P.; Sancini, G.; Moschini, E.; Farina, F.; Gualtieri, M.; Rohr, A.; Miserocchi, G.; Palestini, P.; Camatini, M. Lung toxicity induced by intratracheal instillation of size-fractionated tire particles. Toxicol. Lett. 2009, 189, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.; Saldiva, P.; Sakae, R.; Böhm, G.; Martins, M. Urban Levels of Air Pollution Increase Lung Responsiveness in Rats. Environ. Res. 1995, 69, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-Y.; Ding, H.; Jiang, L.-N.; Chen, S.-W.; Zheng, J.-P.; Qiu, M.; Zhou, Y.-X.; Chen, Q.; Guan, W.-J. Association between Air Pollutants and Asthma Emergency Room Visits and Hospital Admissions in Time Series Studies: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0138146. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Thevenot, P.; Saravia, J.; Ahlert, T.; Cormier, S.A. Radical-Containing Particles Activate Dendritic Cells and Enhance Th17 Inflammation in a Mouse Model of Asthma. Am. J. Respir. Cell Mol. Biol. 2011, 45, 977–983. [Google Scholar] [CrossRef]
- Diaz-Sanchez, D.; Dotson, A.R.; Takenaka, H.; Saxon, A. Diesel exhaust particles induce local IgE production in vivo and alter the pattern of IgE messenger RNA isoforms. J. Clin. Investig. 1994, 94, 1417–1425. [Google Scholar] [CrossRef]
- Diaz-Sanchez, D.; Tsien, A.; Fleming, J.; Saxon, A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J. Immunol. 1997, 158, 2406–2413. [Google Scholar]
- Diaz-Sanchez, D.; Penichet-Garcia, M.; Saxon, A. Diesel exhaust particles directly induce activated mast cells to degranulate and increase histamine levels and symptom severity. J. Allergy Clin. Immunol. 2000, 106, 1140–1146. [Google Scholar] [CrossRef]
- Castañeda, A.R.; Bein, K.; Smiley-Jewell, S.; Pinkerton, K.E. Fine particulate matter (PM2.5) enhances allergic sensitization in BALB/cmice. J. Toxicol. Environ. Health Part A 2017, 80, 197–207. [Google Scholar] [CrossRef]
- Yang, S.-I. Particulate matter and childhood allergic diseases. Korean J. Pediatr. 2018, 62, 22–29. [Google Scholar] [CrossRef]
- Joubert, A.I.; Geppert, M.; Johnson, L.; Mills-Goodlet, R.; Michelini, S.; Korotchenko, E.; Duschl, A.; Weiss, R.; Horejs-Höck, J.; Himly, M. Mechanisms of Particles in Sensitization, Effector Function and Therapy of Allergic Disease. Front. Immunol. 2020, 11, 1334. [Google Scholar] [CrossRef] [PubMed]
- Gruzieva, O.; Bergström, A.; Hulchiy, O.; Kull, I.; Lind, T.; Melén, E.; Moskalenko, V.; Pershagen, G.; Bellander, T. Exposure to Air Pollution from Traffic and Childhood Asthma Until 12 Years of Age. Epidemiology 2013, 24, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, C.; DyBuncio, A.; Becker, A.; Chan-Yeung, M.; Brauer, M. Traffic-related air pollution and incident asthma in a high-risk birth cohort. Occup. Environ. Med. 2010, 68, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, E.; Standl, M.; Cyrys, J.; Berdel, D.; von Berg, A.; Bauer, C.P.; Krämer, U.; Sugiri, D.; Lehmann, I.; Koletzko, S.; et al. A longitudinal analysis of associations between traffic-related air pollution with asthma, allergies and sensitization in the GINIplus and LISAplus birth cohorts. PeerJ 2013, 1, e193. [Google Scholar] [CrossRef] [PubMed]
- Gehring, U.; Wijga, A.H.; Hoek, G.; Bellander, T.; Berdel, D.; Brueske, I.; Fuertes, E.; Gruzieva, O.; Heinrich, J.; Hoffmann, B.; et al. Exposure to air pollution and development of asthma and rhinoconjunctivitis throughout childhood and adolescence: A population-based birth cohort study. Lancet Respir. Med. 2015, 3, 933–942. [Google Scholar] [CrossRef]
- Jung, D.Y.; Leem, J.H.; Kim, H.C.; Kim, J.H.; Hwang, S.S.; Lee, J.Y.; Kim, B.J.; Hong, Y.C.; Hong, S.J.; Kwon, H.J. Effect of traffic-related air pollution on allergic disease: Results of the children’s health and environmental research. Allergy Asthma Immunol. Res. 2015, 7, 359–366. [Google Scholar] [CrossRef]
- Gauderman, W.J.; Avol, E.; Gilliland, F.; Vora, H.; Thomas, D.; Berhane, K.; McConnell, R.; Kuenzli, N.; Lurmann, F.; Rappaport, E.; et al. The effect of air pollution on lung development from 10 to 18 years of age. N. Engl. J. Med. 2004, 351, 1057–1067. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Kim, J.; Han, Y.; Jeon, B.-H.; Cheong, H.-K.; Ahn, K. Short-term effects of weather and air pollution on atopic dermatitis symptoms in children: A panel study in Korea. PLoS ONE 2017, 12, e0175229. [Google Scholar] [CrossRef]
- Jung, C.-R.; Chen, W.-T.; Tang, Y.-H.; Hwang, B.-F. Fine particulate matter exposure during pregnancy and infancy and incident asthma. J. Allergy Clin. Immunol. 2019, 143, 2254–2262.e5. [Google Scholar] [CrossRef]
- Mölter, A.; Agius, R.; De Vocht, F.; Lindley, S.; Gerrard, W.; Custovic, A.; Simpson, A. Effects of long-term exposure to PM10 and NO2 on asthma and wheeze in a prospective birth cohort. J. Epidemiol. Community Health 2014, 68, 21–28. [Google Scholar] [CrossRef]
- Wu, N.; Lu, B.; Chen, J.; Li, X. Size distributions of particle-generated hydroxyl radical (·OH) in surrogate lung fluid (SLF) solution and their potential sources. Environ. Pollut. 2021, 268, 115582. [Google Scholar] [CrossRef]
- Pardo, M.; Xu, F.; Shemesh, M.; Qiu, X.; Barak, Y.; Zhu, T.; Rudich, Y. Nrf2 protects against diverse PM2.5 components-induced mitochondrial oxidative damage in lung cells. Sci. Total. Environ. 2019, 669, 303–313. [Google Scholar] [CrossRef]
- Risom, L.; Møller, P.; Loft, S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. Mol. Mech. Mutagen. 2005, 592, 119–137. [Google Scholar] [CrossRef]
- Vidrio, E.; Jung, H.; Anastasio, C. Generation of hydroxyl radicals from dissolved transition metals in surrogate lung fluid solutions. Atmos. Environ. 2008, 42, 4369–4379. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.K.; Sioutas, C.; Miguel, A.H.; Kumagai, Y.; Schmitz, D.A.; Singh, M.; Eiguren-Fernandez, A.; Froines, J.R. Redox activity of airborne particulate matter at different sites in the Los Angeles Basin. Environ. Res. 2005, 99, 40–47. [Google Scholar] [CrossRef]
- Harmon, A.C.; Hebert, V.Y.; Cormier, S.; Subramanian, B.; Reed, J.R.; Backes, W.L.; Dugas, T.R. Particulate matter containing environmentally persistent free radicals induces AhR-dependent cytokine and reactive oxygen species production in human bronchial epithelial cells. PLoS ONE 2018, 13, e0205412. [Google Scholar] [CrossRef]
- Pardo, M.; Shafer, M.M.; Rudich, A.; Schauer, J.J.; Rudich, Y. Single Exposure to near Roadway Particulate Matter Leads to Confined Inflammatory and Defense Responses: Possible Role of Metals. Environ. Sci. Technol. 2015, 49, 8777–8785. [Google Scholar] [CrossRef] [PubMed]
- Kopf, P.; Walker, M.K. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases reactive oxygen species production in human endothelial cells via induction of cytochrome P4501A1. Toxicol. Appl. Pharmacol. 2010, 245, 91–99. [Google Scholar] [CrossRef]
- Costa, C.; Catania, S.; De Pasquale, R.; Stancanelli, R.; Scribano, G.; Melchini, A. Exposure of human skin to benzo[a]pyrene: Role of CYP1A1 and aryl hydrocarbon receptor in oxidative stress generation. Toxicology 2010, 271, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Loyens, A.; Coddeville, P.; Guidice, J.-M.L.; Marchetti, P.; et al. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 2018, 243, 1434–1449. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Naidoo, R.N.; Robins, T.G.; Mentz, G.; Li, H.; London, S.J.; Batterman, S. GSTM1andGSTP1gene variants and the effect of air pollutants on lung function measures in South African children. Am. J. Ind. Med. 2012, 55, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Gawda, A.; Majka, G.; Nowak, B.; Marcinkiewicz, J. Air pollution, oxidative stress, and exacerbation of autoimmune diseases. Cent. Eur. J. Immunol. 2016, 3, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-N.; Xu, Z.; Wu, G.-C.; Mao, Y.-M.; Liu, L.-N.; Wu, Q.-; Dan, Y.-L.; Tao, S.-S.; Zhang, Q.; Sam, N.B.; et al. Emerging role of air pollution in autoimmune diseases. Autoimmun. Rev. 2019, 18, 607–614. [Google Scholar] [CrossRef]
- Lambert, A.L.; Trasti, F.S.; Mangum, J.B.; Everitt, J.I. Effect of Preexposure to Ultrafine Carbon Black on Respiratory Syncytial Virus Infection in Mice. Toxicol. Sci. 2003, 72, 331–338. [Google Scholar] [CrossRef]
- Brandt, E.B.; Myers, J.M.B.; Acciani, T.H.; Ryan, P.H.; Sivaprasad, U.; Ruff, B.; Lemasters, G.K.; Bernstein, D.I.; Lockey, J.E.; LeCras, T.D.; et al. Exposure to allergen and diesel exhaust particles potentiates secondary allergen-specific memory responses, promoting asthma susceptibility. J. Allergy Clin. Immunol. 2015, 136, 295–303. [Google Scholar] [CrossRef]
- Deo, S.S.; Mistry, K.J.; Kakade, A.M.; Niphadkar, P.V. Role played by Th2 type cytokines in IgE mediated allergy and asthma. Lung India 2010, 27, 66–71. [Google Scholar] [CrossRef]
- Paul, W.E.; Zhu, J. How are TH2-type immune responses initiated and amplified? Nat. Rev. Immunol. 2010, 10, 225–235. [Google Scholar] [CrossRef]
- Fu, H.; Liu, X.; Li, W.; Zu, Y.; Zhou, F.; Shou, Q.; Ding, Z. PM2.5 Exposure Induces Inflammatory Response in Macrophages via the TLR4/COX-2/NF-κB Pathway. Inflammation 2020, 43, 1948–1958. [Google Scholar] [CrossRef]
- Gawda, A.; Majka, G.; Nowak, B.; Śróttek, M.; Walczewska, M.; Marcinkiewicz, J. Air particulate matter SRM 1648a primes macrophages to hyperinflammatory response after LPS stimulation. Inflamm. Res. 2018, 67, 765–776. [Google Scholar] [CrossRef]
- Orazzo, F.; Nespoli, L.; Ito, K.; Tassinari, D.; Giardina, D.; Funis, M.; Cecchi, A.; Trapani, C.; Forgeschi, G.; Vignini, M.; et al. Air pollution, aeroallergens, and emergency room visits for acute respiratory diseases and gastroenteric disorders among young children in six Italian cities. Environ. Health Perspect. 2009, 117, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- Leikauf, G.D.; Kim, S.; Jang, A.S. Mechanisms of ultrafine particle-induced respiratory health effects. Exp. Mol. Med. 2020, 52, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Tang, W.-Y.; Herbstman, J.; Tang, D.; Levin, L.; Miller, R.; Ho, S.-M. Relation of DNA Methylation of 5′-CpG Island of ACSL3 to Transplacental Exposure to Airborne Polycyclic Aromatic Hydrocarbons and Childhood Asthma. PLoS ONE 2009, 4, e4488. [Google Scholar] [CrossRef]
- Ahn, K. The role of air pollutants in atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 993–999. [Google Scholar] [CrossRef]
- Yoon, W.S.; Ryu, S.R.; Lee, S.S.; Chae, Y.S.; Kim, E.J.; Choi, J.H.; Oh, S.; Park, S.H.; Choung, J.T.; Yoo, Y.; et al. Suppression of Inflammation by Recombinant Salmonella typhimurium Harboring CCL22 MicroRNA. DNA Cell Biol. 2012, 31, 290–297. [Google Scholar] [CrossRef]
- Herberth, G.; Bauer, M.; Gasch, M.; Hinz, D.; Röder, S.; Olek, S.; Kohajda, T.; Rolle-Kampczyk, U.; Von Bergen, M.; Sack, U.; et al. Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J. Allergy Clin. Immunol. 2014, 133, 543–550.e4. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Characteristics | Ultrafine Particles (PM0.1) | Fine Particles (PM2.5) | Coarse Mode Particles (PM10) |
|---|---|---|---|---|
| 1. | Diameter | ≤ 0.1 μm | 0.1–2.5 μm | 2.5–10 μm |
| 2. | Sources | Diesel and automobile exhaust, emissions from the combustion of gas stove, vented gas dryer and candle, electric motors, residential burning | Emissions from the combustion of gasoline, oil, diesel fuel, wood burning, and coal burning | Abraded soil, dust from road and construction sites, landfills and agriculture, wildfires and brush/waste burning, industrial sources, fungi and bacteria, endotoxins, and pollen |
| 3. | Atmospheric half-life | Minutes to hours | Days to weeks | Minutes to days |
| 4. | Ability to travel (km) | 1 to 10 | 100 to 1000 | 1 to 100 |
| 5. | Redox activity | High | Medium | Low |
| 6. | Transition metal | Low | High | Medium |
| 7. | Polyaromatic hydrocarbon | High | Low | Low |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagappan, A.; Park, S.B.; Lee, S.-J.; Moon, Y. Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders. Toxics 2021, 9, 18. https://doi.org/10.3390/toxics9020018
Nagappan A, Park SB, Lee S-J, Moon Y. Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders. Toxics. 2021; 9(2):18. https://doi.org/10.3390/toxics9020018
Chicago/Turabian StyleNagappan, Arulkumar, Su Bum Park, Su-Jun Lee, and Yuseok Moon. 2021. "Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders" Toxics 9, no. 2: 18. https://doi.org/10.3390/toxics9020018
APA StyleNagappan, A., Park, S. B., Lee, S.-J., & Moon, Y. (2021). Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders. Toxics, 9(2), 18. https://doi.org/10.3390/toxics9020018