Biological Evaluation of DNA Biomarkers in a Chemically Defined and Site-Specific Manner



Abstract
1. Introduction
2. Discussions on Individual Modifications
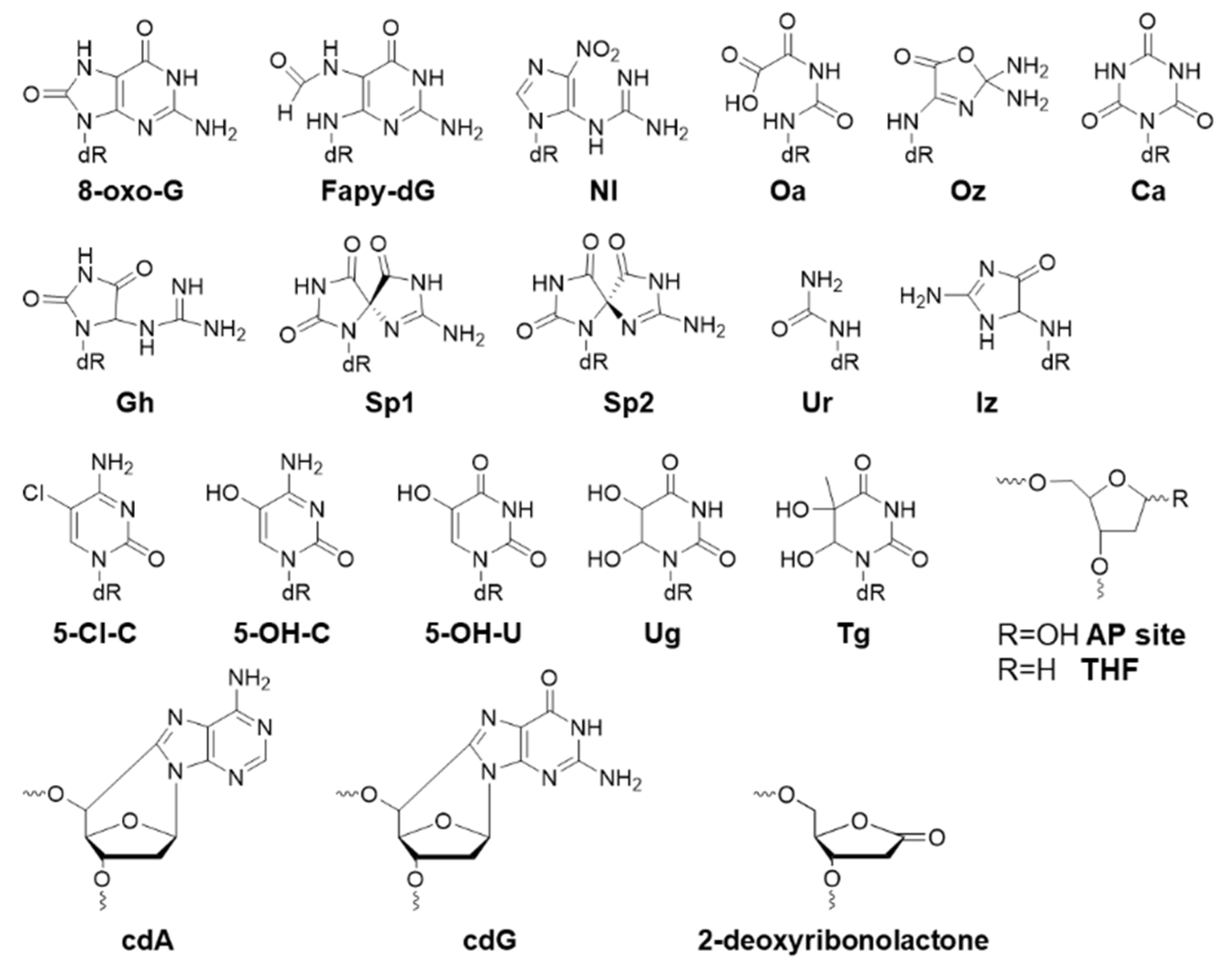
2.1. Oxidative Biomarkers
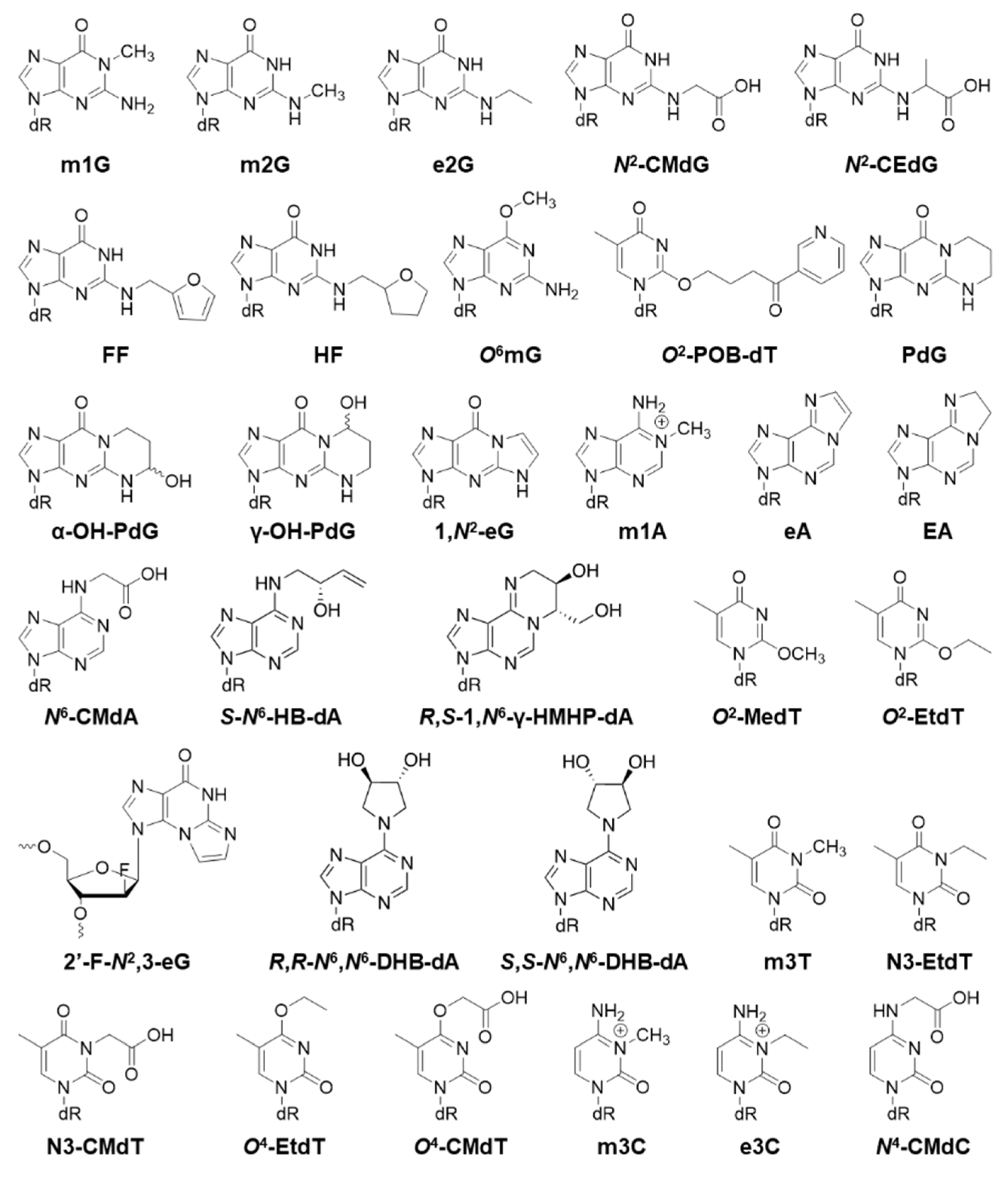
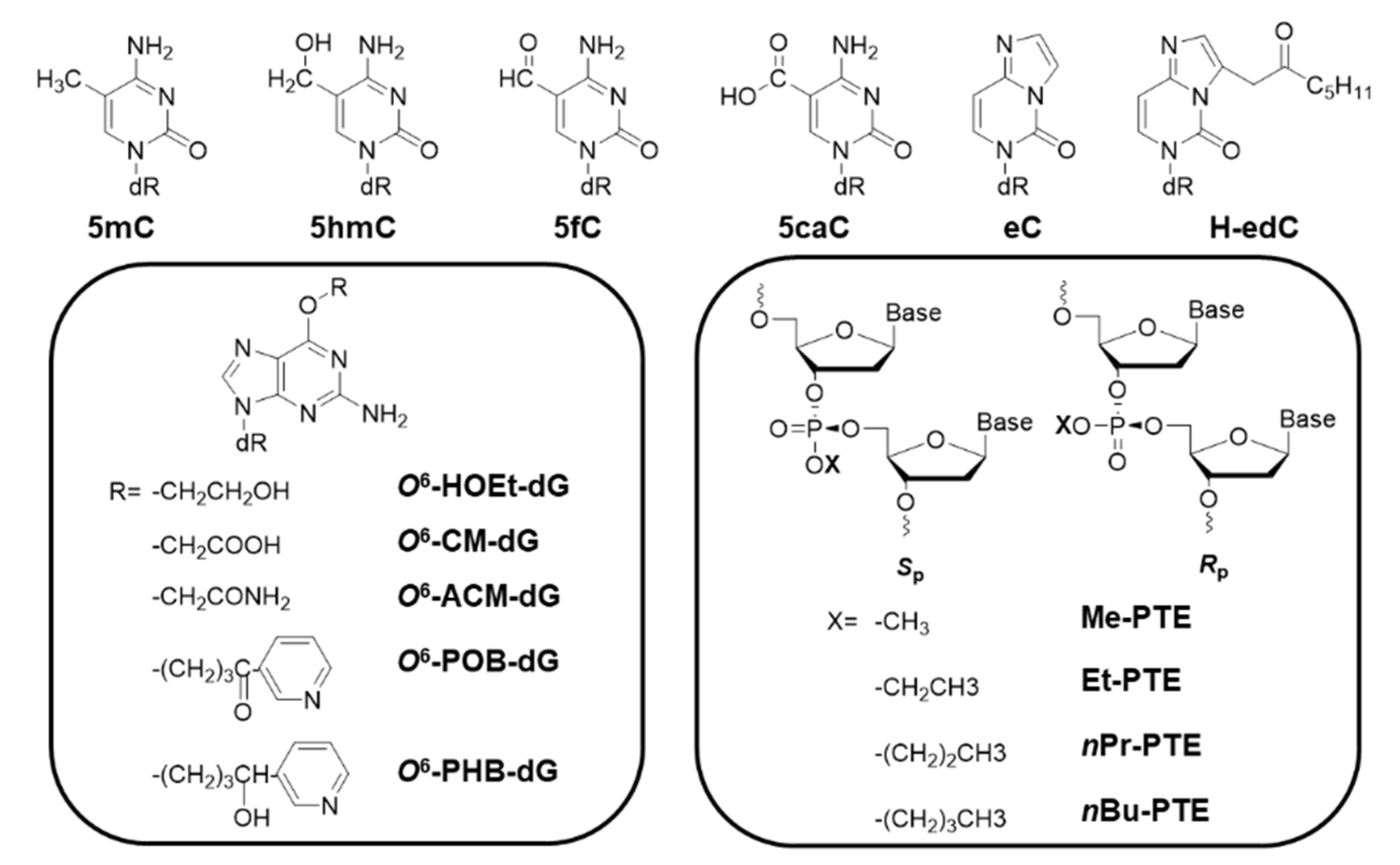
2.2. Alkyl Biomarkers
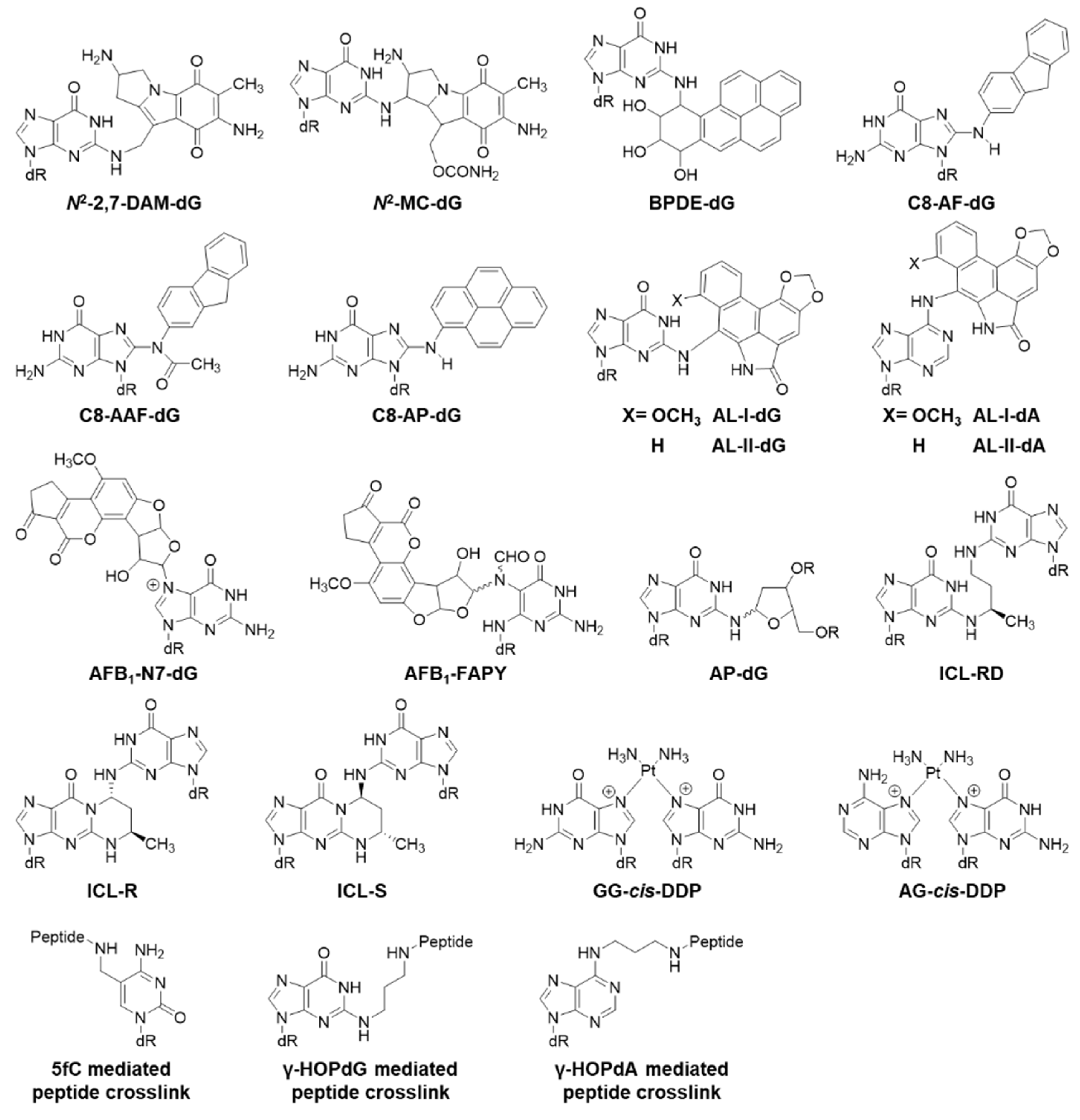
2.3. Bulky Lesions
2.4. Crosslinked Lesions
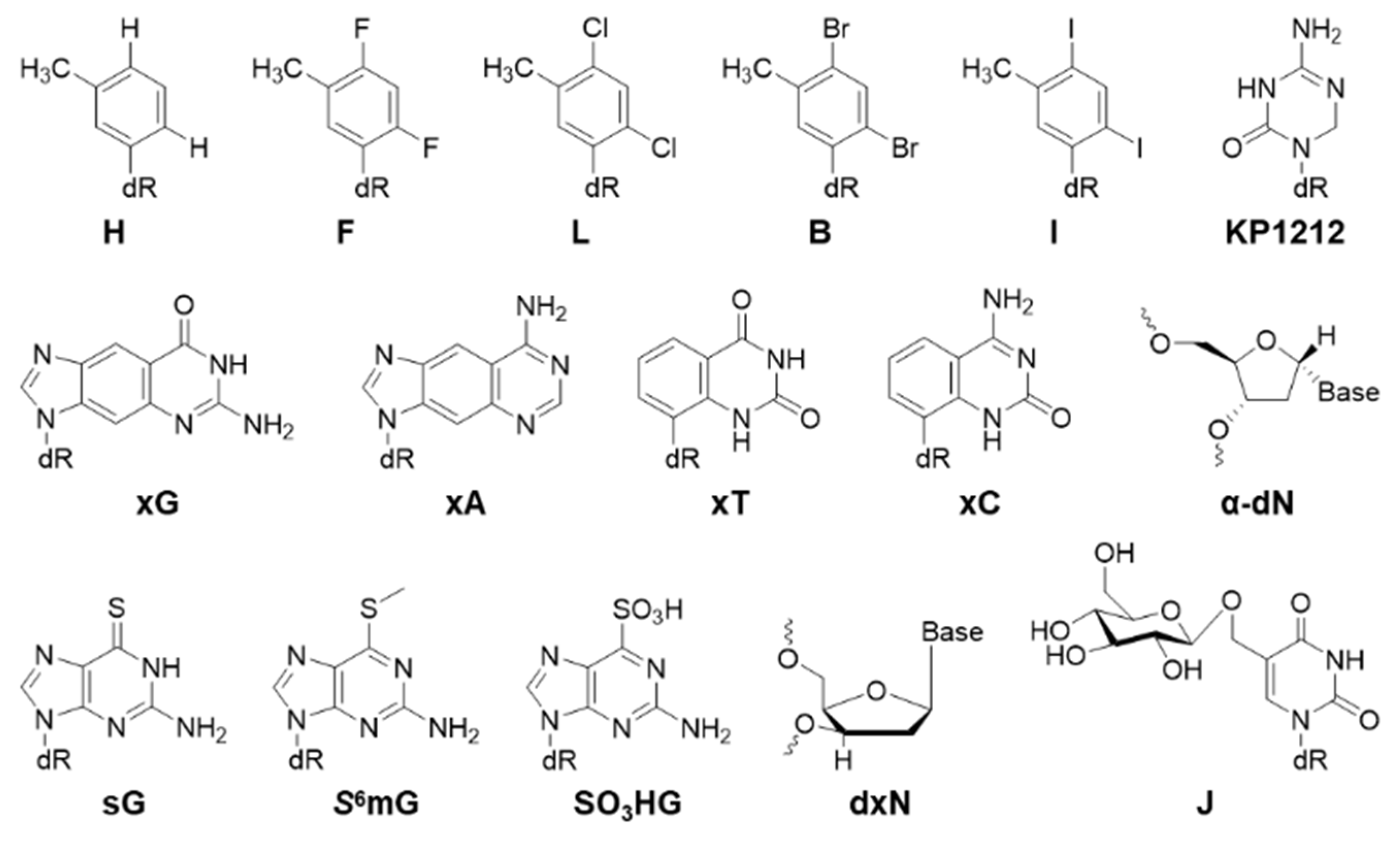
2.5. Other Nucleotide Analogs
3. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hwa Yun, B.; Guo, J.; Bellamri, M.; Turesky, R.J. DNA adducts: Formation, biological effects, and new biospecimens for mass spectrometric measurements in humans. Mass Spectrom. Rev. 2018, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.C.; Essigmann, J.M. Biological properties of single chemical-DNA adducts: A twenty year perspective. Chem. Res. Toxicol. 2008, 21, 232–252. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.C. Some current perspectives on chemical carcinogenesis in humans and experimental animals: Presidential Address. Cancer Res. 1978, 38, 1479–1496. [Google Scholar]
- Yu, Y.; Wang, P.; Cui, Y.; Wang, Y. Chemical Analysis of DNA Damage. Anal. Chem. 2018, 90, 556–576. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Wang, Y. Mass Spectrometry-Based Quantitative Strategies for Assessing the Biological Consequences and Repair of DNA Adducts. Acc. Chem. Res. 2016, 49, 205–213. [Google Scholar] [CrossRef]
- Shrivastav, N.; Li, D.; Essigmann, J.M. Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 2010, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.C.; Essigmann, J.M. Assays for determining lesion bypass efficiency and mutagenicity of site-specific DNA lesions in vivo. Meth. Enzymol. 2006, 408, 1–15. [Google Scholar] [PubMed]
- Chang, S.; Fedeles, B.I.; Wu, J.; Delaney, J.C.; Li, D.; Zhao, L.; Christov, P.P.; Yau, E.; Singh, V.; Jost, M.; et al. Next-generation sequencing reveals the biological significance of the N2,3-ethenoguanine lesion in vivo. Nucleic Acids Res. 2015, 43, 5489–5500. [Google Scholar] [CrossRef]
- Delaney, J.C.; Essigmann, J.M. Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli. Proc. Natl. Acad. Sci. USA 2004, 101, 14051–14056. [Google Scholar] [CrossRef]
- Loechler, E.L.; Green, C.L.; Essigmann, J.M. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. Natl. Acad. Sci. USA 1984, 81, 6271–6275. [Google Scholar] [CrossRef]
- Green, C.L.; Loechler, E.L.; Fowler, K.W.; Essigmann, J.M. Construction and characterization of extrachromosomal probes for mutagenesis by carcinogens: Site-specific incorporation of O6-methylguanine into viral and plasmid genomes. Proc. Natl. Acad. Sci. USA 1984, 81, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.-Y.; Johnson, F.; Grollman, A.P.; Moriya, M. Genotoxic mechanism for the major acrolein-derived deoxyguanosine adduct in human cells. Chem. Res. Toxicol. 2002, 15, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.-Y.; Chan, G.; Miller, H.; Huang, Y.; Torres, M.C.; Johnson, F.; Moriya, M. Mutagenesis by acrolein-derived propanodeoxyguanosine adducts in human cells. Biochemistry 2002, 41, 13826–13832. [Google Scholar] [CrossRef] [PubMed]
- Livneh, Z.; Cohen, I.S.; Paz-Elizur, T.; Davidovsky, D.; Carmi, D.; Swain, U.; Mirlas-Neisberg, N. High-resolution genomic assays provide insight into the division of labor between TLS and HDR in mammalian replication of damaged DNA. DNA Repair (Amst.) 2016, 44, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ziv, O.; Diamant, N.; Shachar, S.; Hendel, A.; Livneh, Z. Quantitative measurement of translesion DNA synthesis in mammalian cells. Methods Mol. Biol. 2012, 920, 529–542. [Google Scholar] [PubMed]
- Huang, H.; Greenberg, M.M. Hydrogen bonding contributes to the selectivity of nucleotide incorporation opposite an oxidized abasic lesion. J. Am. Chem. Soc. 2008, 130, 6080–6081. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Greenberg, M.M. The formamidopyrimidines: Purine lesions formed in competition with 8-oxopurines from oxidative stress. Acc. Chem. Res. 2012, 45, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Millsap, A.D.; DeLeon, A.; Rizzo, C.J.; Basu, A.K. Translesion Synthesis of the N2-2′-Deoxyguanosine Adduct of the Dietary Mutagen IQ in Human Cells: Error-Free Replication by DNA Polymerase κ and Mutagenic Bypass by DNA Polymerases η, ζ, and Rev1. Chem. Res. Toxicol. 2016, 29, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef]
- Minko, I.G.; Kozekov, I.D.; Kozekova, A.; Harris, T.M.; Rizzo, C.J.; Lloyd, R.S. Mutagenic potential of DNA-peptide crosslinks mediated by acrolein-derived DNA adducts. Mutat. Res. 2008, 637, 161–172. [Google Scholar] [CrossRef]
- Fernandes, P.H.; Wang, H.; Rizzo, C.J.; Lloyd, R.S. Site-specific mutagenicity of stereochemically defined 1,N2-deoxyguanosine adducts of trans-4-hydroxynonenal in mammalian cells. Environ. Mol. Mutagen. 2003, 42, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Benasutti, M.; Ezzedine, Z.D.; Loechler, E.L. Construction of an Escherichia coli vector containing the major DNA adduct of activated benzo[a]pyrene at a defined site. Chem. Res. Toxicol. 1988, 1, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Grueneberg, D.A.; Ojwang, J.O.; Benasutti, M.; Hartman, S.; Loechler, E.L. Construction of a human shuttle vector containing a single nitrogen mustard interstrand, DNA-DNA cross-link at a unique plasmid location. Cancer Res. 1991, 51, 2268–2272. [Google Scholar] [PubMed]
- Fuchs, R.P.; Fujii, S. Translesion DNA synthesis and mutagenesis in prokaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012682. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.P.; Fujii, S. Translesion synthesis in Escherichia coli: Lessons from the NarI mutation hot spot. DNA Repair (Amst.) 2007, 6, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Pagès, V. Single-strand gap repair involves both RecF and RecBCD pathways. Curr. Genet. 2016, 62, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Pagès, V.; Fuchs, R.P. Inserting Site-Specific DNA Lesions into Whole Genomes. Methods Mol. Biol. 2018, 1672, 107–118. [Google Scholar] [PubMed]
- Henderson, P.T.; Delaney, J.C.; Gu, F.; Tannenbaum, S.R.; Essigmann, J.M. Oxidation of 7,8-dihydro-8-oxoguanine affords lesions that are potent sources of replication errors in vivo. Biochemistry 2002, 41, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Delaney, S.; Neeley, W.L.; Delaney, J.C.; Essigmann, J.M. The substrate specificity of MutY for hyperoxidized guanine lesions in vivo. Biochemistry 2007, 46, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Pande, P.; Haraguchi, K.; Jiang, Y.-L.; Greenberg, M.M.; Basu, A.K. Unlike catalyzing error-free bypass of 8-oxodGuo, DNA polymerase λ is responsible for a significant part of Fapy·dG-induced G → T mutations in human cells. Biochemistry 2015, 54, 1859–1862. [Google Scholar] [CrossRef]
- Patro, J.N.; Wiederholt, C.J.; Jiang, Y.L.; Delaney, J.C.; Essigmann, J.M.; Greenberg, M.M. Studies on the replication of the ring opened formamidopyrimidine, Fapy·dG in Escherichia coli. Biochemistry 2007, 46, 10202–10212. [Google Scholar] [CrossRef] [PubMed]
- Neeley, W.L.; Delaney, J.C.; Henderson, P.T.; Essigmann, J.M. In vivo bypass efficiencies and mutational signatures of the guanine oxidation products 2-aminoimidazolone and 5-guanidino-4-nitroimidazole. J. Biol. Chem. 2004, 279, 43568–43573. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.C.; Smeester, L.; Wong, C.; Frick, L.E.; Taghizadeh, K.; Wishnok, J.S.; Drennan, C.L.; Samson, L.D.; Essigmann, J.M. AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nat. Struct. Mol. Biol. 2005, 12, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.T.; Delaney, J.C.; Muller, J.G.; Neeley, W.L.; Tannenbaum, S.R.; Burrows, C.J.; Essigmann, J.M. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry 2003, 42, 9257–9262. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.T.; Neeley, W.L.; Delaney, J.C.; Gu, F.; Niles, J.C.; Hah, S.S.; Tannenbaum, S.R.; Essigmann, J.M. Urea lesion formation in DNA as a consequence of 7,8-dihydro-8-oxoguanine oxidation and hydrolysis provides a potent source of point mutations. Chem. Res. Toxicol. 2005, 18, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Wang, J.; Cao, H.; Sun, R.; Wang, Y. High-throughput analysis of the mutagenic and cytotoxic properties of DNA lesions by next-generation sequencing. Nucleic Acids Res. 2011, 39, 5945–5954. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Swanson, A.L.; Dai, X.; Yuan, B.; Wang, J.; Wang, Y. Translesion synthesis of 8,5′-cyclopurine-2′-deoxynucleosides by DNA polymerases η, ι, and ζ. J. Biol. Chem. 2013, 288, 28548–28556. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Jiang, Y.; Wang, Y.; Wang, Y. Efficient formation of the tandem thymine glycol/8-oxo-7,8-dihydroguanine lesion in isolated DNA and the mutagenic and cytotoxic properties of the tandem lesions in Escherichia coli cells. Chem. Res. Toxicol. 2010, 23, 11–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fedeles, B.I.; Freudenthal, B.D.; Yau, E.; Singh, V.; Chang, S.; Li, D.; Delaney, J.C.; Wilson, S.H.; Essigmann, J.M. Intrinsic mutagenic properties of 5-chlorocytosine: A mechanistic connection between chronic inflammation and cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4571–E4580. [Google Scholar] [CrossRef]
- Kreutzer, D.A.; Essigmann, J.M. Oxidized, deaminated cytosines are a source of C --> T transitions in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 3578–3582. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, K.M.; Jiang, Y.L.; Kow, Y.W.; Goodman, M.F.; Greenberg, M.M. Mutagenic effects of 2-deoxyribonolactone in Escherichia coli. An abasic lesion that disobeys the A-rule. Biochemistry 2004, 43, 6723–6733. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, N.; Fedeles, B.I.; Li, D.; Delaney, J.C.; Frick, L.E.; Foti, J.J.; Walker, G.C.; Essigmann, J.M. A chemical genetics analysis of the roles of bypass polymerase DinB and DNA repair protein AlkB in processing N2-alkylguanine lesions in vivo. PLoS ONE 2014, 9, e94716. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; You, C.; Andersen, N.; Jiang, Y.; Moriya, M.; O’Connor, T.R.; Wang, Y. The roles of DNA polymerases κ and ι in the error-free bypass of N2-carboxyalkyl-2′-deoxyguanosine lesions in mammalian cells. J. Biol. Chem. 2011, 286, 17503–17511. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Cao, H.; Jiang, Y.; Hong, H.; Wang, Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 8679–8684. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.C.; Essigmann, J.M. Context-dependent mutagenesis by DNA lesions. Chem. Biol. 1999, 6, 743–753. [Google Scholar] [CrossRef]
- Delaney, J.C.; Essigmann, J.M. Effect of sequence context on O6-methylguanine repair and replication in vivo. Biochemistry 2001, 40, 14968–14975. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Leng, J.; Wang, Y. DNA replication studies of N-nitroso compound-induced O6-alkyl-2′-deoxyguanosine lesions in Escherichia coli. J. Biol. Chem. 2019, 294, 3899–3908. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, P.; Li, L.; Williams, N.L.; Ji, D.; Zahurancik, W.J.; You, C.; Wang, J.; Suo, Z.; Wang, Y. Replication studies of carboxymethylated DNA lesions in human cells. Nucleic Acids Res. 2017, 45, 7276–7284. [Google Scholar] [CrossRef] [PubMed]
- Frick, L.E.; Delaney, J.C.; Wong, C.; Drennan, C.L.; Essigmann, J.M. Alleviation of 1,N6-ethanoadenine genotoxicity by the Escherichia coli adaptive response protein AlkB. Proc. Natl. Acad. Sci. USA 2007, 104, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Pollack, M.; Yang, I.-Y.; Kim, H.-Y.H.; Blair, I.A.; Moriya, M. Translesion DNA Synthesis across the heptanone--etheno-2′-deoxycytidine adduct in cells. Chem. Res. Toxicol. 2006, 19, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-C.; Seneviratne, U.I.; Wu, J.; Tretyakova, N.; Essigmann, J.M. 1,3-Butadiene-Induced Adenine DNA Adducts Are Genotoxic but Only Weakly Mutagenic When Replicated in Escherichia coli of Various Repair and Replication Backgrounds. Chem. Res. Toxicol. 2017, 30, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, P.; Li, L.; You, C.; Wang, Y. Cytotoxic and mutagenic properties of minor-groove O2-alkylthymidine lesions in human cells. J. Biol. Chem. 2018, 293, 8638–8644. [Google Scholar] [CrossRef] [PubMed]
- Weerasooriya, S.; Jasti, V.P.; Bose, A.; Spratt, T.E.; Basu, A.K. Roles of translesion synthesis DNA polymerases in the potent mutagenicity of tobacco-specific nitrosamine-derived O2-alkylthymidines in human cells. DNA Repair (Amst.) 2015, 35, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Jasti, V.P.; Spratt, T.E.; Basu, A.K. Tobacco-specific nitrosamine-derived O2-alkylthymidines are potent mutagenic lesions in SOS-induced Escherichia coli. Chem. Res. Toxicol. 2011, 24, 1833–1835. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Wang, P.; Wang, Y. Cytotoxic and mutagenic properties of regioisomeric O2-, N3- and O4-ethylthymidines in bacterial cells. Carcinogenesis 2014, 35, 2002–2006. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, J.; Li, L.; Wang, P.; You, C.; Williams, N.L.; Wang, Y. Translesion synthesis of O4-alkylthymidine lesions in human cells. Nucleic Acids Res. 2016, 44, 9256–9265. [Google Scholar]
- Li, D.; Fedeles, B.I.; Singh, V.; Peng, C.S.; Silvestre, K.J.; Simi, A.K.; Simpson, J.H.; Tokmakoff, A.; Essigmann, J.M. Tautomerism provides a molecular explanation for the mutagenic properties of the anti-HIV nucleoside 5-aza-5,6-dihydro-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA 2014, 111, E3252–E3259. [Google Scholar] [CrossRef]
- Ji, D.; You, C.; Wang, P.; Wang, Y. Effects of tet-induced oxidation products of 5-methylcytosine on DNA replication in mammalian cells. Chem. Res. Toxicol. 2014, 27, 1304–1309. [Google Scholar] [CrossRef]
- Xing, X.-W.; Liu, Y.-L.; Vargas, M.; Wang, Y.; Feng, Y.-Q.; Zhou, X.; Yuan, B.-F. Mutagenic and cytotoxic properties of oxidation products of 5-methylcytosine revealed by next-generation sequencing. PLoS ONE 2013, 8, e72993. [Google Scholar] [CrossRef]
- Ji, D.; Wang, Y. Facile enzymatic synthesis of base J-containing oligodeoxyribonucleotides and an analysis of the impact of base J on DNA replication in cells. PLoS ONE 2014, 9, e103335. [Google Scholar] [CrossRef]
- Wu, J.; Wang, P.; Wang, Y. Cytotoxic and mutagenic properties of alkyl phosphotriester lesions in Escherichia coli cells. Nucleic Acids Res. 2018, 46, 4013–4021. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Surugihalli, C.; Pande, P.; Champeil, E.; Basu, A.K. Comparative Error-Free and Error-Prone Translesion Synthesis of N2-2′-Deoxyguanosine Adducts Formed by Mitomycin C and Its Metabolite, 2,7-Diaminomitosene, in Human Cells. Chem. Res. Toxicol. 2016, 29, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Attaluri, S.; Bonala, R.R.; Yang, I.-Y.; Lukin, M.A.; Wen, Y.; Grollman, A.P.; Moriya, M.; Iden, C.R.; Johnson, F. DNA adducts of aristolochic acid II: Total synthesis and site-specific mutagenesis studies in mammalian cells. Nucleic Acids Res. 2010, 38, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.A.; Iyer, R.S.; Stone, M.P.; Harris, T.M.; Essigmann, J.M. Mutational properties of the primary aflatoxin B1-DNA adduct. Proc. Natl. Acad. Sci. USA 1996, 93, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Smela, M.E.; Hamm, M.L.; Henderson, P.T.; Harris, C.M.; Harris, T.M.; Essigmann, J.M. The aflatoxin B1 formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2002, 99, 6655–6660. [Google Scholar] [CrossRef] [PubMed]
- Watt, D.L.; Utzat, C.D.; Hilario, P.; Basu, A.K. Mutagenicity of the 1-nitropyrene-DNA adduct N-(deoxyguanosin-8-yl)-1-aminopyrene in mammalian cells. Chem. Res. Toxicol. 2007, 20, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Bonala, R.; Johnson, F.; Grollman, A.P.; Moriya, M. Y-family DNA polymerase-independent gap-filling translesion synthesis across aristolochic acid-derived adenine adducts in mouse cells. DNA Repair (Amst.) 2016, 46, 55–60. [Google Scholar] [CrossRef]
- Hashimoto, K.; Cho, Y.; Yang, I.-Y.; Akagi, J.; Ohashi, E.; Tateishi, S.; de Wind, N.; Hanaoka, F.; Ohmori, H.; Moriya, M. The Vital Role of Polymerase ζ and REV1 in Mutagenic, but Not Correct, DNA Synthesis across Benzo[a]pyrene-dG and Recruitment of Polymerase ζ by REV1 to Replication-stalled Site. J. Biol. Chem. 2012, 287, 9613–9622. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lao, Y.; Yang, I.-Y.; Hecht, S.S.; Moriya, M. Replication-coupled repair of crotonaldehyde/acetaldehyde-induced guanine-guanine interstrand cross-links and their mutagenicity. Biochemistry 2006, 45, 12898–12905. [Google Scholar] [CrossRef]
- Price, N.E.; Li, L.; Gates, K.S.; Wang, Y. Replication and repair of a reduced 2′-deoxyguanosine-abasic site interstrand cross-link in human cells. Nucleic Acids Res. 2017, 45, 6486–6493. [Google Scholar] [CrossRef]
- Naser, L.J.; Pinto, A.L.; Lippard, S.J.; Essigmann, J.M. Chemical and biological studies of the major DNA adduct of cis-diamminedichloroplatinum(II), cis-[Pt(NH3)2(d(GpG)], built into a specific site in a viral genome. Biochemistry 1988, 27, 4357–4367. [Google Scholar] [CrossRef] [PubMed]
- Yarema, K.J.; Lippard, S.J.; Essigmann, J.M. Mutagenic and genotoxic effects of DNA adducts formed by the anticancer drug cis-diamminedichloroplatinum(II). Nucleic Acids Res. 1995, 23, 4066–4072. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ji, S.; Fu, I.; Naldiga, S.; Shao, H.; Basu, A.K.; Broyde, S.; Tretyakova, N.Y. 5-Formylcytosine mediated DNA-protein cross-links block DNA replication and induce mutations in human cells. Nucleic Acids Res. 2018, 46, 6455–6469. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Delaney, J.C.; Essigmann, J.M.; Kool, E.T. Probing the active site tightness of DNA polymerase in subangstrom increments. Proc. Natl. Acad. Sci. USA 2005, 102, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.C.; Gao, J.; Liu, H.; Shrivastav, N.; Essigmann, J.M.; Kool, E.T. Efficient replication bypass of size-expanded DNA base pairs in bacterial cells. Angew. Chem. Int. Ed. Engl. 2009, 48, 4524–4527. [Google Scholar] [CrossRef] [PubMed]
- Amato, N.J.; Zhai, Q.; Navarro, D.C.; Niedernhofer, L.J.; Wang, Y. In vivo detection and replication studies of α-anomeric lesions of 2′-deoxyribonucleosides. Nucleic Acids Res. 2015, 43, 8314–8324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, P.; Amato, N.J.; Wang, Y. Cytotoxic and Mutagenic Properties of C3’-Epimeric Lesions of 2′-Deoxyribonucleosides in Escherichia coli Cells. Biochemistry 2017, 56, 3725–3732. [Google Scholar] [CrossRef]
- Yuan, B.; Wang, Y. Mutagenic and cytotoxic properties of 6-thioguanine, S6-methylthioguanine, and guanine-S6-sulfonic acid. J. Biol. Chem. 2008, 283, 23665–23670. [Google Scholar] [CrossRef]
- Yuan, B.; O’Connor, T.R.; Wang, Y. 6-Thioguanine and S6-methylthioguanine are mutagenic in human cells. ACS Chem. Biol. 2010, 5, 1021–1027. [Google Scholar] [CrossRef]
- Reuven, N.B.; Tomer, G.; Livneh, Z. The mutagenesis proteins UmuD’ and UmuC prevent lethal frameshifts while increasing base substitution mutations. Mol. Cell 1998, 2, 191–199. [Google Scholar] [CrossRef]
- Stein, S.; Lao, Y.; Yang, I.-Y.; Hecht, S.S.; Moriya, M. Genotoxicity of acetaldehyde- and crotonaldehyde-induced 1,N2-propanodeoxyguanosine DNA adducts in human cells. Mutat. Res. 2006, 608, 1–7. [Google Scholar] [CrossRef]
- Zhai, Q.; Wang, P.; Cai, Q.; Wang, Y. Syntheses and characterizations of the in vivo replicative bypass and mutagenic properties of the minor-groove O2-alkylthymidine lesions. Nucleic Acids Res. 2014, 42, 10529–10537. [Google Scholar] [CrossRef]
- Fedeles, B.I.; Essigmann, J.M. Impact of DNA lesion repair, replication and formation on the mutational spectra of environmental carcinogens: Aflatoxin B1 as a case study. DNA Repair (Amst.) 2018, 71, 12–22. [Google Scholar] [CrossRef]
- Chawanthayatham, S.; Valentine, C.C.; Fedeles, B.I.; Fox, E.J.; Loeb, L.A.; Levine, S.S.; Slocum, S.L.; Wogan, G.N.; Croy, R.G.; Essigmann, J.M. Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, E3101–E3109. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef]
- Stratton, M.R. Exploring the genomes of cancer cells: Progress and promise. Science 2011, 331, 1553–1558. [Google Scholar] [CrossRef]
- Kim, M.Y.; Zhou, X.; Delaney, J.C.; Taghizadeh, K.; Dedon, P.C.; Essigmann, J.M.; Wogan, G.N. AlkB influences the chloroacetaldehyde-induced mutation spectra and toxicity in the pSP189 supF shuttle vector. Chem. Res. Toxicol. 2007, 20, 1075–1083. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidative Lesion | Bypass Efficiency | Mutation | Cell |
| 8-oxo-G | 88% [28,29] | G>T 3% (44%, MutY-) [28,29] | E. coli |
| G>T 8% [30] | Human | ||
| Fapy-dG | 31–43% (TXN sequences) [31] | G>T 1.2–1.9% (0.7–2.1%, MutM-/MutY-) [31] | E. coli |
| G>T 10% [30] | Human | ||
| NI | 7% (57%, SOS) [32] | G>C 8.9%, G>A 19%, G>T 22% (G>C 2.5%, G>A 13%, G>T, 18%, SOS) [32] | E. coli |
| Oa | 52% [28], 51% [33], 108% (118% MutY-) [29] | G>T 97% [28], 99% [33], 97% (no change, MutY-) [29] | E. coli |
| Oz | 57% [28] | G>T 86% [28] | E. coli |
| Ca | 65% [28] | G>T 95% [28] | E. coli |
| Gh | 75% [34], 20% (30% MutY-) [29] | G>C 98%, G>T 2% [34], G>T 40%, G>C 57%, G>A 3% (no change, MutY-) [29] | E. coli |
| Sp1 | 9% [34], 19% (38%, MutY-) [29] | G>C 72%, G>T 27% [34], G>T 78%, G>C 19%, G>A 1% (no change, MutY-) [29] | E. coli |
| Sp2 | 9% [34], 17% (30%, MutY-) [29] | G>C 57%, G>T 41% [34], G>T 49%, G>C 48%, G>A 3% (no change, MutY-) [29] | E. coli |
| Ur | 11% [35], 10% (10% MutY-) [29] | G>T 99% [35], G>T 54%, G>C 35%, G>A 9% (no change, MutY-) [29] | E. coli |
| Iz | 60% (71%, SOS) [32] | G>C 88%, G>A 2%, G>T 1.1% (G>C 75%, G>A 3.4%, G>T 5.5%, SOS) [32] | E. coli |
| Cyclo-dG | 11% (6% pol V-) [36] | G>A 20% [36] | E.coli |
| S-cdG | 4% [37] | G>T 35%, G>A 20% [37] | Human |
| Cyclo-dA | 31% (13% pol V-) [36] | A>T 11% [36] | E. coli |
| S-cdA | 6% [37] | A>T 12% [37] | Human |
| Tg | 96% [38] | E. coli | |
| 5ClC | 75% (75% AlkB-) [39] | C>T 5% (same in AlkB-) [39] | E. coli |
| 5-OH-C | C>T 0.05%, C>G 0.001% [40] | E. coli | |
| 5-OH-U | C>T 83% [40] | E. coli | |
| Ug | C>T 80% [40] | E. coli | |
| THF AP site | 6% [28], 5.8% [32], 4% (4% MutY-) [29] | AP>T 50%, AP>C 26%, AP>A 7%, -1 del 13% (no change, MutY-) [29] | E. coli |
| 2-deoxyribonolactone | 5%, (3% pol II-), (1% pol V-) [41] | T 35%, C 42%, A 12%, G 8%, 5′ T (T 42%, C 38%, A 6%, G 14%, 5′ C) [41] | E. coli |
| Alkyl Modification | Bypass Efficiency | Mutation | Cell |
| m1G | 15% (3%, AlkB-) [9], 20% (2%, AlkB-) [33] | G>T 3% (G>T 57%, G>A 17%, G>C 6%, AlkB-) [9], G>T 4%, G>A 2% (G>T 52%, G>A 20%, G>C 4%, AlkB-) [33] | E. coli |
| m2G | 90% (84% AlkB-; 98% DinB-; 96% AlkB- and DinB-) [42] | G>A 3% (2.7%, AlkB-; 3%, DinB-; 3%, AlkB- and DinB-) [42] | E. coli |
| e2G | 100% (98% AlkB-), (106% DinB-), (99% AlkB- and DinB-) [42] | G>A 2%, G>C 1% (G>A 2.3%, AlkB-), (G>A 2%, AlkB- and DinB-) [42] | E. coli |
| N2-CMdG | 100% [43] | Not mutagenic [43] | Mouse |
| R-N2-CEdG | 39% (13% pol V-) [44] | Not mutagenic [44] | E. coli |
| 100% [43] | Not mutagenic (G>A 23%, G>T 15%, pol κ-) [43] | Mouse | |
| S-N2-CEdG | 75% (28% pol V-) [44] | Not mutagenic [44] | E. coli |
| 99% [43] | Not mutagenic (G>A 23%, G>T 15%, pol κ-) [43] | Mouse | |
| FF | 101% (100% AlkB-), (28% DinB-), (36% AlkB- and DinB-) [42] | G>C 1%, (G>A 1%, G>T 1%, AlkB-DinB-) [42] | E. coli |
| HF | 92% (88% AlkB-), (28% DinB-), (40% AlkB- and DinB-) [42] | G>C 2% [42] | E. coli |
| O6mG | G>A 99% [45,46] | E. coli | |
| O6-POB-dG | 70% [47] | G>A 90%, G>T 2.5% [47] | E. coli |
| O6-PHB-dG | 40% [47] | G>A 95% [47] | E. coli |
| O6-CM-dG | 10% [47] | G>A 10% [47] | E. coli |
| 40% [48] | G>A 6% [48] | Human | |
| O6-ACM-dG | 2% [47] | G>A 30% [47] | E. coli |
| O6-HOEt-dG | 15% [47] | G>A 40% [47] | E. coli |
| PdG | 25% [12] | G>T 6% [12] | Human |
| α-OH-PdG | 17% [12] | G>T 11% [12] | Human |
| γ-OH-PdG | 73% [12] | Not mutagenic [12] | Human |
| 1,N2-eG | 4% (2% AlkB-) (1.8% AlkB-DinB-) [8] | G>A 6%, G>T 6%, G>C 2%, −1/2 del 5% (G>A 13%, G>T 13%, G>C 1%, −1/2 del 9%, AlkB-), (same in AlkB-DinB-) [8] | E. coli |
| 2′-F-N2,3-eG | 21% (26% AlkB-) (14% AlkB-DinB-) [8] | G>A 30% (30% AlkB-), (30% AlkB-DinB-) [8] | E. coli |
| m1A | 100% (12%, AlkB-) [9] | A>T 0.06% (0.61%, AlkB-) [9] | E. coli |
| eA | 85% (5% AlkB-) [33], 130% (9% AlkB-) [49] | <0.5% (A>T 25%, A>G 5%, A>C 5%, AlkB-) [33], A>C 1%, A>T 1% (A>T 22%, A>C 8%, A>G 7%, AlkB-) [49] | E. coli |
| 17% [50] | Human | ||
| EA | 135% (14% AlkB-) [49] | A>C 1%, A>G 0.5%, A>T 0.5% (A>C 2%, A>G 1%, A>T 1%, AlkB-) [49] | E. coli |
| N6-CMdA | 98% [36] | Not mutagenic [36] | E. coli |
| 65% (35% pol k-) [48] | Not mutagenic [48] | Human | |
| S-N6-HB-dA | 120% [51] | Not mutagenic [51] | E. coli |
| R,R-N6,N6-DHB-dA | 100% [51] | <1% [51] | E. coli |
| S,S-N6,N6-DHB-dA | 60% [51] | A>G 1% [51] | E. coli |
| R,S-1,N6-γ-HMHP-dA | 10% [51] | A>T 2% [51] | E. coli |
| O2-MedT | 60% [52], 55% [53] | T>A 1%, T>G 1% [52], T>A 56% [53] | Human |
| 5% [54] | T>A 10%, T>G 10% [54] | E. coli | |
| O2-EtdT | 21% (5% pol V-) [55] | T>C 35%, T>A 15%, T>G 5% (T>C 10%, pol V-) [55] | E. coli |
| 45% [52] | T>A 5%, T>G 3% [52] | Human | |
| O2-nPrdT | 35% [52] | T>A 12%, T>G 5% [52] | Human |
| O2-iPrdT | 35% [52] | T>A 4%, T>G 1% [52] | Human |
| O2-nBudT | 30% [52] | T>A 13%, T>G 6% [52] | Human |
| O2-iBudT | 15% [52] | T>A 4%, T>G 2% [52] | Human |
| O2-sBudT | 15% [52] | T>A 4%, T>G 2% [52] | Human |
| O2-POB-dT | 3% [54] | 12% T>A, 38% T>G [54] | E. coli |
| 26% [53] | T>A 47% [53] | Human | |
| m3T | 6%, (4% AlkB-) [9] | T>A 32%, T>C 6%, T>G 2% (T>A 47%, T>C 9%, T>G 2%, AlkB-) [9] | E. coli |
| N3-EtdT | 17% (3% pol V-) [55] | T>C 15%, T>A 21%, T>G 3% (Not mutagenic, pol V-) [55] | E. coli |
| N3-CMdT | 55% [36] | T>A 66% [36] | E. coli |
| 40% [48] | T>A 81% [48] | Human | |
| O4-CMdT | 49% [36] | T>C 86% [36] | E. coli |
| 40% [48] | T>C 68% (25% pol ζ-) [48] | Human | |
| O4-MedT | 32% [56] | T>C 58% [56] | Human |
| O4-EtdT | 76% [55] | T>C 84% (Not mutagenic, pol V-) [55] | E. coli |
| 33% [56] | T>C 82% [56] | Human | |
| O4-nPrdT | 35% [56] | T>C 42% [56] | Human |
| O4-iPrdT | 30% [56] | T>C 44% [56] | Human |
| O4-nBudT | 32% [56] | T>C 29% [56] | Human |
| O4-iBudT | 24% [56] | T>C 42% [56] | Human |
| O4-R-sBudT | 20% [56] | T>C 25% [56] | Human |
| O4-S-sBudT | 22% [56] | T>C 25% [56] | Human |
| m3C | 100% (10% AlkB-) [9], 113% (14% AlkB-) [57], 98% (5% AlkB-; 115% DinB-; 7.5% AlkB-DinB-) [42], 100% (15% AlkB-) [39] | C>T 1% (C>T 14%, C>A 14%, C>G 2%, AlkB-) [9], Not mutagenic (C>T 55%, C>A 30%, C>G 1%, AlkB-) [57], Not mutagenic (C>T 41%, C>A 41%, C>G 4%, AlkB-) [42], Not mutagenic (C>T 52%, C>A 30%, AlkB-) [39] | E. coli |
| e3C | 96%, (9% AlkB-) [9] | Not mutagenic (C>T 17%, C>A 11%, C>G 2%, AlkB-) [9] | E. coli |
| N4-CMdC | 83% [36] | Not mutagenic [36] | E. coli |
| 80% [48] | Not mutagenic [48] | Human | |
| 5mC | 100% (100% AlkB-) [39] | Not mutagenic (same in AlkB-) [39] | E. coli |
| 100% [58] | Not mutagenic [58] | Human | |
| 5hmC | 100% [59] | Not mutagenic [59] | E. coli |
| 98% [58] | Not mutagenic [58] | Human | |
| 5fC | 100% [59] | Not mutagenic [59] | E. coli |
| 74% [58] | Not mutagenic [58] | Human | |
| 5caC | 100% [59] | Not mutagenic [59] | E. coli |
| 72% [58] | Not mutagenic [58] | Human | |
| eC | 24% (13% AlkB-) [33] | C>A 24%, C>T 11% (C>A 49%, C>T 31%, AlkB-) [33] | E. coli |
| H-edC | 1% [50] | C>G 40% [50] | E. coli |
| 10% [50] | C>A 60%, C>T 32% [50] | Human | |
| 5hmU | 80% [60] | Not mutagenic [60] | Human |
| Sp-Me-PTE | 110% (Ada-, decreases from 140% to 70%) [61] | TT>GT 50%, TT>GC 15% [61] | E. coli |
| Rp-Me-PTE | 30% [61] | Not mutagenic [61] | E. coli |
| Sp-Et-PTE | 190% [61] | Not mutagenic [61] | E. coli |
| Rp-Et-PTE | 40% [61] | Not mutagenic [61] | E. coli |
| Sp-nPr-PTE | 160% [61] | Not mutagenic [61] | E. coli |
| Rp-nPr-PTE | 70% [61] | Not mutagenic [61] | E. coli |
| Sp-nBu-PTE | 100% [61] | Not mutagenic [61] | E. coli |
| Bulky Lesion | Bypass Efficiency | Mutation | Cell |
| N2-MC-dG | 38% [62] | G>T 18% [62] | Human |
| N2-2,7-DAM-dG | 27% [62] | G>T 10% [62] | Human |
| AL-II-dG | 9% [63] | G>T 9% [63] | Mouse |
| AFB1-N7-dG | G>T 1.5% [64] | E. coli | |
| AFB1-FAPY | G>T 14% [65] | E. coli | |
| C8-AP-dG | 51% [66] | Not mutagenic [66] | Human |
| C8-AAF-dG | 13% [66] | Not mutagenic [66] | Human |
| C8-AF-dG | 97% [66] | Not mutagenic [66] | Human |
| AL-I-dA | 100% (5% Rev3L-) [67] | A>T 50% (Not mutagenic, Rev3L-) [67] | Mouse |
| AL-II-dA | 5% [63] | A>T 22% [63] | Mouse |
| BPDE-dG | (40% Rev1-); (13% Rev3L-) [68] | G>T 73%, G>A 12%; (G>T 32%, G>A 18%, Rev1-); (G>T 6%, Rev3L-) [68] | Mouse |
| Crosslinked Lesion | Bypass Efficiency | Mutation | Cell |
| ICL-RD | 43% [69] | 5′-G>T 3% [69] | E. coli |
| ICL-R | 38% [69] | 5′-G>T 3% [69] | E. coli |
| ICL-S | 53% [69] | 5′-G>T 3% [69] | E. coli |
| AP-dG (dG strand) | 38% (43% Pol η-), (13% Pol ι-), (2% Pol κ-), (5% Pol ζ-) [70] | G>A 2-5%, G>T 1–2%, G>C 1% [70] | Human |
| AP-dG (AP strand) | 18% (25% Pol η-), (4% Pol ι-), (1% Pol κ-), (5% Pol ζ-) [70] | AP>T 74%, AP>C 10-20%, AP>G 4–6%, AP>A 1–2% [70] | Human |
| 1,2-GG-cis-DDP | 11% [71]; 5% (30% SOS) [72] | <0.25% (G>T 1.3%, SOS) [72] | E. coli |
| 1,2-AG-cis-DDP | 22% (32% SOS) [72] | <0.2% (A>T 4.4%, SOS) [72] | E. coli |
| 1,3-GTG-cis-DDP | 13% (14% SOS) [72] | <0.7% [72] | E. coli |
| γ-HOPdG mediated peptide crosslink | G>T 5%, G>C 3% [20] | Human | |
| γ-HOPdA mediated peptide crosslink | Not mutagenic [20] | Human | |
| 5fC mediated peptide crosslink | C>T 7%, C>G 1%, C del 2% [73] | Human | |
| Other Nucleotide Analog | Bypass Efficiency | Mutation | Cell |
| H | 5% [74] | T>A 41%, T>C 5%, T>G 4%, −1 del 13% [74] | E. coli |
| F | 13% [74] | T>A 9%, T>C 1%, T>G 1% [74] | E. coli |
| L | 20% [74] | T>A 5% [74] | E. coli |
| B | 12% [74] | T>A 24% [74] | E. coli |
| I | 10% [74] | T>A 46%, T>C 1%, T>G 1%, −1 del 6% [74] | E. coli |
| KP1212 | 128% [57] | C>T 10% [57] | E. coli |
| xG | 11% (45% SOS) [75] | G>A 95% [75] | E. coli |
| xA | 80% (108% SOS) [75] | <1% [75] | E. coli |
| xT | 73% (102% SOS) [75] | T>A 73% [75] | E. coli |
| xC | 29% (53% SOS) [75] | C>A 10% [75] | E. coli |
| α-dG | 3% [76] | G>A 60%, G>C 6% [76] | E. coli |
| α-dA | 20% [76] | Not mutagenic [76] | E. coli |
| α-dT | 1% [76] | Not mutagenic [76] | E. coli |
| α-dC | 1% [76] | C>A 72% [76] | E. coli |
| dxG | 25% [77] | Not mutagenic [77] | E. coli |
| dxA | 75% [77] | A>G 10% [77] | E. coli |
| dxT | 150% [77] | Not mutagenic [77] | E. coli |
| dxC | 125% (CXT), 175%(GXG) [77] | Not mutagenic [77] | E. coli |
| sG | 98% [78] | G>A 11% [78] | E. coli |
| 98% [79] | G>A 8% [79] | Human | |
| S6mG | 91% [78] | G>A 94% [78] | E. coli |
| 95% [79] | G>A 40% [79] | Human | |
| SO3HG | 87% [78] | G>A 77% [78] | E. coli |
| 2′-F-G | 99% [8] | Not mutagenic [8] | E. coli |
| J | 52% [60] | Not mutagenic [60] | Human |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, K.; Delaney, J.C.; Zhou, X.; Li, D. Biological Evaluation of DNA Biomarkers in a Chemically Defined and Site-Specific Manner. Toxics 2019, 7, 36. https://doi.org/10.3390/toxics7020036
Bian K, Delaney JC, Zhou X, Li D. Biological Evaluation of DNA Biomarkers in a Chemically Defined and Site-Specific Manner. Toxics. 2019; 7(2):36. https://doi.org/10.3390/toxics7020036
Chicago/Turabian StyleBian, Ke, James C. Delaney, Xianhao Zhou, and Deyu Li. 2019. "Biological Evaluation of DNA Biomarkers in a Chemically Defined and Site-Specific Manner" Toxics 7, no. 2: 36. https://doi.org/10.3390/toxics7020036
APA StyleBian, K., Delaney, J. C., Zhou, X., & Li, D. (2019). Biological Evaluation of DNA Biomarkers in a Chemically Defined and Site-Specific Manner. Toxics, 7(2), 36. https://doi.org/10.3390/toxics7020036