
Antioxidants Protect against Arsenic Induced Mitochondrial Cardio-Toxicity



Abstract
:1. Introduction
1.1. Arsenic and Cardiovascular Disease
1.2. Mitochondria in Cardiovascular Disease
1.3. Oxidative Stress
1.4. Antioxidant Phytonutrients
1.5. Mitochondria
1.5.1. Energy Production
1.5.2. Calcium Storage
1.5.3. Apoptosis
1.5.4. Membrane Potential
1.5.5. Source of ROS
1.5.6. Arsenic and Cysteine Thiol Binding
1.5.7. ROS and the Nrf2 Pathway
1.5.8. Arsenic-Induced Mitochondrial Toxicity
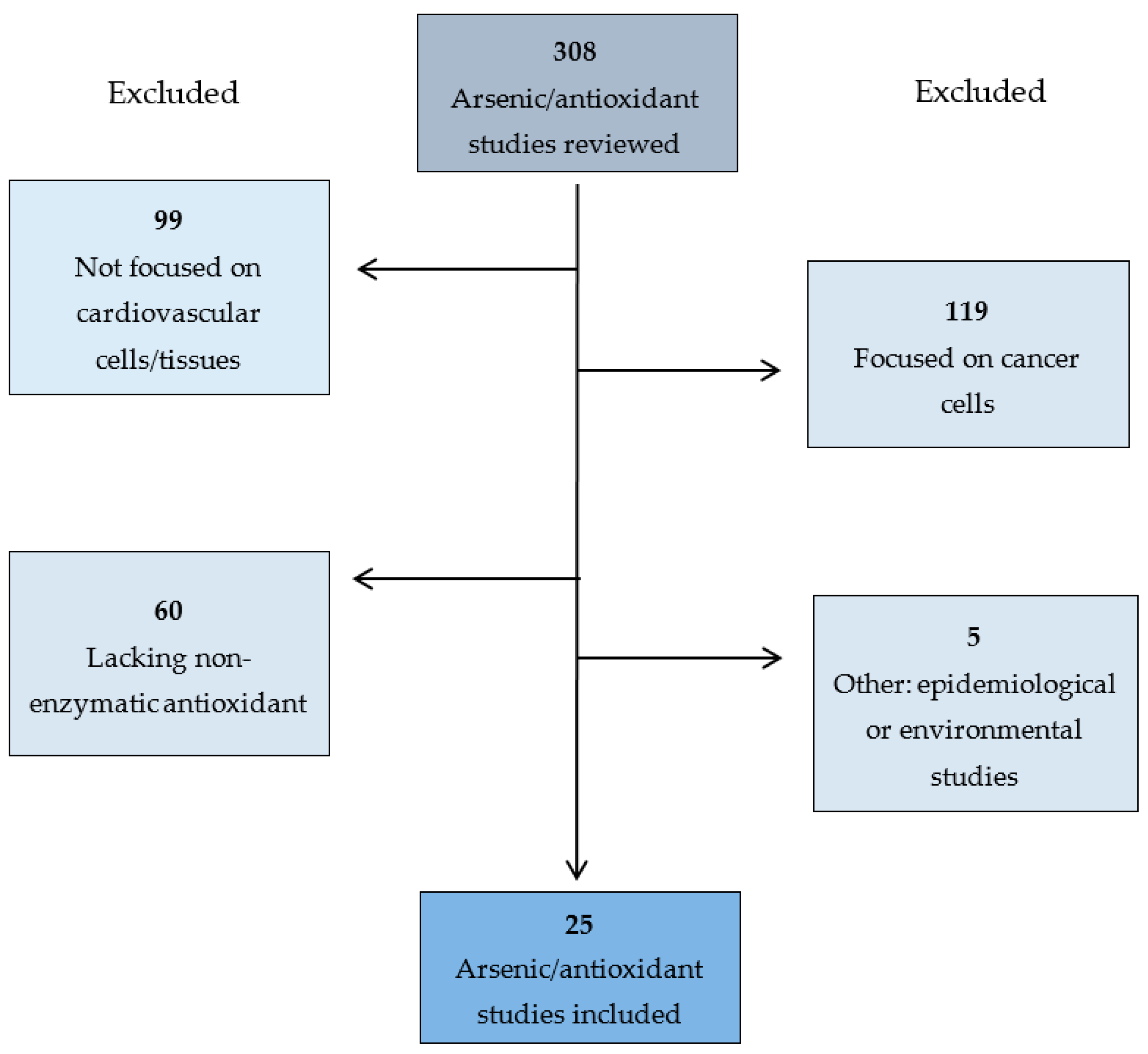
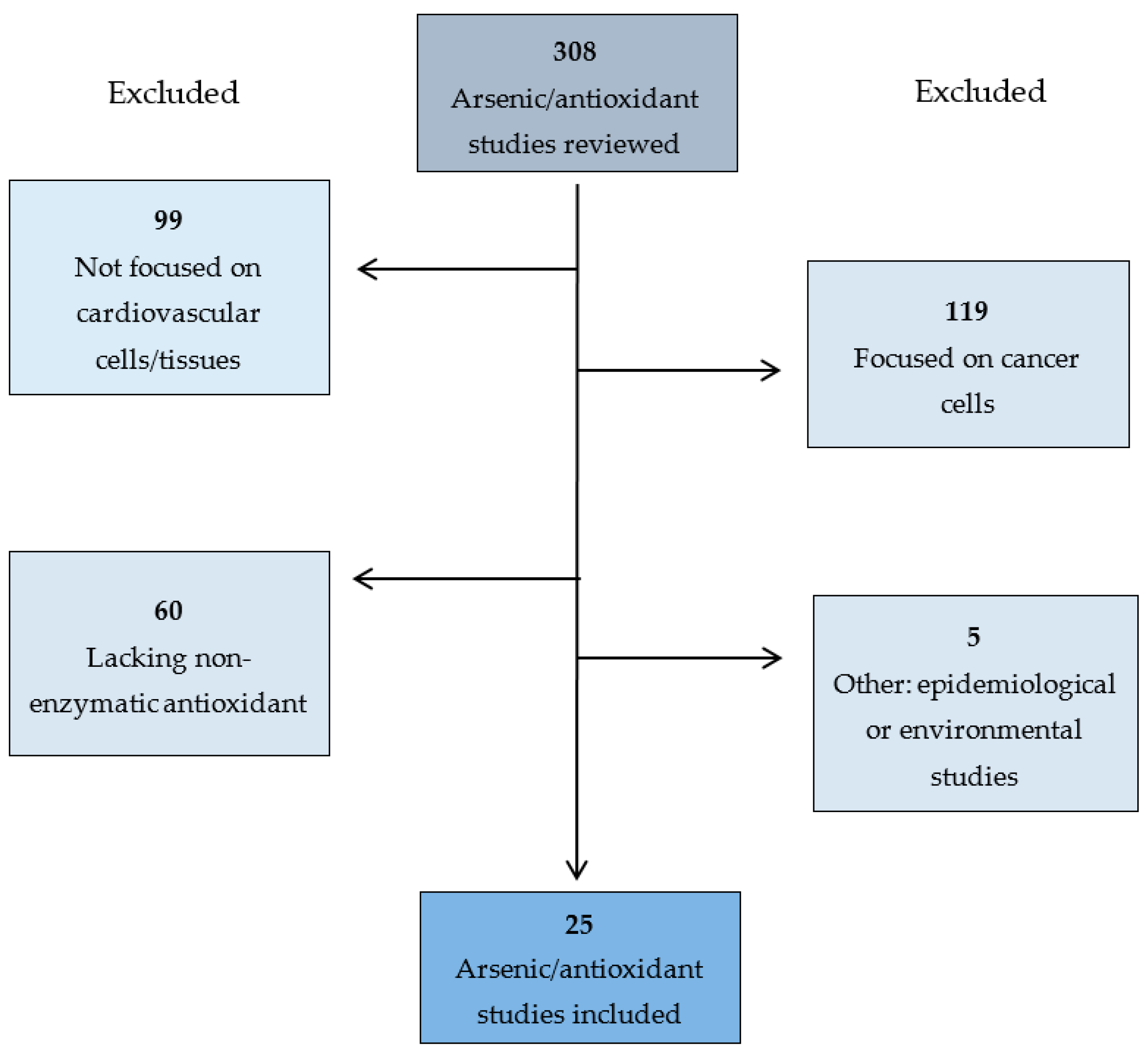
2. Methods
3. Results
3.1. Arsenic
3.1.1. Dose and Duration
3.1.2. Cardiovascular Structure and Function
3.1.3. Cardiac Nrf2
3.1.4. ROS
3.1.5. Apoptosis
3.1.6. Calcium Overload
3.1.7. Mitochondrial Function
3.1.8. Antioxidants
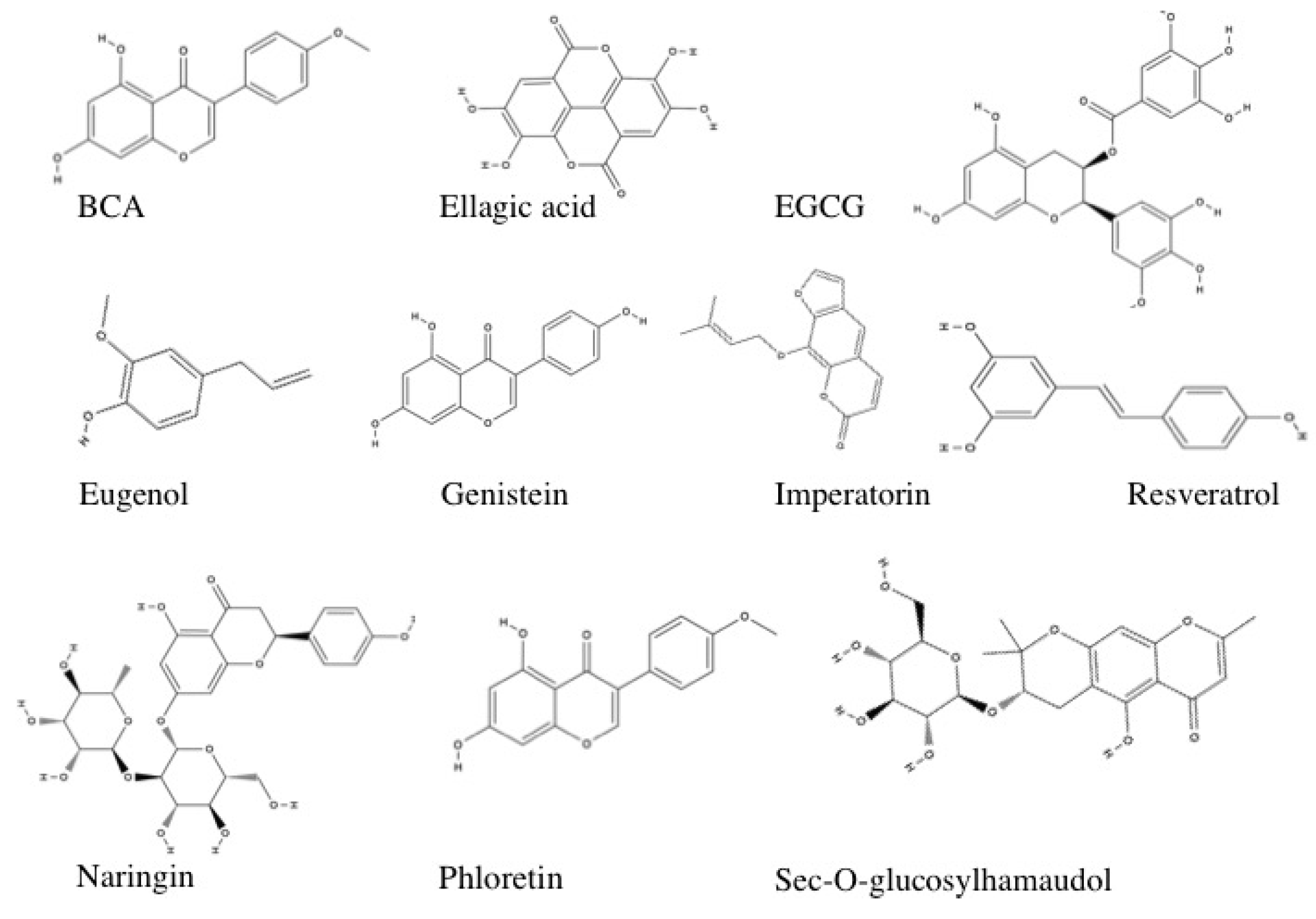
3.1.9. Polyphenols
Biochanin A
Boerhavia Diffusa
Ellagic Acid
EGCG
Eugenol
Genistein
Grape Seed and Skin Extract
Imperatorin and sec-O-glucosylhamaudol
Malus domestica Apple Peel Extract
Naringin
Phloretin
Resveratrol
Resveratrol and Genistein
Silybum Marianum
Sorbus Phnuashanesis (Hante) Hedl
Trichosanthes Dioca
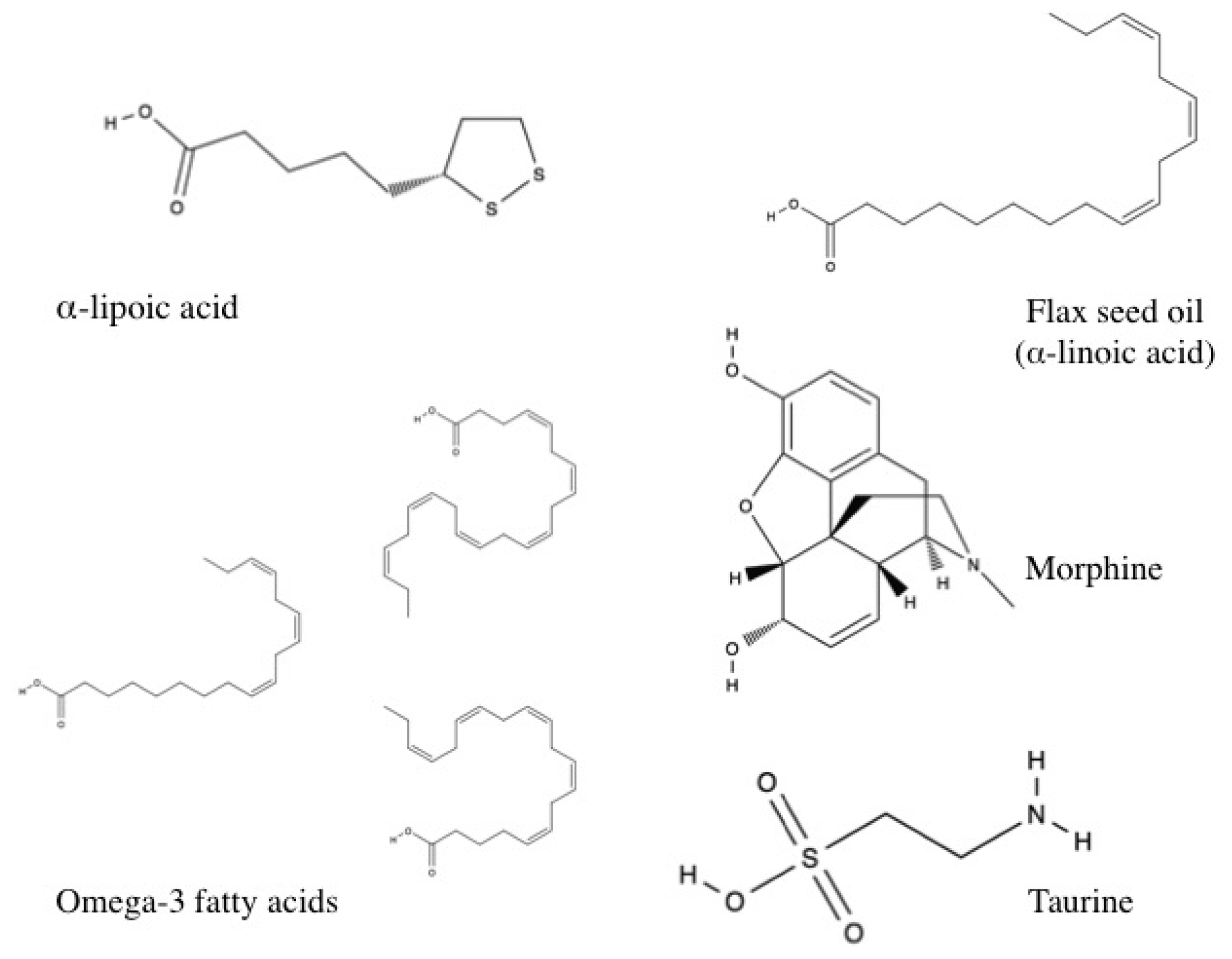
3.1.10. Other Antioxidants
α-lipoic Acid
Flax Seed Oil
Morphine
Omega-3 Fatty Acid
Selenium
Taurine
4. Discussion
4.1. Identifying Inconsistencies
4.2. Study Comparisons
4.3. Limitations
4.4. Future Directions
5. Conclusions
Supplementaly Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mandal, B.K.; Suzuki, K.T. Arsenic round the world: A review. Talanta 2002, 58, 201–235. [Google Scholar] [CrossRef]
- Chen, C.J.; Hsueh, Y.M.; Lai, M.S.; Shyu, M.P.; Chen, S.Y.; Wu, M.M.; Kuo, T.L.; Tai, T.Y. Increased prevalence of hypertension and long-term arsenic exposure. Hypertension 1995, 25, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chiou, H.Y.; Chiang, M.H.; Lin, L.J.; Tai, T.Y. Dose-response relationship between ischemic heart disease mortality and long-term arsenic exposure. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Abhyankar, L.N.; Jones, M.R.; Guallar, E.; Navas-Acien, A. Arsenic exposure and hypertension: A systematic review. Environ. Health Perspect. 2012, 120, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Babazono, A.; Yamamoto, E.; Kurumatani, N.; Mino, Y.; Ogawa, T.; Kishi, Y.; Aoyama, H. Ingested arsenic and internal cancer: A historical cohort study followed for 33 years. Am. J. Epidemiol. 1995, 141, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhou, X.; Liu, W.; Sun, X.; Chen, C.; Hudson, L.G.; Liu, K.J. Arsenite-induced ros/rns generation causes zinc loss and inhibits the activity of poly(adp-ribose) polymerase-1. Free Radic. Biol. Med. 2013, 61, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mandal, A.K.; Saito, H.; Pulliam, J.F.; Lee, E.Y.; Ke, Z.J.; Lu, J.; Ding, S.; Li, L.; Shelton, B.J.; et al. Arsenic and chromium in drinking water promote tumorigenesis in a mouse colitis-associated colorectal cancer model and the potential mechanism is ros-mediated wnt/β-catenin signaling pathway. Toxicol. Appl. Pharmacol. 2012, 262, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Nasr, R.; Lallemand-Breitenbach, V.; Zhu, J.; Guillemin, M.C.; de Thé, H. Therapy-induced pml/rara proteolysis and acute promyelocytic leukemia cure. Clin. Cancer Res. 2009, 15, 6321–6326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lu, H.; Li, W.; Hu, R.; Chen, Z. Identification of arsenic direct-binding proteins in acute promyelocytic leukaemia cells. Int. J. Mol. Sci. 2015, 16, 26871–26879. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. USA 2007, 104, 12288–12293. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules—From biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Mustacich, D.; Coon, A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic. Biol. Med. 2000, 29, 312–322. [Google Scholar] [CrossRef]
- Urig, S.; Becker, K. On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin. Cancer Biol. 2006, 16, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Ducas, R.A.; Seftel, M.D.; Ducas, J.; Seifer, C. Monomorphic ventricular tachycardia caused by arsenic trioxide therapy for acute promyelocytic leukaemia. J. R. Coll. Phys. Edinb. 2011, 41, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Mumford, J.L.; Wu, K.; Xia, Y.; Kwok, R.; Yang, Z.; Foster, J.; Sanders, W.E. Chronic arsenic exposure and cardiac repolarization abnormalities with qt interval prolongation in a population-based study. Environ. Health Perspect. 2007, 115, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Vizzardi, E.; Zanini, G.; Antonioli, E.; D’Aloia, A.; Raddino, R.; Cas, L.D. Qt prolongation: A case of arsenical pericardial and pleural effusion. Cardiovasc. Toxicol. 2008, 8, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Ohnsorge, P. Influence of mitochondrion-toxic agents on the cardiovascular system. Regul. Toxicol. Pharmacol. 2013, 67, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fang, H.; Huang, Z.; Shang, W.; Hou, T.; Cheng, A.; Cheng, H. Imaging ros signaling in cells and animals. J. Mol. Med. (Berl.) 2013, 91, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Freinbichler, W.; Colivicchi, M.A.; Stefanini, C.; Bianchi, L.; Ballini, C.; Misini, B.; Weinberger, P.; Linert, W.; Varešlija, D.; Tipton, K.F.; et al. Highly reactive oxygen species: Detection, formation, and possible functions. Cell. Mol. Life Sci. 2011, 68, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J.; Yim, M.B.; Chock, P.B. Free radicals and disease. Arch. Biochem. Biophys. 2002, 397, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P. Molecular insight of arsenic-induced carcinogenesis and its prevention. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Ajila, C.M.; Brar, S.K.; Verma, M.; Tyagi, R.D.; Godbout, S.; Valéro, J.R. Extraction and analysis of polyphenols: Recent trends. Crit. Rev. Biotechnol. 2011, 31, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Goldenthal, M.J. Understanding the impact of mitochondrial defects in cardiovascular disease: A review. J. Card. Fail. 2002, 8, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Goldenthal, M.J. The mitochondrial organelle and the heart. Rev. Esp. Cardiol. 2002, 55, 1293–1310. [Google Scholar] [CrossRef]
- Antico Arciuch, V.G.; Alippe, Y.; Carreras, M.C.; Poderoso, J.J. Mitochondrial kinases in cell signaling: Facts and perspectives. Adv. Drug Deliv. Rev. 2009, 61, 1234–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Fan, H.S.; Zhao, C.Y.; Lu, J.; Ikoma, T.; Tanaka, J.; Zhang, X.D. Competitive adsorption of bovine serum albumin and lysozyme on characterized calcium phosphates by polyacrylamide gel electrophoresis method. J. Mater. Sci. Mater. Med. 2007, 18, 2243–2249. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Binu, P.; Priya, N.; Abhilash, S.; Vineetha, R.C.; Nair, R.H. Studies on curative efficacy of monoterpene eugenol on anti- leukemic drug arsenic trioxide induced cardiotoxicity. Biomed. Pharmacother. 2017, 91, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, C.; Gao, R.; Ge, M.; Zhu, Y.; Zhang, Z. The protective role of resveratrol against arsenic trioxide-induced cardiotoxicity. Evid. Based Complement. Alternat. Med. 2013, 2013, 407839. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Chen, X.; Yang, T.; Yin, Y.; Ge, M.; Luo, M.; Chen, D.; Qian, X. A rapid fluorescent screening method for cellular sensitivity to anti-cancer compound. Cytotechnology 2012, 64, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Quig, D. Cysteine metabolism and metal toxicity. Altern. Med. Rev. 1998, 3, 262–270. [Google Scholar] [PubMed]
- He, X.; Ma, Q. Induction of metallothionein i by arsenic via metal-activated transcription factor 1: Critical role of c-terminal cysteine residues in arsenic sensing. J. Biol. Chem. 2009, 284, 12609–12621. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Kobayashi, Y.; Cui, X.; Kanno, S.; Hayakawa, T.; Shraim, A. The accumulation and toxicity of methylated arsenicals in endothelial cells: Important roles of thiol compounds. Toxicol. Appl. Pharmacol. 2004, 198, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, H.; Wang, Y.; Shao, Y.; Wang, B.; Xing, M. Regulation of autophagy factors by oxidative stress and cardiac enzymes imbalance during arsenic or/and copper induced cardiotoxicity in gallus gallus. Ecotoxicol. Environ. Saf. 2017, 148, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Bhattacharyya, S.S.; Bhattacharjee, N.; Pathak, S.; Boujedaini, N.; Belon, P.; Khuda-Bukhsh, A.R. Ascorbic acid combats arsenic-induced oxidative stress in mice liver. Ecotoxicol. Environ. Saf. 2009, 72, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Deneke, S.M. Thiol-based antioxidants. Curr. Top. Cell. Regul. 2000, 36, 151–180. [Google Scholar] [PubMed]
- Zhao, P.; Guo, Y.; Zhang, W.; Chai, H.; Xing, H.; Xing, M. Neurotoxicity induced by arsenic in gallus gallus: Regulation of oxidative stress and heat shock protein response. Chemosphere 2017, 166, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of nrf2/ho-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Jyrkkänen, H.K.; Levonen, A.L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 2012, 52, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.; Banerjee, T.D.; Welch, B.; Khalili, R.; Dagda, R.K.; Angermann, J. Monomethylarsonous acid, but not inorganic arsenic, is a mitochondria-specific toxicant in vascular smooth muscle cells. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2016, 35, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, V.P.; Soumya, R.S.; Raghu, K.G. Phloretin ameliorates arsenic trioxide induced mitochondrial dysfunction in h9c2 cardiomyoblasts mediated via alterations in membrane permeability and etc complexes. Eur. J. Pharmacol. 2015, 754, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Choi, J.Y.; Lee, H.J.; Byun, C.J.; Park, J.H.; Cho, H.S.; Cho, S.J.; Jo, S.A.; Jo, I. The green tea component (-)-epigallocatechin-3-gallate sensitizes primary endothelial cells to arsenite-induced apoptosis by decreasing c-jun n-terminal kinase-mediated catalase activity. PLoS ONE 2015, 10, e0138590. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shan, H.; Zhao, J.; Hong, Y.; Bai, Y.; Sun, I.; Pan, Z.; Zhang, Y.; Yang, B.; Du, Z. L-type calcium current (ica,l) and inward rectifier potassium current (ik1) are involved in qt prolongation induced by arsenic trioxide in rat. Cell. Physiol. Biochem. 2010, 26, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Sharma, Y.; Irshad, M.; Gupta, S.D.; Dogra, T.D. Arsenic-induced cell death in liver and brain of experimental rats. Basic Clin. Pharmacol. Toxicol. 2006, 98, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.L.; Liu, Z.; Qi, Z.J.; Huang, Y.P.; Gao, X.Q.; Zhang, Y.Y. (-)-Epigallocatechin-3-gallate (egcg) attenuates arsenic-induced cardiotoxicity in rats. Food Chem. Toxicol. 2016, 93, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Griesmacher, A.; Kindhauser, M.; Andert, S.E.; Schreiner, W.; Toma, C.; Knoebl, P.; Pietschmann, P.; Prager, R.; Schnack, C.; Schernthaner, G. Enhanced serum levels of thiobarbituric-acid-reactive substances in diabetes mellitus. Am. J. Med. 1995, 98, 469–475. [Google Scholar] [CrossRef]
- Li, Q.; Pogwizd, S.M.; Prabhu, S.D.; Zhou, L. Inhibiting Na+/K+ atpase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS ONE 2014, 9, e93928. [Google Scholar] [CrossRef] [PubMed]
- Mathews, V.V.; Paul, M.V.; Abhilash, M.; Manju, A.; Abhilash, S.; Nair, R.H. Myocardial toxicity of acute promyelocytic leukaemia drug-arsenic trioxide. Eur. Rev. Med. Pharmacol. Sci. 2013, 17 (Suppl. 1), 34–38. [Google Scholar] [PubMed]
- Varghese, M.V.; Abhilash, M.; Alex, M.; Sauganth Paul, M.V.; Prathapan, A.; Raghu, K.G.; Harikumaran Nair, R. Attenuation of arsenic trioxide induced cardiotoxicity through flaxseed oil in experimental rats. Redox Rep. 2017, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, A.A.; Olapour, S.; Varzi, H.N.; Khodayar, M.J.; Dianat, M.; Mohammadian, B.; Yaghooti, H. Ellagic acid protects against arsenic trioxide-induced cardiotoxicity in rat. Hum. Exp. Toxicol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jalaludeen, A.M.; Lee, W.Y.; Kim, J.H.; Jeong, H.Y.; Ki, K.S.; Kwon, E.G.; Song, H. Therapeutic efficacy of biochanin a against arsenic-induced renal and cardiac damage in rats. Environ. Toxicol. Pharmacol. 2015, 39, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, M.; Prabu, S.M. Silibinin potentially attenuates arsenic-induced oxidative stress mediated cardiotoxicity and dyslipidemia in rats. Cardiovasc. Toxicol. 2014, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.V.; Abhilash, M.; Paul, M.V.; Alex, M.; Nair, R.H. Omega-3 fatty acid protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Cardiovasc. Toxicol. 2017, 17, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Kumazaki, M.; Ando, H.; Kakei, M.; Ushijima, K.; Taniguchi, Y.; Yoshida, M.; Yamato, S.; Washino, S.; Koshimizu, T.A.; Fujimura, A. Alpha-lipoic acid protects against arsenic trioxide-induced acute qt prolongation in anesthetized guinea pigs. Eur. J. Pharmacol. 2013, 705, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kumazaki, M.; Ando, H.; Sasaki, A.; Koshimizu, T.A.; Ushijima, K.; Hosohata, K.; Oshima, Y.; Fujimura, A. Protective effect of alpha-lipoic acid against arsenic trioxide-induced acute cardiac toxicity in rats. J. Pharmacol. Sci. 2011, 115, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Amini-Khoei, H.; Hosseini, M.J.; Momeny, M.; Rahimi-Balaei, M.; Amiri, S.; Haj-Mirzaian, A.; Khedri, M.; Jahanabadi, S.; Mohammadi-Asl, A.; Mehr, S.E.; et al. Morphine attenuated the cytotoxicity induced by arsenic trioxide in h9c2 cardiomyocytes. Biol. Trace Elem. Res. 2016, 173, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Sun, J.; Li, H.; Wang, L.; Wei, Y.; Wang, Y.; Zhu, Y.; Huo, H.; Tan, Y. Differential mechanistic investigation of protective effects from imperatorin and sec-o-glucosylhamaudol against arsenic trioxide-induced cytotoxicity in vitro. Toxicol In Vitro 2016, 37, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Z.; Shu, Z.; Li, Z.; Ning, Y.; Yun, K.; Bai, H.; Liu, R.; Liu, W. Effect and mechanism of sorbus pohuashanensis (hante) hedl. Flavonoids protect against arsenic trioxide-induced cardiotoxicity. Biomed. Pharmacother. 2017, 88, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sfaxi, I.; Charradi, K.; Limam, F.; El May, M.V.; Aouani, E. Grape seed and skin extract protects against arsenic trioxide induced oxidative stress in rat heart. Can. J. Physiol. Pharmacol. 2015, 94, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, C.; Zhang, Y.; Hang, P.; Liu, Y.; Pan, Z.; Wang, N.; Du, Z. Genistein ameliorates adverse cardiac effects induced by arsenic trioxide through preventing cardiomyocytes apoptosis. Cell. Physiol. Biochem. 2013, 31, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chen, M.; Meng, J.; Yu, L.; Tu, Y.; Wan, L.; Fang, K.; Zhu, W. Arsenic trioxide and resveratrol show synergistic anti-leukemia activity and neutralized cardiotoxicity. PLoS ONE 2014, 9, e105890. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, V.P.; Girija, S.; Soumya, R.S.; Raghu, K.G. Polyphenol-rich apple (Malus domestica L.) peel extract attenuates arsenic trioxide induced cardiotoxicity in h9c2 cells via its antioxidant activity. Food Funct. 2014, 5, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Das, S.K.; Haldar, P.K. Arsenic induced myocardial toxicity in rats: Alleviative effect of trichosanthes dioica fruit. J. Diet. Suppl. 2014, 11, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Haldar, P.K. Trichosanthes dioica root alleviates arsenic induced myocardial toxicity in rats. J. Environ. Pathol. Toxicol. Oncol. 2013, 32, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Das, J.; Manna, P.; Sil, P.C. Taurine prevents arsenic-induced cardiac oxidative stress and apoptotic damage: Role of nf-kappa b, p38 and jnk mapk pathway. Toxicol. Appl. Pharmacol. 2009, 240, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Adil, M.; Kandhare, A.D.; Ghosh, P.; Bodhankar, S.L. Sodium arsenite-induced myocardial bruise in rats: Ameliorative effect of naringin via tgf-β/smad and nrf/ho pathways. Chem. Biol. Interact. 2016, 253, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Krohn, R.M.; Lemaire, M.; Negro Silva, L.F.; Lemarié, C.; Bolt, A.; Mann, K.K.; Smits, J.E. High-selenium lentil diet protects against arsenic-induced atherosclerosis in a mouse model. J. Nutr. Biochem. 2016, 27, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Li, G.Y.; Liu, Y.; Chai, L.M.; Chen, J.X.; Zhang, Y.; Du, Z.M.; Lu, Y.J.; Yang, B.F. Resveratrol protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Br. J. Pharmacol. 2008, 154, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, V.P.; Prathapan, A.; Soumya, R.S.; Raghu, K.G. Arsenic trioxide toxicity in h9c2 myoblasts—Damage to cell organelles and possible amelioration with boerhavia diffusa. Cardiovasc. Toxicol. 2013, 13, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. Camp signal transduction in the heart: Understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 2009, 158, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Kurian, G.A.; Paddikkala, J. Role of mitochondrial enzymes and sarcoplasmic atpase in cardioprotection mediated by aqueous extract of Desmodium gangeticum (L.) dc root on ischemic reperfusion injury. Indian J. Pharm. Sci. 2010, 72, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Duan, X.; Dong, D.; Bai, C.; Li, X.; Sun, G.; Li, B. Activation of the nrf2 pathway by inorganic arsenic in human hepatocytes and the role of transcriptional repressor bach1. Oxidative Med. Cell. Longev. 2013, 2013, 984546. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Qu, W.; Reece, J.M.; Kumagai, Y.; Waalkes, M.P. Transcription factor nrf2 activation by inorganic arsenic in cultured keratinocytes: Involvement of hydrogen peroxide. Exp. Cell. Res. 2003, 290, 234–245. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Chen, W.; Eblin, K.E.; Gandolfi, J.A.; Zhang, D.D. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol. Appl. Pharmacol. 2007, 225, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Wang, X.; Chang, Q.; Hitron, A.; Zhang, Z.; Xu, M.; Chen, G.; Luo, J.; Jiang, B.; Fang, J.; et al. Arsenic promotes angiogenesis in vitro via a heme oxygenase-1-dependent mechanism. Toxicol. Appl. Pharmacol. 2010, 244, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Abiko, Y.; Shinkai, Y.; Sumi, D.; Kumagai, Y. Reduction of arsenic-induced cytotoxicity through nrf2/ho-1 signaling in hepg2 cells. J. Toxicol. Sci. 2010, 35, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Sumi, D.; Abe, K.; Himeno, S. Arsenite retards the cardiac differentiation of rat cardiac myoblast h9c2 cells. Biochem. Biophys. Res. Commun. 2013, 436, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.H.; Yu, C.L.; Chang, L.W.; Yu, H.S. Low concentrations of arsenic induce vascular endothelial growth factor and nitric oxide release and stimulate angiogenesis in vitro. Chem. Res. Toxicol. 2003, 16, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor nf-kappab pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Ho, I.C.; Lee, T.C. Oxidative stress mediates sodium arsenite-induced expression of heme oxygenase-1, monocyte chemoattractant protein-1, and interleukin-6 in vascular smooth muscle cells. Toxicol. Sci. 2005, 85, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Martin-Pardillos, A.; Sosa, C.; Sorribas, V. Arsenic increases pi-mediated vascular calcification and induces premature senescence in vascular smooth muscle cells. Toxicol. Sci. 2013, 131, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.M.; Shin, Y.S.; Kang, S.; Noh, J.Y.; Kim, K.; Chung, S.M.; Yun, Y.P.; Chung, J.H. Potentiation of vasoconstriction and pressor response by low concentration of monomethylarsonous acid (mma(iii)). Toxicol. Lett. 2011, 205, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, M.; Devesa, V.; Vélez, D. Differential toxicity and gene expression in caco-2 cells exposed to arsenic species. Toxicol. Lett. 2013, 218, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Wieder, T.; Stärck, L.; Essmann, F.; Schulze-Osthoff, K.; Dörken, B.; Daniel, P.T. Arsenic trioxide triggers a regulated form of caspase-independent necrotic cell death via the mitochondrial death pathway. Oncogene 2005, 24, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Correa, F.; Buelna-Chontal, M.; Hernández-Reséndiz, S.; García-Niño, W.R.; Roldán, F.J.; Soto, V.; Silva-Palacios, A.; Amador, A.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Curcumin maintains cardiac and mitochondrial function in chronic kidney disease. Free Radic. Biol. Med. 2013, 61, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. Lc3 and autophagy. Autophagosome Phagosome 2008, 445, 77–88. [Google Scholar]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Arsenic trioxide inhibits the growth of calu-6 cells via inducing a g2 arrest of the cell cycle and apoptosis accompanied with the depletion of gsh. Cancer Lett. 2008, 270, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Partridge, M.A.; Huang, S.X.; Hernandez-Rosa, E.; Davidson, M.M.; Hei, T.K. Arsenic induced mitochondrial dna damage and altered mitochondrial oxidative function: Implications for genotoxic mechanisms in mammalian cells. Cancer Res. 2007, 67, 5239–5247. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [PubMed]
- Cassady, J.M.; Zennie, T.M.; Chae, Y.H.; Ferin, M.A.; Portuondo, N.E.; Baird, W.M. Use of a mammalian cell culture benzo(a)pyrene metabolism assay for the detection of potential anticarcinogens from natural products: Inhibition of metabolism by biochanin a, an isoflavone from Trifolium pratense L. Cancer Res. 1988, 48, 6257–6261. [Google Scholar] [PubMed]
- Hyson, D.A. A comprehensive review of apples and apple components and their relationship to human health. Adv. Nutr. Int. Rev. J. 2011, 2, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Abe, L.T.; Lajolo, F.M.; Genovese, M.I. Potential dietary sources of ellagic acid and other antioxidants among fruits consumed in brazil: Jabuticaba (myrciaria jaboticaba (vell.) berg). J. Sci. Food Agric. 2012, 92, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Kannan, M.M.; Quine, S.D.; Sangeetha, T. Protective efficacy of ellagic acid on glycoproteins, hematological parameters, biochemical changes, and electrolytes in myocardial infarcted rats. J. Biochem. Mol. Toxicol. 2012, 26, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Devika, P.T.; Stanely Mainzen Prince, P. (-)Epigallocatechingallate protects the mitochondria against the deleterious effects of lipids, calcium and adenosine triphosphate in isoproterenol induced myocardial infarcted male wistar rats. J. Appl. Toxicol. 2008, 28, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Devika, P.T.; Stanely Mainzen Prince, P. (-)Epigallocatechin-gallate (egcg) prevents mitochondrial damage in isoproterenol-induced cardiac toxicity in albino wistar rats: A transmission electron microscopic and in vitro study. Pharmacol. Res. 2008, 57, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Nègre-Salvayre, A.; Salvayre, R. Quercetin prevents the cytotoxicity of oxidized ldl on lymphoid cell lines. Free Radic. Biol. Med. 1992, 12, 101–106. [Google Scholar] [CrossRef]
- Rossi, L.; Lippe, G.; Marchese, E.; De Martino, A.; Mavelli, I.; Rotilio, G.; Ciriolo, M.R. Decrease of cytochrome c oxidase protein in heart mitochondria of copper-deficient rats. Biometals 1998, 11, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, M.; Prabu, S.M. Silibinin potentially protects arsenic-induced oxidative hepatic dysfunction in rats. Toxicol. Mech. Methods 2012, 22, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.S.; Kumar, J.K.; Luqman, S.; Shanker, K.; Gupta, M.M.; Khanuja, S.P. Recent advances in plant hepatoprotectives: A chemical and biological profile of some important leads. Med. Res. Rev. 2008, 28, 746–772. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Cellular basis of drug-induced torsades de pointes. Br. J. Pharmacol. 2008, 154, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Berquin, I.M.; Edwards, I.J.; Chen, Y.Q. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett. 2008, 269, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Gülçin, I.; Beydemir, S.; Alici, H.A.; Elmastaş, M.; Büyükokuroğlu, M.E. In vitro antioxidant properties of morphine. Pharmacol. Res. 2004, 49, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, A.; Marsch, L.A.; Joseph, H.; Portenoy, R.K. Opioids and the treatment of chronic pain: Controversies, current status, and future directions. Exp. Clin. Psychopharmacol. 2008, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Costa-Malaquias, A.; Almeida, M.B.; Monteiro, J.R.S.; de Matos Macchi, B.; do Nascimento, J.L.M.; Crespo-Lopez, M.E. Morphine protects against methylmercury intoxication: A role for opioid receptors in oxidative stress? PLoS ONE 2014, 9, e110815. [Google Scholar] [CrossRef] [PubMed]
- Kanesaki, T.; Saeki, M.; Ooi, Y.; Suematsu, M.; Matsumoto, K.; Sakuda, M.; Saito, K.; Maeda, S. Morphine prevents peroxynitrite-induced death of human neuroblastoma sh-sy5y cells through a direct scavenging action. Eur. J. Pharmacol. 1999, 372, 319–324. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.S.; Park, C.; Jung, E.B.; Choi, D.H.; Kim, T.Y.; Moon, S.K.; Park, R. Morphine prevents glutamate-induced death of primary rat neonatal astrocytes through modulation of intracellular redox. Immunopharmacol. Immunotoxicol. 2004, 26, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Tan, K.S.; Wei, S.J.; Wu, H.M.; Xu, Z.; Wilson, B.; Lu, R.B.; Hong, J.S.; Flood, P.M. Microglia-mediated neurotoxicity is inhibited by morphine through an opioid receptor-independent reduction of nadph oxidase activity. J. Immunol. 2007, 179, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Huh, J.; Wang, H.; Kang, Y.; Lou, J.; Xu, Z. Mitochondrial events responsible for morphine’s cardioprotection against ischemia/reperfusion injury. Toxicol. Appl. Pharmacol. 2016, 290, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Mechanisms of action of (n-3) fatty acids. J. Nutr. 2012, 142, 592S–599S. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Khairallah, R.J.; Dabkowski, E.R. Update on lipids and mitochondrial function: Impact of dietary n-3 polyunsaturated fatty acids. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Jong, C.J.; Azuma, J.; Schaffer, S. Mechanism underlying the antioxidant activity of taurine: Prevention of mitochondrial oxidant production. Amino Acids 2012, 42, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Saini, H.K.; Zhang, M.; Elimban, V.; Dhalla, N.S. Mapk activation and apoptotic alterations in hearts subjected to calcium paradox are attenuated by taurine. Cardiovasc. Res. 2006, 72, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Sole, M.J.; Jeejeebhoy, K.N. Conditioned nutritional requirements and the pathogenesis and treatment of myocardial failure. Curr. Opin. Clin. Nutr. Metab. Care 2000, 3, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Baldwin, L.A. Inorganics and hormesis. Crit. Rev. Toxicol. 2003, 33, 215–304. [Google Scholar] [CrossRef] [PubMed]
- Brglez Mojzer, E.; Knez Hrnčič, M.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction methods, antioxidative action, bioavailability and anticarcinogenic effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [PubMed]
- Croft, K.D. Dietary polyphenols: Antioxidants or not? Arch. Biochem. Biophys. 2016, 595, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Gebel, T.W. Genotoxicity of arsenical compounds. Int. J. Hyg. Environ. Health 2001, 203, 249–262. [Google Scholar] [CrossRef]
- Wang, C.H.; Jeng, J.S.; Yip, P.K.; Chen, C.L.; Hsu, L.I.; Hsueh, Y.M.; Chiou, H.Y.; Wu, M.M.; Chen, C.J. Biological gradient between long-term arsenic exposure and carotid atherosclerosis. Circulation 2002, 105, 1804–1809. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Tondel, M.; Ahmad, S.A.; Chowdhury, I.A.; Faruquee, M.H.; Axelson, O. Hypertension and arsenic exposure in bangladesh. Hypertension 1999, 33, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Verret, W.J.; Chen, Y.; Ahmed, A.; Islam, T.; Parvez, F.; Kibriya, M.G.; Graziano, J.H.; Ahsan, H. A randomized, double-blind placebo-controlled trial evaluating the effects of vitamin E and selenium on arsenic-induced skin lesions in bangladesh. J. Occup. Environ. Med. 2005, 47, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Mahata, J.; Argos, M.; Verret, W.; Kibriya, M.G.; Santella, R.M.; Ahsan, H. Effect of selenium and vitamin E supplementation on plasma protein carbonyl levels in patients with arsenic-related skin lesions. Nutr. Cancer 2008, 60, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, M.G.; Jasmine, F.; Argos, M.; Verret, W.J.; Rakibuz-Zaman, M.; Ahmed, A.; Parvez, F.; Ahsan, H. Changes in gene expression profiles in response to selenium supplementation among individuals with arsenic-induced pre-malignant skin lesions. Toxicol. Lett. 2007, 169, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Krohn, R.M.; Raqib, R.; Akhtar, E.; Vandenberg, A.; Smits, J.E. A high-selenium lentil dietary intervention in bangladesh to counteract arsenic toxicity: Study protocol for a randomized controlled trial. Trials 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Tenore, G.C.; Caruso, D.; Buonomo, G.; D’Avino, M.; Campiglia, P.; Marinelli, L.; Novellino, E. A healthy balance of plasma cholesterol by a novel annurca apple-based nutraceutical formulation: Results of a randomized trial. J. Med. Food 2017, 20, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Chen, N.G.; Shi, Z.; Qiu, J.; He, C.; Chen, M. Recent advances in anticancer activities and drug delivery systems of tannins. Med. Res. Rev. 2017, 37, 665–701. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, S.S.; Solati-Hashjin, M.; Samadikuchaksaraei, A.; Kalantarinejad, R.; Asadi-Eydivand, M.; Abu Osman, N.A. Epigallocatechin gallate/layered double hydroxide nanohybrids: Preparation, characterization, and in vitro anti-tumor study. PLoS ONE 2015, 10, e0136530. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, H. Lignans and human health. Crit. Rev. Clin. Lab. Sci. 2007, 44, 483–525. [Google Scholar] [CrossRef] [PubMed]
- Sanna, V.; Siddiqui, I.A.; Sechi, M.; Mukhtar, H. Resveratrol-loaded nanoparticles based on poly(epsilon-caprolactone) and poly(d,l-lactic-co-glycolic acid)-poly(ethylene glycol) blend for prostate cancer treatment. Mol. Pharm. 2013, 10, 3871–3881. [Google Scholar] [CrossRef] [PubMed]
- Carletto, B.; Berton, J.; Ferreira, T.N.; Dalmolin, L.F.; Paludo, K.S.; Mainardes, R.M.; Farago, P.V.; Favero, G.M. Resveratrol-loaded nanocapsules inhibit murine melanoma tumor growth. Colloids Surf. B Biointerfaces 2016, 144, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Li, X.; Lu, X.; Jiang, C.; Hu, Y.; Li, Q.; You, Y.; Fu, Z. Enhanced growth inhibition effect of resveratrol incorporated into biodegradable nanoparticles against glioma cells is mediated by the induction of intracellular reactive oxygen species levels. Colloids Surf. B Biointerfaces 2009, 72, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Narayanan, S.; Sethuraman, S.; Krishnan, U.M. Novel resveratrol and 5-fluorouracil coencapsulated in pegylated nanoliposomes improve chemotherapeutic efficacy of combination against head and neck squamous cell carcinoma. Biomed. Res. Int. 2014, 2014, 424239. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Li, A.; Jia, Z.; Yuan, Y.; Dai, H.; Li, H. Transferrin modified peg-pla-resveratrol conjugates: In vitro and in vivo studies for glioma. Eur. J. Pharmacol. 2013, 718, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sassi, N.; Mattarei, A.; Azzolini, M.; Bernardi, P.; Szabo, I.; Paradisi, C.; Zoratti, M.; Biasutto, L. Mitochondria-targeted resveratrol derivatives act as cytotoxic pro-oxidants. Curr. Pharm. Des. 2014, 20, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.; Brownlow, B.; Elbayoumi, T. Mitochondria-specific pro-apoptotic activity of genistein lipidic nanocarriers. Mol. Pharm. 2013, 10, 3789–3800. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.K.; Skulas-Ray, A.C.; Fleming, J.A.; Link, C.J.; Mukherjea, R.; Krul, E.S.; Kris-Etherton, P.M. Effects of isoflavone-containing soya protein on ex vivo cholesterol efflux, vascular function and blood markers of cvd risk in adults with moderately elevated blood pressure: A dose-response randomised controlled trial. Br. J. Nutr. 2017, 117, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The antioxidant transcription factor nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Aggarwal, D.; Migrino, R.Q.; Joseph, J.; McAllister, D.; Konorev, E.A.; Antholine, W.E.; Zielonka, J.; Srinivasan, S.; Avadhani, N.G.; et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by mito-q. Biophys. J. 2009, 96, 1388–1398. [Google Scholar] [CrossRef] [PubMed]

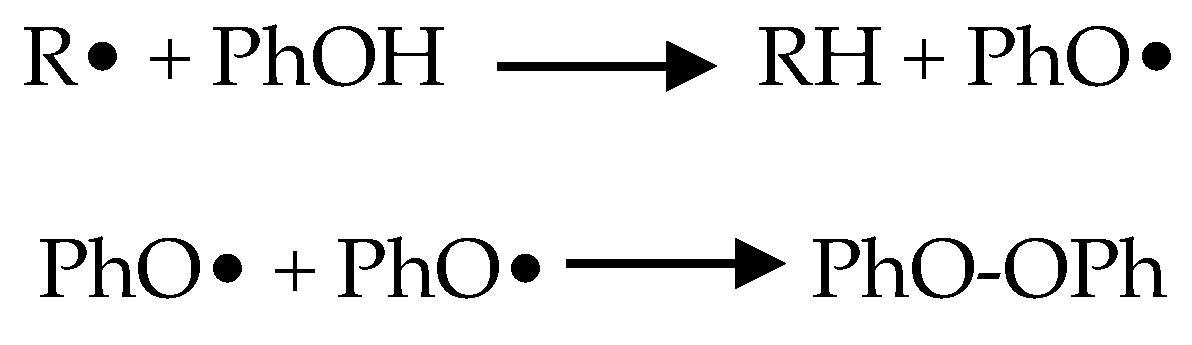

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Vivo Studies Stratified by Arsenic Exposure Duration | |||||
| Duration | Arsenic Dose | Type | Method | Model | Citation |
| 2 h | 0.15 μM, 1.5 μM, 5 μM | As2O3 | IV 1 | Wistar rat | [59] |
| 1.5 μM | As2O3 | IV | Guinea pig | [58] | |
| 6–10 days | 0.8 mg/kg | As2O3 | IV | Wistar rat | [64] |
| 1 mg/kg | As2O3 | IV | Balb/c mouse | [72] | |
| 3 mg/kg | As2O3 | IV | Wistar rat | [33] | |
| 10 mg/kg | NaAsO2 | Oral | Wistar rat | [68] | |
| 10 mg/kg | NaAsO2 | Oral | Wistar rat | [67] | |
| 10–29 days | 1 mg/kg | As2O3 | IV | Balb/c mouse | [62] |
| 2.5 mg/kg | As2O3 | IP 2 | Wistar rat | [63] | |
| 5 mg/kg | As2O3 | IP | Wistar rat | [54] | |
| 5 mg/kg | As2O3 | Oral | SD rat 3 | [70] | |
| 5 mg/kg | NaAsO2 | Oral | Wistar rat | [56] | |
| 200 ppb | NaAsO2 | Oral | APO E-/- | [71] | |
| 30–56 days | 2 mg/kg | NaAsO2 | Oral | SD rat | [69] |
| 4 mg/kg | As2O3 | Oral intubation | Wistar rat | [57] | |
| 4 mg/kg | As2O3 | Oral intubation | Wistar rat | [57] | |
| 4 mg/kg | As2O3 | Oral | Wistar rat | [32] | |
| 5 mg/kg | As2O3 | IV | SD rat | [59] | |
| 10 mg/kg | NaAsO2 | Oral | SD rat | [55] | |
| 50 mg/kg | NaAsO2 | Oral | SD rat | [49] | |
| In Vitro Studies | |||||
| 1 μM | NaAsO2 | H9c2 4 | [49] | ||
| 1 μM, 2 μM | As2O3 | H9c2 | [60] | ||
| 2 μM/mL | As2O3 | H9c2 | [61] | ||
| 4 μM | As2O3 | H9c2 | [62] | ||
| 5 μM | As2O3 | NRLVM 5 | [65] | ||
| 5 μM | As2O3 | H9c2 | [66] | ||
| 5 μM | As2O3 | H9c2 | [45] | ||
| 5 μM | NaAsO2 | Primary myocytes | [69] | ||
| 5 μM, 7.5 μM, 10 μM | As2O3 | H9c2 | [73] | ||
| 5 μM, 6 μM, 12 μM | As2O3 | NRLVM | [64] | ||
| 10 μM | As2O3 | H9c2 | [57] | ||
| 10 μM | NaAsO2 | H9c2 | [72] | ||
| Polyphenolic Antioxidants | Classification | Source |
| Biochanin A [55] | Flavonoid | Cabbage, alfalfa |
| Boerhavia diffusa [73] | Flavonoid | B. diffusa |
| Ellagic acid [54] | Phenol | Berries, walnuts |
| EGCG [49] | Catechin | Green tea |
| Eugenol [32] | Phenol | Clove |
| Genistein [65] | Flavonoid | Soy |
| Grape seed and skin extract [63] | Flavonoid, stilbene | Grapes |
| Imperatorin [61] | Flavonoid | Radix Saposhininkovaie |
| Sec-O-glucosylhamandol [61] | Flavonoid | Radix Saposhininkovaie |
| Malus domestica apple peel [66] | Flavonoid | Apples |
| Naringin [70] | Flavonoid | Citrus fruit |
| Phloretin [45] | Flavonoid | Apples |
| Resveratrol [33] | Stilbene | Red wine |
| Silybum marianum [56] | Flavonoid | Milk thistle |
| Sorbus phnuashanesis (Hante) Hedl [62] | Flavonoid | Chinese herb |
| Trichosanthes dioca [68] | Flavonoid | T. dioica |
| Other Antioxidants | Type | Source |
| α-lipoic acid [59] | Organosulfur compound | Spinach, broccoli |
| Flax seed oil [53] | α-linoleic acid | Flax seeds |
| Morphine [60] | Opioid | Poppy seeds |
| Omega-3 fatty acid [57] | Polyunsaturated fatty acid | Fish oil |
| Selenium [71] | Essential trace element | Lentils |
| Taurine [69] | Sulfonic acid | Amino acid cysteine |
| Arsenic Induced Effect | Restored with Antioxidant | Citation |
|---|---|---|
| Heart | ||
| Arsenic deposition in heart | Eugenol | [32] |
| Grape seed and skin extract | [63] | |
| EGCG | [49] | |
| Omega-3 | [57] | |
| Flax seed oil | [53] | |
| Resveratrol | [33] | |
| QT interval prolongation | Naringin | [70] |
| Eugenol | [32] | |
| Genistein | [64] | |
| α-lipoic acid | [58] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [72] | |
| Increased QTc interval | Naringin | [70] |
| Ellagic acid | [54] | |
| α-lipoic acid | [58] | |
| ST-T wave change | Sorbus phnuashanesis | [62] |
| Increased RR interval | Naringin | [70] |
| Increased QRS interval | Naringin | [70] |
| Inhibited IKS currents | α-lipoic acid | [58] |
| Reduced amperage of IK | α-lipoic acid | [58] |
| Decreased heart rate | Naringin | [70] |
| Eugenol | [32] | |
| Genistein | [64] | |
| Decreased cardiac output | Genistein | [64] |
| Decreased CAMP | Resveratrol | [33] |
| Structural changes in cardiac tissue | Naringin | [70] |
| Eugenol | [32] | |
| Ellagic acid | [54] | |
| Grape seed and skin extract 1 | [63] | |
| EGCG | [49] | |
| Omega-3 | [57] | |
| Flax seed oil | [53] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol 1 | [33] | |
| Resveratrol 1 | [72] | |
| Atherosclerotic plaque formation | Selenium | [71] |
| Increased ALP activity | Naringin | [70] |
| Silybum marianum | [56] | |
| Blood Plasma | ||
| Elevated triglycerides | Biochanin A | [55] |
| Grape seed and skin extract | [63] | |
| Increased total cholesterol | Naringin | [70] |
| T. dioica root | [68] | |
| T. dioica fruit | [67] | |
| Taurine | [69] | |
| Silybum marianum | [56] | |
| Grape seed and skin extract | [63] | |
| Increased LDL cholesterol | Naringin | [70] |
| Biochanin A | [55] | |
| Silybum marianum | [56] | |
| Decreased HDL cholesterol | T. dioica root | [68] |
| T. dioica fruit | [67] | |
| Decreased phospholipids | Silybum marianum | [56] |
| Increased atherogenic Coefficient (AC) | Biochanin A | [55] |
| Increased cardiac risk ratio | Biochanin A | [55] |
| Increased free fatty acids | Silybum marianum | [56] |
| Increased lipase activity | GSSE | [63] |
| Increased CPK | T. dioica root | [68] |
| T. dioica fruit | [67] | |
| Increased CK-MB | Naringin | [70] |
| Eugenol | [32] | |
| Ellagic Acid | [54] | |
| Silybum marianum | [56] | |
| EGCG | [49] | |
| Omega 3 | [57] | |
| Flax seed oil | [53] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [33] | |
| Increased serum troponin | Ellagic acid | [54] |
| Elevated LDH | Naringin | [70] |
| T. dioica root | [68] | |
| T. dioica fruit | [67] | |
| Eugenol | [32] | |
| Biochanin A | [55] | |
| Silybum marianum | [56] | |
| EGCG | [49] | |
| Flax seed oil | [53] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [33] | |
| Resveratrol | [72] | |
| Increased AST activity | Naringin | [70] |
| α-lipoic acid | [59] | |
| Silybum marianum | [56] | |
| EGCG | [49] | |
| Resveratrol | [33] | |
| Increased ALT activity | Naringin | [70] |
| Silybum marianum | [56] | |
| Increased ALP activity | Silybum marianum | [56] |
| Increased CRP | Grape seed and skin extract | [63] |
| Increased CK | Sorbus phnuashanesis | [62] |
| Resveratrol | [33] | |
| Antioxidants | ||
| Downregulated NRf2 | Naringin | [70] |
| Silybum marianum (liver) | [56] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [33] | |
| Increased Nrf2 level | sec-O-glucosylhamaudol 2 | [61] |
| Imperatorin 2 | [61] | |
| Upregulation of Keap-1 | Silybum marianum (liver) | [56] |
| Decreased SOD activity | Naringin | [70] |
| T. dioica root | [68] | |
| T. dioica fruit | [67] | |
| Resveratrol | [65] | |
| Genistein | [65] | |
| Taurine | [69] | |
| Biochanin A | [55] | |
| Silybum marianum | [56] | |
| Grape seed and skin extract | [63] | |
| EGCG | [49] | |
| Omega-3 | [57] | |
| Flax seed oil | [53] | |
| Boerhavia diffusa | [73] | |
| Malus domestica L. Peel | [66] | |
| Phloretin | [45] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [72] | |
| Decreased TR activity | Phloretin | [45] |
| Decreased GPx activity | T. dioica root | [68] |
| T. dioica fruit | [67] | |
| Eugenol | [32] | |
| Taurine | [69] | |
| Silybum marianum | [56] | |
| Grape seed and skin extract | [63] | |
| EGCG | [49] | |
| Omega-3 | [57] | |
| Flax seed oil | [53] | |
| Malus domestica L. Peel | [66] | |
| Phloretin | [45] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [72] | |
| Increased GPx activity | Ellagic acid | [54] |
| Decreased GR activity | T. dioica root | [68] |
| T. dioica fruit | [67] | |
| Taurine | [69] | |
| Silybum marianum | [56] | |
| Malus domestica L. Peel | [66] | |
| Decreased GST activity | T. dioica root | [68] |
| T. dioica fruit | [67] | |
| Eugenol | [32] | |
| Taurine | [69] | |
| Silybum marianum | [56] | |
| Omega-3 | [57] | |
| Flax seed oil | [53] | |
| Decreased G6PD activity | Silybum marianum | [56] |
| Downregulated HO-1 | Naringin | [70] |
| Silybum marianum (liver) | [56] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [33] | |
| Elevated HO-1 expression | Imperatorin | [61] |
| Sec-O-glucosylhamaudol 2 | [61] | |
| Elevated NQ01 expression | Imperatorin | [61] |
| Sec-O-glucosylhamaudol | [61] | |
| Decreased catalase activity | T. dioica root | [68] |
| T. dioica fruit | [67] | |
| Taurine | [69] | |
| Biochanin A 3 | [55] | |
| Silybum marianum | [56] | |
| Grape seed and skin extract | [63] | |
| Omega-3 | [57] | |
| Flax seed oil | [53] | |
| Boerhavia diffusa | [73] | |
| Malus domestica L. Peel | [66] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [72] | |
| Decreased GSH levels | Naringin | [70] |
| T. dioica root | [68] | |
| T. dioica fruit | [67] | |
| Eugenol | [32] | |
| Resveratrol | [65] | |
| Taurine | [69] | |
| Biochanin A | [55] | |
| Silybum marianum | [56] | |
| Omega-3 | [57] | |
| Flax seed oil | [53] | |
| Phloretin | [45] | |
| No significant change in GSH | Ellagic acid | [54] |
| Short term GSH elevation followed by decrease | Malus domestica L. Peel | [66] |
| Increased GSSG | Selenium | [71] |
| T. dioica root | [68] | |
| T. dioica fruit | [67] | |
| Decreased GSH/GSSG ratio | Taurine | [69] |
| Resveratrol | [33] | |
| ROS | ||
| Elevated H2O2 | Grape seed and skin extract | [63] |
| Elevated mitochondrial ROS | Morphine | [60] |
| EGCG | [49] | |
| Malus domestica L. Peel | [66] | |
| Phloretin | [45] | |
| Elevated (H2O2, ONOO−, OH−) | Naringin | [70] |
| Morphine | [60] | |
| Resveratrol | [65] | |
| Genistein | [65] | |
| Imperatorin | [61] | |
| Phloretin | [45] | |
| Sorbus phnuashanesis | [62] | |
| Boerhavia diffusa | [73] | |
| Malus domestica L. Peel | [66] | |
| Resveratrol | [33] | |
| Resveratrol | [72] | |
| Lipid peroxidation (elevated MDA) | Naringin | [70] |
| Eugenol | [32] | |
| Ellagic acid | [54] | |
| Biochanin A 3 | [55] | |
| Grape seed and skin extract | [63] | |
| EGCG | [49] | |
| Taurine | [69] | |
| Omega-3 | [57] | |
| Increased 8-OHdG | α-lipoic acid | [59] |
| Resveratrol | [33] | |
| Elevated TBARS | T. dioica root | [68] |
| T. dioica fruit | [67] | |
| Silybum marianum | [56] | |
| Flax seed oil | [53] | |
| Increased XO | Malus domestica L. Peel | [66] |
| Phloretin | [45] | |
| Increased NOX activity (NOX2 and NOX4) | Silybum marianum | [56] |
| No change in NO | Resveratrol | [33] |
| Increased NO content | Naringin | [70] |
| Apoptosis | ||
| LDH release | Taurine | [69] |
| Imperatorin | [61] | |
| Sec-O-glucosylhamaudol | [61] | |
| EGCG | [49] | |
| Omega-3 | [57] | |
| Boerhavia diffusa | [73] | |
| Malus domestica L. Peel | [66] | |
| Phloretin | [45] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [33] | |
| Resveratrol | [72] | |
| Decreased cell viability | Morphine | [60] |
| Resveratrol | [65] | |
| Genistein | [65] | |
| Taurine | [69] | |
| Imperatorin | [61] | |
| Sec-O-glucosylhamaudol | [61] | |
| EGCG | [49] | |
| Boerhavia diffusa | [73] | |
| Malus domestica L. Peel | [66] | |
| Phloretin | [45] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [72] | |
| Apoptosis | Naringin | [70] |
| Genistein | [64] | |
| Resveratrol + Genistein | [65] | |
| Taurine | [69] | |
| Imperatorin | [61] | |
| Sec-O-glucosylhamaudol | [61] | |
| EGCG | [49] | |
| Omega-3 | [57] | |
| Boerhavia diffusa | [73] | |
| Malus domestica L. Peel | [66] | |
| Phloretin | [45] | |
| Sorbus phnuashanesis | [62] | |
| Resveratrol | [72] | |
| DNA fragmentation | Morphine | [60] |
| T. dioica root | [68] | |
| T. dioica fruit | [67] | |
| Genistein | [65] | |
| Silybum marianum | [56] | |
| Omega 3 | [33] | |
| Resveratrol | [72] | |
| Decreased cell growth via DNA synthesis | Morphine | [60] |
| Increased caspase-3 cleavage | Genistein | [65] |
| EGCG | [49] | |
| Elevated caspase-3 activity | Morphine | [60] |
| Genistein | [64] | |
| Taurine | [69] | |
| Imperatorin 3 | [61] | |
| Sec-O-glucosylhamaudol 3 | [61] | |
| EGCG | [49] | |
| Sorbus phnuashanesis | [62] | |
| Malus domestica L. Peel | [66] | |
| Phloretin | [45] | |
| Resveratrol | [72] | |
| Elevated caspase-8 activity | Sorbus phnuashanesis | [62] |
| Elevated caspase-9 activity | Sorbus phnuashanesis | [62] |
| Elevated cytochrome-c | Taurine | [69] |
| Proteolysis of PARP | Taurine | [69] |
| Increased BAX | Morphine | [60] |
| Taurine | [69] | |
| Increased Bad | Taurine | [69] |
| Increased PUMA | Morphine | [60] |
| Decreased Bcl2 | Morphine | [60] |
| Taurine | [69] | |
| Decreased Bcl-xL | Morphine 3 | [60] |
| Taurine | [69] | |
| Decreased Bcl2/BAX ratio | Sorbus phnuashanesis | [62] |
| Decreased CIAP1, CIAP2, XIAP | Morphine 3 | [60] |
| Decreased Survivin | Morphine | [60] |
| Decreased P-Akt/Akt | Sorbus phnuashanesis | [62] |
| Upregulated TGF-β | Naringin | [70] |
| Increased SMAD3 | Naringin | [70] |
| Calcium and Membrane potential | ||
| Decreased NF-κB activity | Morphine | [60] |
| Phosphorylated NF-κB | Taurine | [69] |
| Calcium accumulation | Eugenol | [32] |
| Genistein | [64] | |
| Taurine | [69] | |
| Imperatorin 3 | [61] | |
| Sec-O-glucosylhamaudol 3 | [61] | |
| Grape seed and skin extract | [63] | |
| EGCG | [49] | |
| Omega-3 | [57] | |
| Boerhavia diffusa | [73] | |
| Malus domestica L. Peel | [66] | |
| Phloretin | [45] | |
| Resveratrol | [72] | |
| Resveratrol | [33] | |
| Increased in Cav1.2 | Genistein | [64] |
| Decreased Ca-ATPase activity | Eugenol | [32] |
| Silybum marianum | [56] | |
| Phloretin | [45] | |
| Decreased Na+/K+ ATPase activity | Naringin | [70] |
| Eugenol | [32] | |
| Silybum marianum | [56] | |
| Decreased transmembrane potential | Resveratrol | [65] |
| Genistein | [65] | |
| Taurine | [69] | |
| Genistein | [64] | |
| Omega-3 | [57] | |
| Phloretin | [45] | |
| Boerhavia diffusa | [73] | |
| Elevated phosphorylation of JNK | Genistein | [64] |
| and p-38 MAPK | Taurine | [69] |
| Mitochondria | ||
| Decreased activity at mito. | Naringin | [70] |
| complex I, III, IV | Phloretin | [45] |
| Decreased activity at mito. | Naringin | [70] |
| complex II | Morphine | [60] |
| Decreased ATP content | Phloretin | [45] |
| Decreased Mg2+ ATPase | Silybum marianum | [56] |
| Altered mitochondrial morphology | Naringin | [70] |
| Silybum marianum | [56] | |
| Phloretin | [45] | |
| Decreased OCR | Phloretin | [45] |
| Mito. swelling and pore opening | Phloretin | [45] |
| Decreased activities of heart mitochondrial enzymes | Silybum marianum | [56] |
| Decreased aconitase activity | Phloretin | [45] |
| Increased LC3-II/LC-31 | Genistein 2 | [65] |
| Resveratrol 2 | [65] | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pace, C.; Dagda, R.; Angermann, J. Antioxidants Protect against Arsenic Induced Mitochondrial Cardio-Toxicity. Toxics 2017, 5, 38. https://doi.org/10.3390/toxics5040038
Pace C, Dagda R, Angermann J. Antioxidants Protect against Arsenic Induced Mitochondrial Cardio-Toxicity. Toxics. 2017; 5(4):38. https://doi.org/10.3390/toxics5040038
Chicago/Turabian StylePace, Clare, Ruben Dagda, and Jeff Angermann. 2017. "Antioxidants Protect against Arsenic Induced Mitochondrial Cardio-Toxicity" Toxics 5, no. 4: 38. https://doi.org/10.3390/toxics5040038
APA StylePace, C., Dagda, R., & Angermann, J. (2017). Antioxidants Protect against Arsenic Induced Mitochondrial Cardio-Toxicity. Toxics, 5(4), 38. https://doi.org/10.3390/toxics5040038