
Mefenamic Acid-Upregulated Nrf2/SQSTM1 Protects Hepatocytes against Oxidative Stress-Induced Cell Damage

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemicals
2.2. Chemical Treatment
2.3. Western Blot and Immunoprecipitation
2.4. RNA Preparation and qRT-PCR Analysis
2.5. Transient Transfections and Luciferase Reporter Gene Assay
2.6. Nuclear Fractionation
2.7. Measurement of Cell Viability, Intracellular ROS, and Mitochondrial Membrane Potential
2.8. Statistical Analysis
3. Results
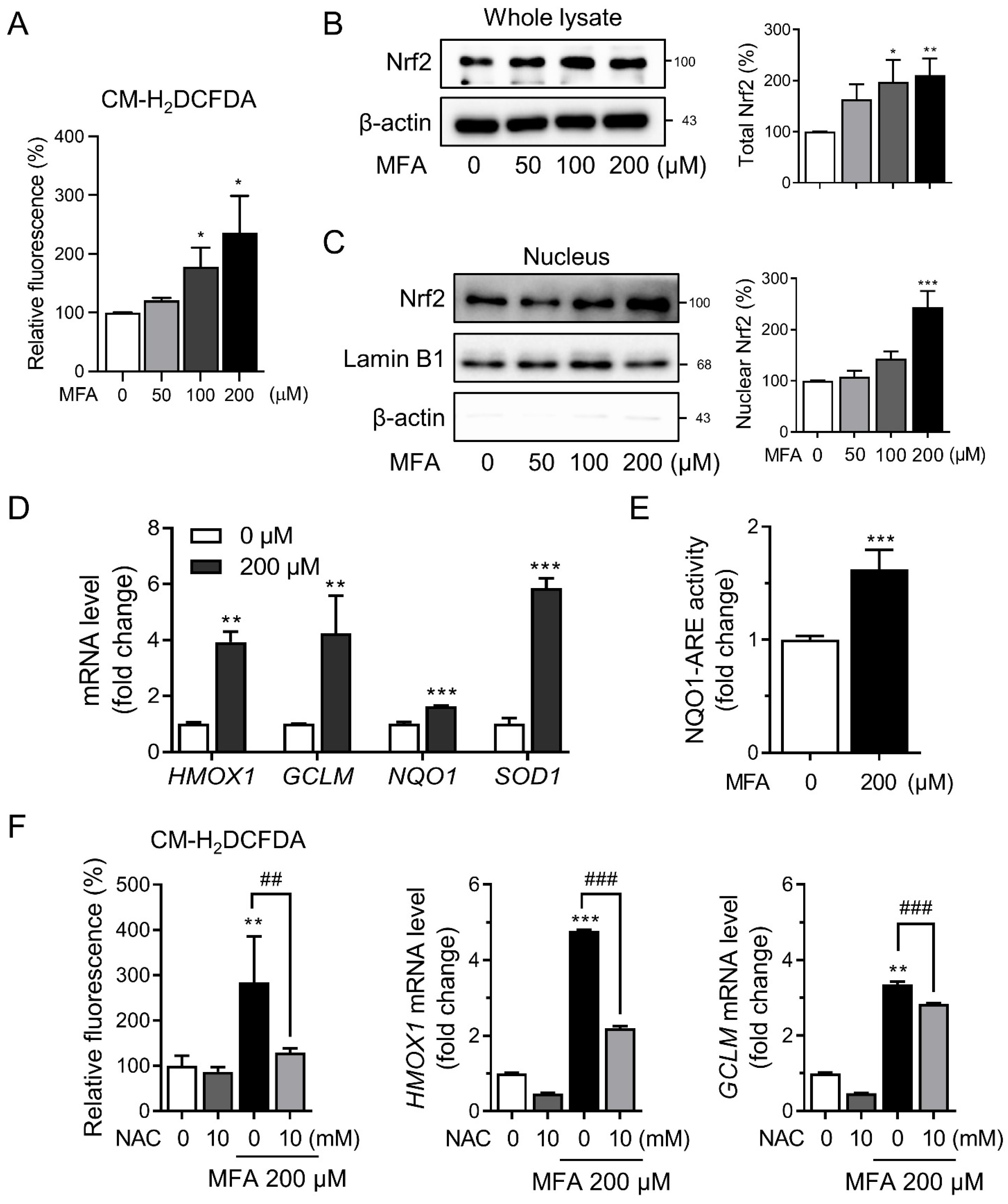
3.1. MFA Activated the Nrf2 Pathway in HepG2 Cells
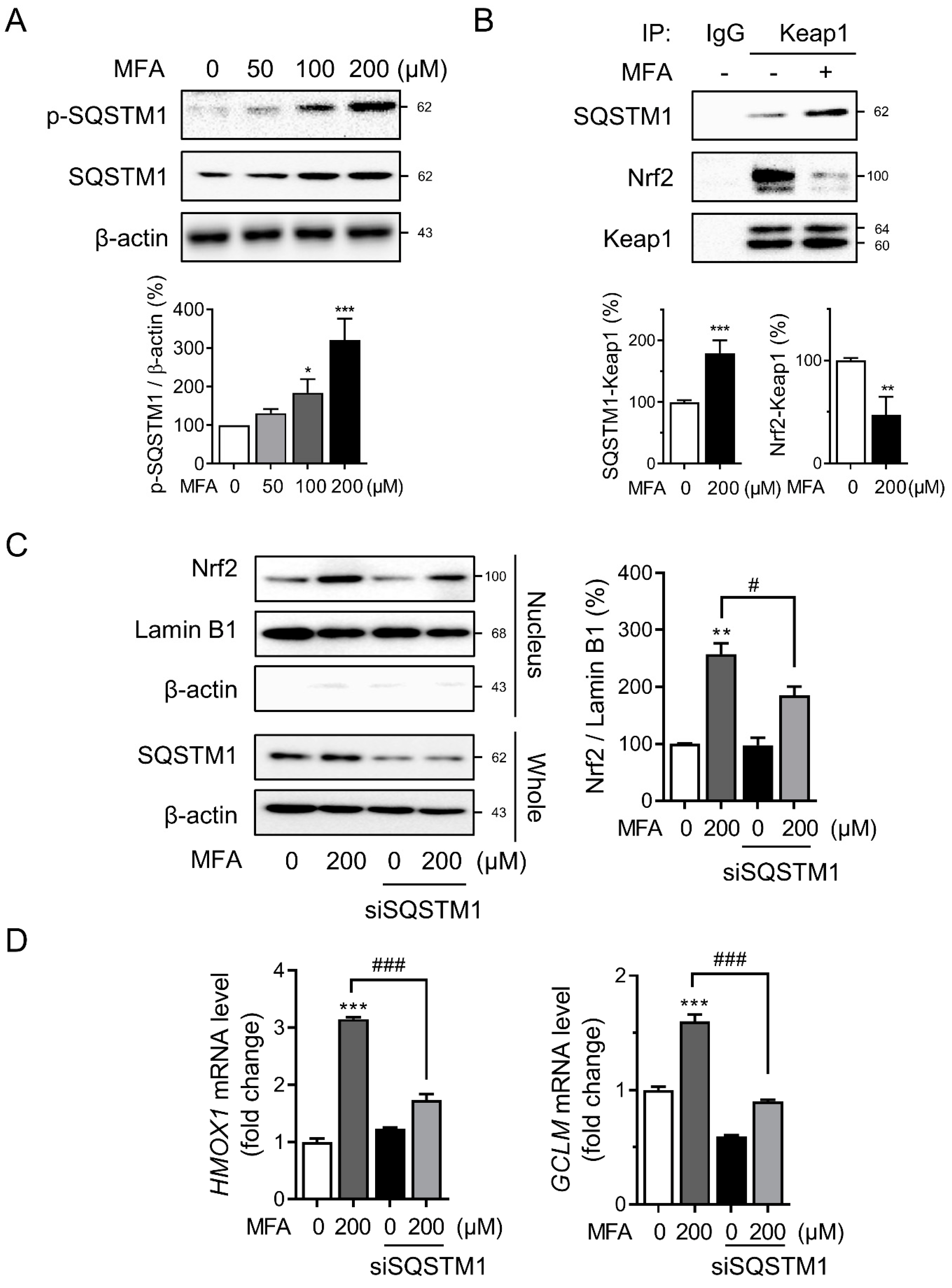
3.2. MFA Increased SQSTM1 Expression in HepG2 Cells
3.3. SQSTM1 Was Required for MFA-Induced Nrf2 Activation
3.4. MFA Alleviated tBHP-Induced Cytotoxicity via SQSTM1-Mediated Nrf2 Activation
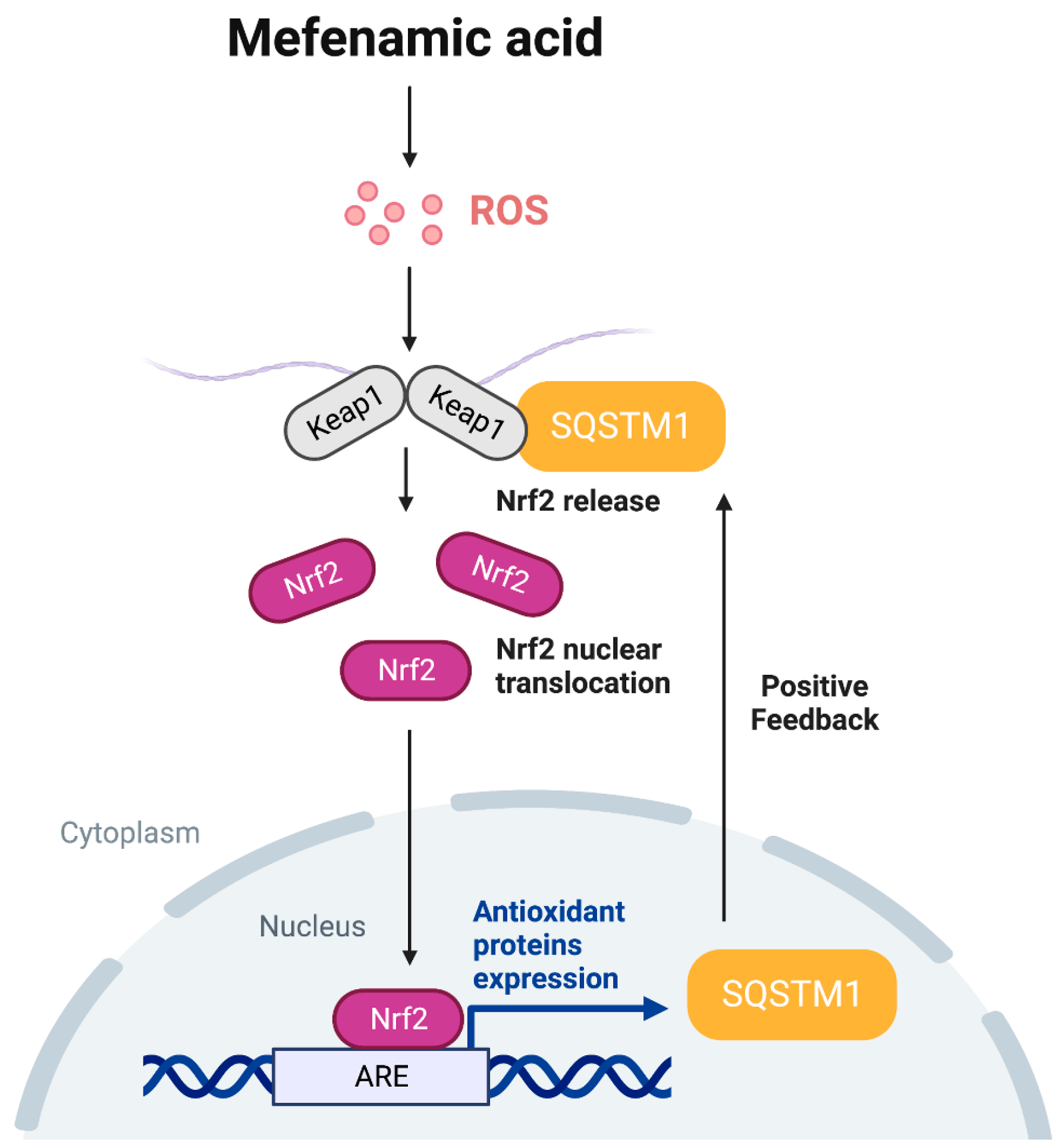
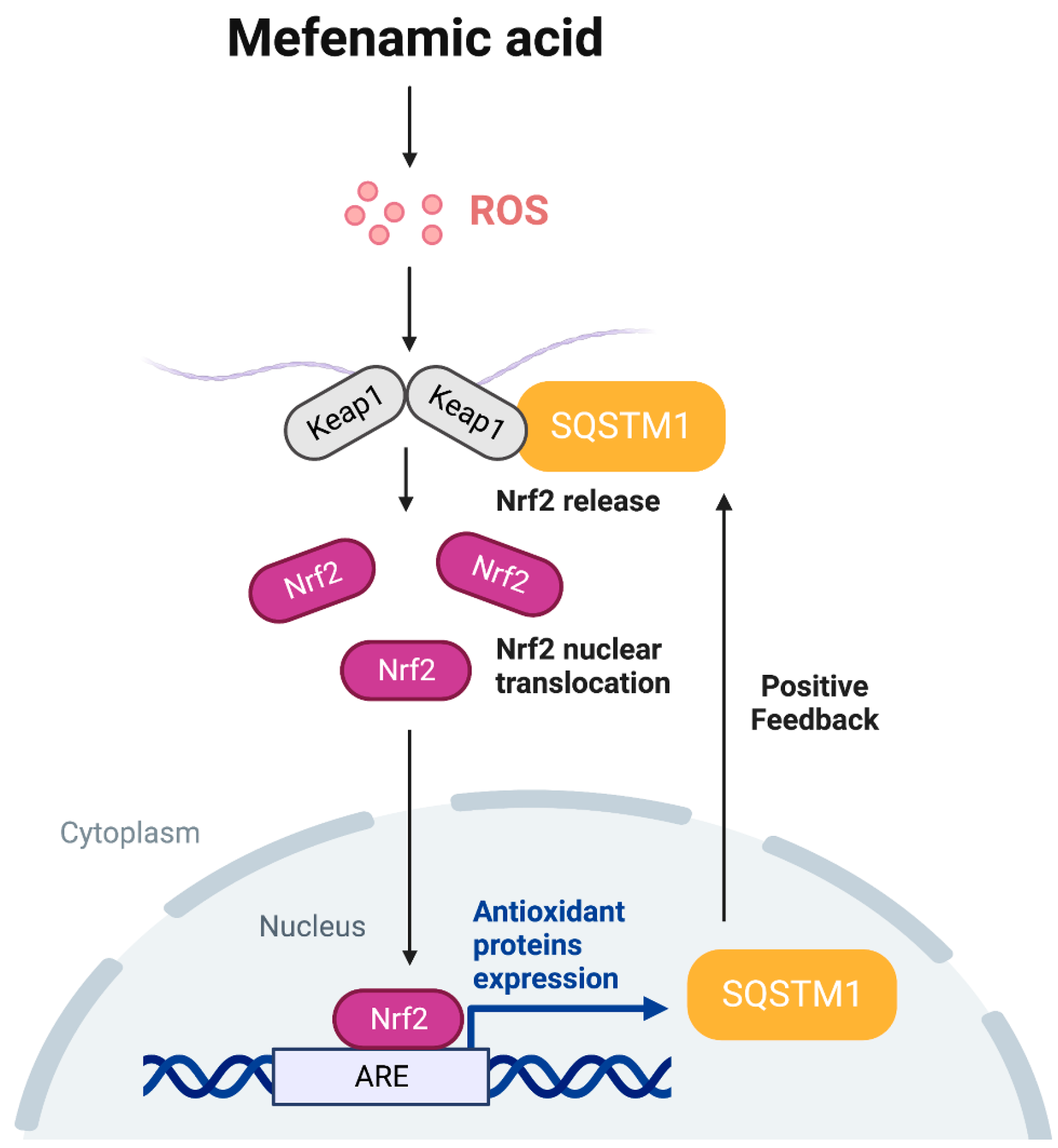
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vial, G.; Dubouchaud, H.; Couturier, K.; Cottet-Rousselle, C.; Taleux, N.; Athias, A.; Galinier, A.; Casteilla, L.; Leverve, X.M. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J. Hepatol. 2011, 54, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Qiu, M.; Liu, Y.; Liu, Y.; Tang, Y.; Teng, X.; Li, S. Nano-selenium protects grass carp hepatocytes against 4-tert-butylphenol-induced mitochondrial apoptosis and necroptosis via suppressing ROS-PARP1 axis. Fish Shellfish Immunol. 2023, 135, 108682. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, X.; Hao, Z.; Yu, M.; Tang, Y.; Teng, X.; Sun, W.; Kang, L. Cadmium exposure caused cardiotoxicity in common carps (Cyprinus carpio L.): miR-9-5p, oxidative stress, energetic impairment, mitochondrial division/fusion imbalance, inflammation, and autophagy. Fish Shellfish Immunol. 2023, 138, 108853. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Teng, X.; Yue, W.; Suo, A.; Zhou, W.; Ding, D. The effect of acute toxicity from tributyltin on Liza haematocheila liver: Energy metabolic disturbance, oxidative stress, and apoptosis. Aquat. Toxicol. 2023, 258, 106506. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M. Nrf2 as a Potential Therapeutic Target for Traumatic Brain Injury. J. Integr. Neurosci. 2023, 22, 81. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Piani, F.; Crescimanno, C.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Modulation of NRF2/KEAP1 Signaling in Preeclampsia. Cells 2023, 12, 1545. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Montanari, E.; Marzioni, D.; Goteri, G. Role of NRF2 in Ovarian Cancer. Antioxidants 2022, 11, 663. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Marzioni, D.; Mazzucchelli, R. Cellular Modulators of the NRF2/KEAP1 Signaling Pathway in Prostate Cancer. Front. Biosci. 2023, 28, 143. [Google Scholar] [CrossRef]
- Ghareghomi, S.; Habibi-Rezaei, M.; Arese, M.; Saso, L.; Moosavi-Movahedi, A.A. Nrf2 Modulation in Breast Cancer. Biomedicines 2022, 10, 2668. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Li, J.; Johnson, D.A.; Stein, T.D.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Johnson, J.A. Nrf2, a multi-organ protector? FASEB J. 2005, 19, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ji, R.; Sun, H.; Peng, J.; Ma, X.; Wang, C.; Fu, Y.; Bao, L.; Jin, Y. Scutellarin ameliorates nonalcoholic fatty liver disease through the PPARγ/PGC-1α-Nrf2 pathway. Free Radic. Res. 2018, 52, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Lin, J.; Wu, D. Sulforaphane induces Nrf2 and protects against CYP2E1-dependent binge alcohol-induced liver steatosis. Biochim. Biophys. Acta 2014, 1840, 209–218. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef]
- Suzuki, M.; Otsuki, A.; Keleku-Lukwete, N.; Yamamoto, M. Overview of redox regulation by Keap1–Nrf2 system in toxicology and cancer. Curr. Opin. Toxicol. 2016, 1, 29–36. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Meunier, L.; Larrey, D. Recent Advances in Hepatotoxicity of Non Steroidal Anti-Inflammatory Drugs. Ann. Hepatol. 2018, 17, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Armagan, G.; Turunc, E.; Kanit, L.; Yalcin, A. Neuroprotection by mefenamic acid against D-serine: Involvement of oxidative stress, inflammation and apoptosis. Free Radic. Res. 2012, 46, 726–739. [Google Scholar] [CrossRef]
- Joo, Y.; Kim, H.S.; Woo, R.S.; Park, C.H.; Shin, K.Y.; Lee, J.P.; Chang, K.A.; Kim, S.; Suh, Y.H. Mefenamic acid shows neuroprotective effects and improves cognitive impairment in in vitro and in vivo Alzheimer’s disease models. Mol. Pharmacol. 2006, 69, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Lee, W.; Park, S.H.; Lee, K.Y.; Choi, Y.J.; Choi, S.; Kang, D.; Kim, S.; Chang, T.S.; Hong, S.S.; et al. Diclofenac impairs autophagic flux via oxidative stress and lysosomal dysfunction: Implications for hepatotoxicity. Redox Biol. 2020, 37, 101751. [Google Scholar] [CrossRef]
- Ghosh, R.; Alajbegovic, A.; Gomes, A.V. NSAIDs and Cardiovascular Diseases: Role of Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2015, 2015, 536962. [Google Scholar] [CrossRef]
- Rai, N.; Sarkar, M.; Raha, S. Piroxicam, a traditional non-steroidal anti-inflammatory drug (NSAID) causes apoptosis by ROS mediated Akt activation. Pharmacol. Rep. 2015, 67, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, X.-F.; Liu, T.-R.; Fan, R.-F.; Xu, Y.-C.; Zhang, X.-Z.; Liu, L.-L. Celecoxib exerts antitumor effects in HL-60 acute leukemia cells and inhibits autophagy by affecting lysosome function. Biomed. Pharmacother. 2016, 84, 1551–1557. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Arauz, J.; Ramos-Tovar, E.; Muriel, P. Redox state and methods to evaluate oxidative stress in liver damage: From bench to bedside. Ann. Hepatol. 2016, 15, 160–173. [Google Scholar] [CrossRef]
- Nishida, K.; Ohta, Y.; Ishiguro, I. Modulating role of endogenous reduced glutathione in tert-butyl hydroperoxide-induced cell injury in isolated rat hepatocytes. Free Radic. Biol. Med. 1997, 23, 453–462. [Google Scholar] [CrossRef]
- Kucera, O.; Endlicher, R.; Rousar, T.; Lotkova, H.; Garnol, T.; Drahota, Z.; Cervinkova, Z. The effect of tert-butyl hydroperoxide-induced oxidative stress on lean and steatotic rat hepatocytes in vitro. Oxid. Med. Cell. Longev. 2014, 2014, 752506. [Google Scholar] [CrossRef]
- Piret, J.P.; Arnould, T.; Fuks, B.; Chatelain, P.; Remacle, J.; Michiels, C. Mitochondria permeability transition-dependent tert-butyl hydroperoxide-induced apoptosis in hepatoma HepG2 cells. Biochem. Pharmacol. 2004, 67, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Imajo, K.; Honda, Y.; Kessoku, T.; Tomeno, W.; Kato, S.; Fujita, K.; Yoneda, M.; Saito, S.; Saigusa, Y.; et al. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci. Rep. 2018, 8, 11365. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. [Google Scholar] [CrossRef] [PubMed]
- Solano-Urrusquieta, A.; Morales-Gonzalez, J.A.; Castro-Narro, G.E.; Cerda-Reyes, E.; Flores-Rangel, P.D.; Fierros-Oceguera, R. NRF-2 and nonalcoholic fatty liver disease. Ann. Hepatol. 2020, 19, 458–465. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1428. [Google Scholar] [CrossRef]
- Cimolai, N. The potential and promise of mefenamic acid. Expert Rev. Clin. Pharmacol. 2013, 6, 289–305. [Google Scholar] [CrossRef]
- Or, S.; Bozkurt, A. Analgesic effect of aspirin, mefenamic acid and their combination in post-operative oral surgery pain. J. Int. Med. Res. 1988, 16, 167–172. [Google Scholar] [CrossRef]
- Hosseinimehr, S.J.; Safavi, Z.; Kangarani Farahani, S.; Noaparst, Z.; Ghasemi, A.; Asgarian-Omran, H. The synergistic effect of mefenamic acid with ionizing radiation in colon cancer. J. Bioenerg. Biomembr. 2019, 51, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.H.; Fatima, M.; Hossain, M.; Mondal, A.C. Evaluation of naproxen-induced oxidative stress, hepatotoxicity and in-vivo genotoxicity in male Wistar rats. J. Pharm. Anal. 2018, 8, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-inducing strategy in anticancer therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Sun, W.; Wang, X.; Long, J.; Liu, X.; Feng, Z.; Liu, J. Punicalagin attenuates palmitate-induced lipotoxicity in HepG2 cells by activating the Keap1-Nrf2 antioxidant defense system. Mol. Nutr. Food Res. 2016, 60, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef]
- Sánchez-Martín, P.; Komatsu, M. p62/SQSTM1–steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef]
- Ni, H.M.; McGill, M.R.; Chao, X.; Du, K.; Williams, J.A.; Xie, Y.; Jaeschke, H.; Ding, W.X. Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J. Hepatol. 2016, 65, 354–362. [Google Scholar] [CrossRef]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Dilution | Company | Catalog # |
|---|---|---|---|
| anti-SQSTM1 | 1:1000 | Cell Signaling Technology | 5114S |
| anti-Keap1 | 1:1000 | Cell Signaling Technology | 4678S |
| anti-p-SQSTM1 | 1:1000 | MBL Life Science | PM074 |
| anti-Nrf2 | 1:1000 | Santa Cruz Biotechnology | sc-365949 |
| anti-β-actin | 1:5000 | Santa Cruz Biotechnology | sc-47778 |
| anti-Lamin B1 | 1:2000 | Santa Cruz Biotechnology | Sc-374015 |
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| SQSTM1 | GACCCACAGGGCTGAAGGAA | CCAGCCGCCTTCATCAGAGA |
| HMOX1 | TCCGATGGGTCCTTACACTC | TAAGGAAGCCAGCCAAGAGA |
| GCLM | CATTTACAGCCTTACTGGGAGG | ATGCAGTCAAATCTGGTGGCA |
| NQO1 | GAAGAGCACTGATCGTACTGGC | GGATACTGAAAGTTCGCAGGG |
| SOD1 | GGTGGGCCAAAGGATGAAGAG | CCACAAGCCAAACGACTTCC |
| ACTB | CACCATTGGCAATGAGCGGTTC | AGGTCTTTGCGGATGTCCACGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.; Mun, Y.; Lee, K.-Y.; Park, J.-M.; Chang, T.-S.; Choi, Y.-J.; Lee, B.-H. Mefenamic Acid-Upregulated Nrf2/SQSTM1 Protects Hepatocytes against Oxidative Stress-Induced Cell Damage. Toxics 2023, 11, 735. https://doi.org/10.3390/toxics11090735
Lee W, Mun Y, Lee K-Y, Park J-M, Chang T-S, Choi Y-J, Lee B-H. Mefenamic Acid-Upregulated Nrf2/SQSTM1 Protects Hepatocytes against Oxidative Stress-Induced Cell Damage. Toxics. 2023; 11(9):735. https://doi.org/10.3390/toxics11090735
Chicago/Turabian StyleLee, Wonseok, Yewon Mun, Kang-Yo Lee, Jung-Min Park, Tong-Shin Chang, You-Jin Choi, and Byung-Hoon Lee. 2023. "Mefenamic Acid-Upregulated Nrf2/SQSTM1 Protects Hepatocytes against Oxidative Stress-Induced Cell Damage" Toxics 11, no. 9: 735. https://doi.org/10.3390/toxics11090735
APA StyleLee, W., Mun, Y., Lee, K.-Y., Park, J.-M., Chang, T.-S., Choi, Y.-J., & Lee, B.-H. (2023). Mefenamic Acid-Upregulated Nrf2/SQSTM1 Protects Hepatocytes against Oxidative Stress-Induced Cell Damage. Toxics, 11(9), 735. https://doi.org/10.3390/toxics11090735