
Pancreas–Liver–Adipose Axis: Target of Environmental Cadmium Exposure Linked to Metabolic Diseases
 , ,
, , 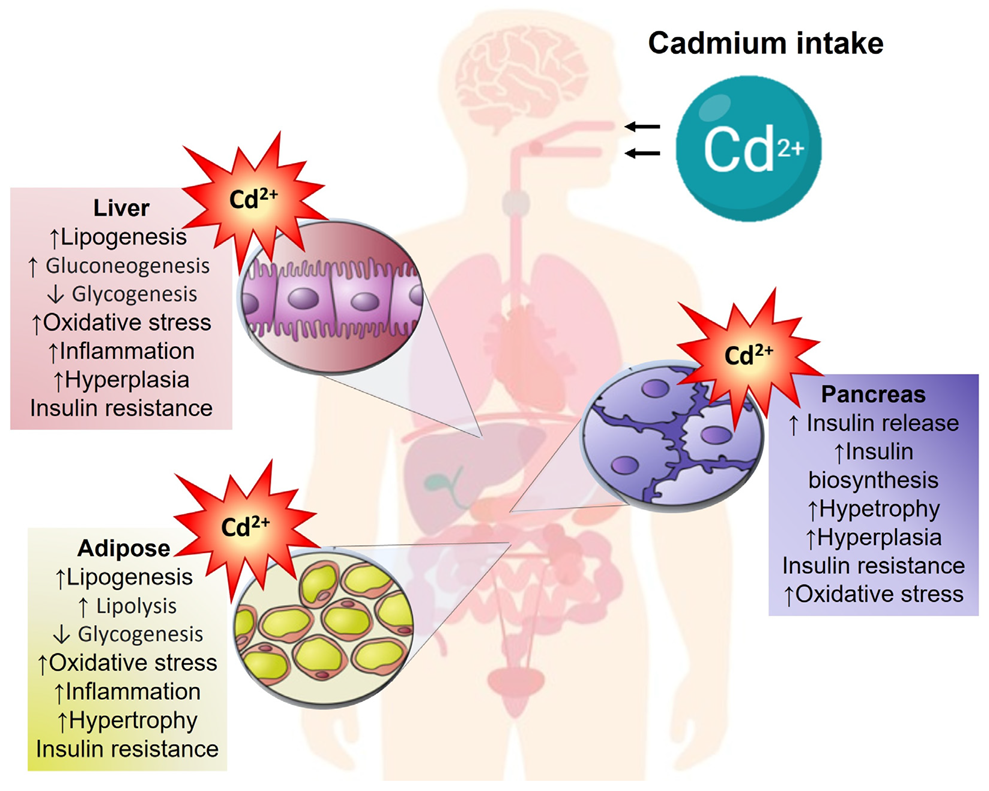{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Minimal Risk Levels of Environmental Cadmium Exposure
3. Cadmium Intake, Absorption, and Distribution
4. Cadmium Toxicity
5. Cadmium and Inflammation
6. Cadmium and Oxidative Stress
7. Antioxidative Defense
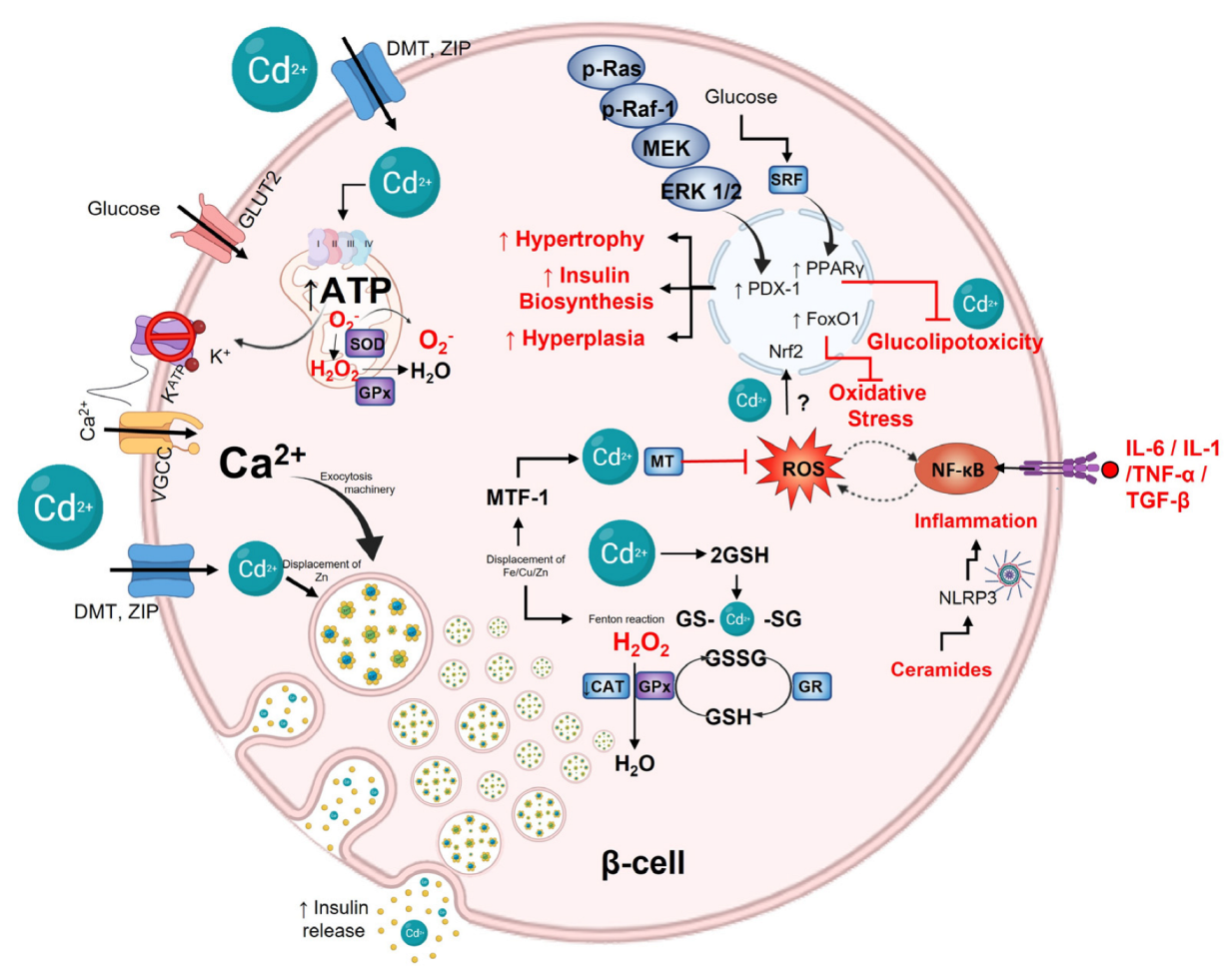
8. Cadmium and Pancreatic β-Cells
9. Protective and Antioxidant β-Cell Capacity
9.1. Nonenzymatic Antioxidant Defense in β-Cell
9.2. Antioxidant Enzymes
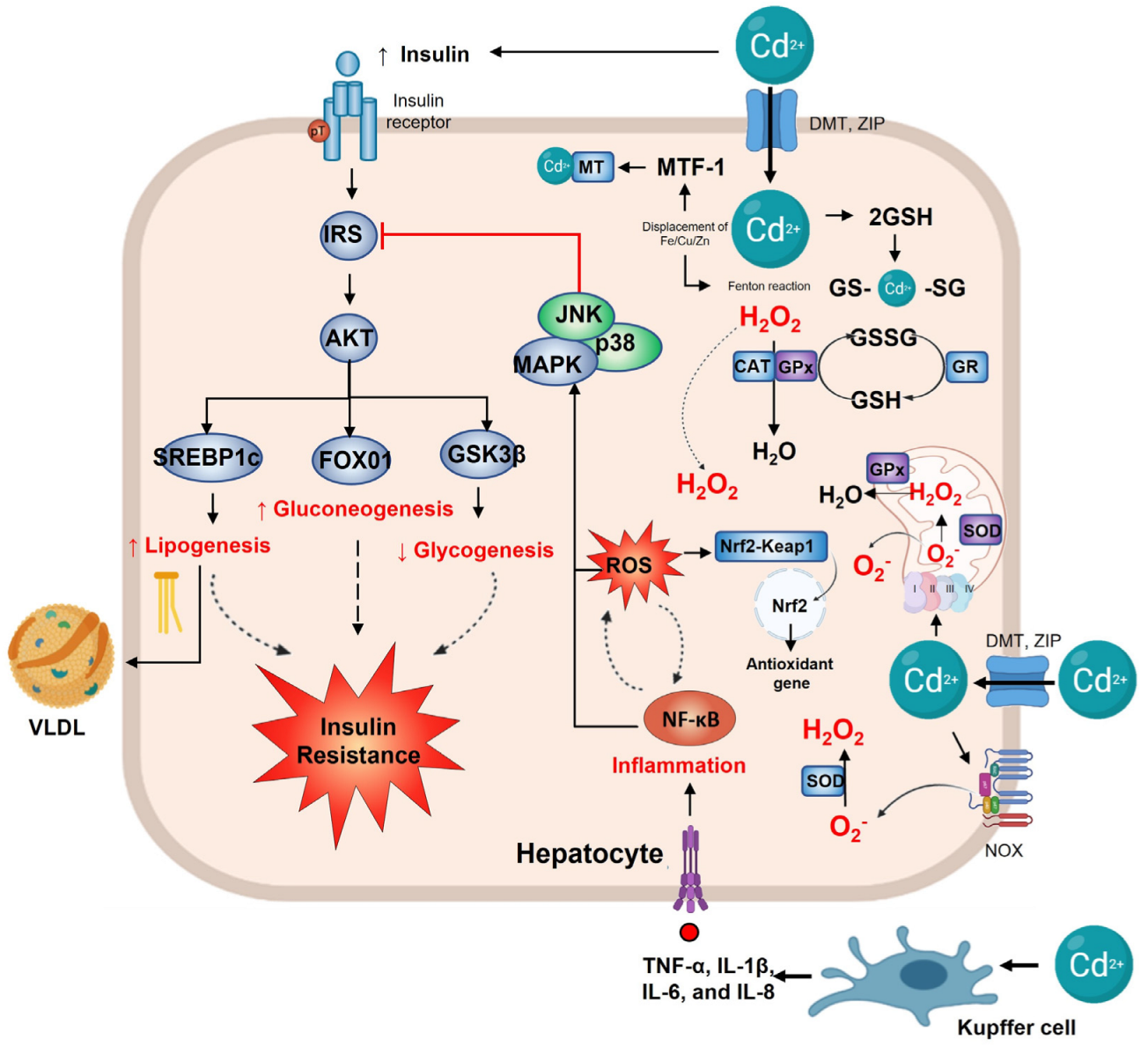
10. Cadmium and Liver
11. Cadmium Impairs Hepatic Glucose Homeostasis
12. Cadmium, De Novo Lipogenesis, and Dyslipidemia
13. Hepatic Antioxidant Defense
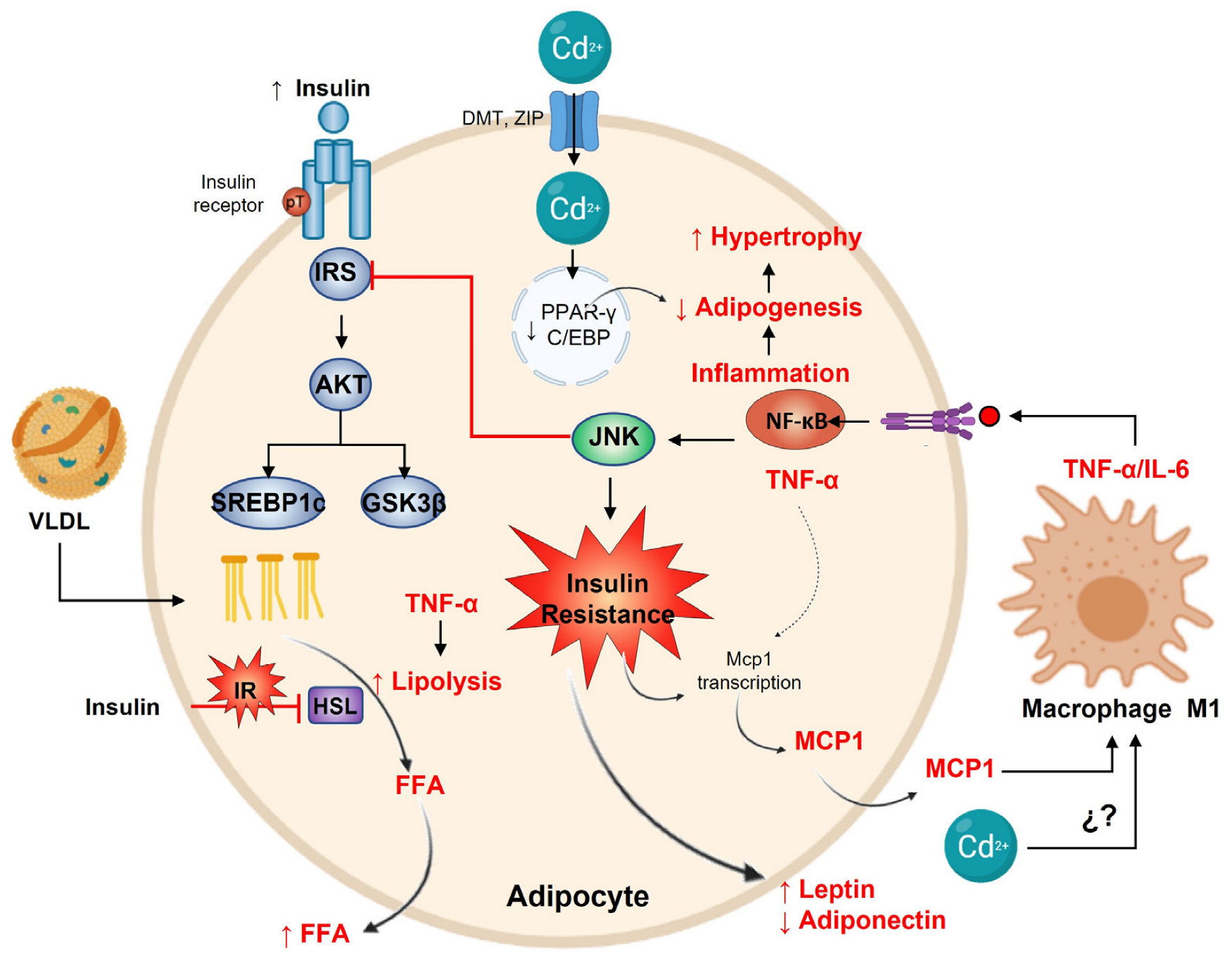
14. Cadmium and Adipose Tissue
15. Adipose Tissue Antioxidant Defense
16. Cadmium and Obesity
17. Cadmium and Diabetes
18. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ATSDR, Agency for Toxic Substance and Disease Registry. Toxicological Profile for Cadmium; Department of Health and Humans Services, Public Health Service, Centers for Disease Control: Atlanta, GA, USA, 2012.
- Nordberg, G.F.; Fowler, B.A.; Nordberg, M. Handbook on the Toxicology of Metals; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Smolders, E.; Mertens, J. Cadmium. In Heavy Metals in Soils; Springer: Dordrecht, The Netherlands, 2013; pp. 283–311. [Google Scholar] [CrossRef]
- Thévenod, F.; Lee, W.-K. Cadmium and Cellular Signaling Cascades: Interactions between Cell Death and Survival Pathways. Arch. Toxicol. 2013, 87, 1743–1786. [Google Scholar] [CrossRef] [PubMed]
- Egger, A.E.; Grabmann, G.; Pechriggl, E.J.; Artner, C.; Bernhard, D. Chemical Imaging and Assessment of Cadmium Distribution in the Human Body. Metallomics 2019, 11, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Gong, W.; Li, B.; Zuo, J.; Pan, L.; Liu, X. Analysis of Heavy Metals in Foodstuffs and an Assessment of the Health Risks to the General Public via Consumption in Beijing, China. Int. J. Environ. Res. Public Health 2019, 16, 909. [Google Scholar] [CrossRef] [PubMed]
- WHO. Chapter 6.3 Cadmium General Description; WHO: Copenhagen, Denmark, 2000. [Google Scholar]
- FAO Joint; WHO Expert Committee on Food Additives. Ninety-First Meeting (Safety Evaluation of Certain Food Additives and Contaminants); World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Asagba, S.O. Cadmium Absorption. In Encyclopedia of Metalloproteins; Springer Science + Business Media: New York, NY, USA, 2013; pp. 332–337. [Google Scholar] [CrossRef]
- Thévenod, F.; Fels, J.; Lee, W.-K.; Zarbock, R. Channels, Transporters and Receptors for Cadmium and Cadmium Complexes in Eukaryotic Cells: Myths and Facts. BioMetals 2019, 32, 469–489. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, M.; Nordberg, G.F. Metallothionein and Cadmium Toxicology-Historical Review and Commentary. Biomolecules 2022, 12, 360. [Google Scholar] [CrossRef] [PubMed]
- Freisinger, E.; Vašák, M. Cadmium in Metallothioneins. Met. Ions Life Sci. 2013, 11, 339–371. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuča, K.; Musílek, K. Redox- and Non-Redox-Metal-Induced Formation of Free Radicals and Their Role in Human Disease. Arch. Toxicol. 2016, 3, 1–37. [Google Scholar] [CrossRef]
- Đukić-Ćosić, D.; Baralić, K.; Javorac, D.; Djordjevic, A.B.; Bulat, Z. An Overview of Molecular Mechanisms in Cadmium Toxicity. Curr. Opin. Toxicol. 2020, 19, 56–62. [Google Scholar] [CrossRef]
- Thévenod, F. Cadmium and Cellular Signaling Cascades: To Be or Not to Be? Toxicol. Appl. Pharmacol. 2009, 238, 221–239. [Google Scholar] [CrossRef]
- Hartwig, A. Cadmium and Cancer. Met. Ions Life Sci. 2013, 11, 491–507. [Google Scholar] [CrossRef]
- Hartwig, A. Mechanisms in Cadmium-Induced Carcinogenicity: Recent Insights. BioMetals 2010, 23, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free Radicals, Metals and Antioxidants in Oxidative Stress-Induced Cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Razzuoli, E.; Mignone, G.; Lazzara, F.; Vencia, W.; Ferraris, M.; Masiello, L.; Vivaldi, B.; Ferrari, A.; Bozzetta, E.; Amadori, M. Impact of Cadmium Exposure on Swine Enterocytes. Toxicol. Lett. 2018, 287, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Hossein-Khannazer, N.; Azizi, G.; Eslami, S.; Alhassan Mohammed, H.; Fayyaz, F.; Hosseinzadeh, R.; Usman, A.B.; Kamali, A.N.; Mohammadi, H.; Jadidi-Niaragh, F.; et al. The Effects of Cadmium Exposure in the Induction of Inflammation. Immunopharmacol. Immunotoxicol. 2019, 42, 1–8. [Google Scholar] [CrossRef]
- Miyahara, T.; Katoh, T.; Watanabe, M.; Mikami, Y.; Uchida, S.; Hosoe, M.; Sakuma, T.; Nemoto, N.; Takayama, K.; Komurasaki, T. Involvement of Mitogen-Activated Protein Kinases and Protein Kinase C in Cadmium-Induced Prostaglandin E2 Production in Primary Mouse Osteoblastic Cells. Toxicology 2004, 200, 159–167. [Google Scholar] [CrossRef]
- Rockwell, P.; Martinez, J.; Papa, L.; Gomes, E. Redox Regulates COX-2 Upregulation and Cell Death in the Neuronal Response to Cadmium. Cell. Signal. 2004, 16, 343–353. [Google Scholar] [CrossRef]
- Cormet-boyaka, E.; Jolivette, K.; Bonnegarde-bernard, A.; Rennolds, J.; Hassan, F.; Mehta, P.; Tridandapani, S.; Webster-marketon, J.; Boyaka, P.N. An NF-ΚB-Independent and Erk1/2-Dependent Mechanism Controls CXCL8/IL-8 Responses of Airway Epithelial Cells to Cadmium. Toxicol. Sci. 2012, 125, 418–429. [Google Scholar] [CrossRef]
- Nair, A.R.; DeGheselle, O.; Smeets, K.; Van Kerkhove, E.; Cuypers, A. Cadmium-Induced Pathologies: Where Is the Oxidative Balance Lost (or Not)? Int. J. Mol. Sci. 2013, 14, 6116. [Google Scholar] [CrossRef]
- Cannino, G.; Ferruggia, E.; Luparello, C.; Rinaldi, A.M. Cadmium and Mitochondria. Mitochondrion 2009, 9, 377–384. [Google Scholar] [CrossRef]
- Dorta, D.J.; Leite, S.; DeMarco, K.C.; Prado, I.M.R.; Rodrigues, T.; Mingatto, F.E.; Uyemura, S.A.; Santos, A.C.; Curti, C. A Proposed Sequence of Events for Cadmium-Induced Mitochondrial Impairment. J. Inorg. Biochem. 2003, 97, 251–257. [Google Scholar] [CrossRef]
- Souza, V.; Flores, K.; Ortiz, L.; Gómez-Quiroz, L.; Gutierrez-Ruiz, M. Liver and Cadmium Toxicity. J. Drug Metab. Toxicol. 2012, S5, 5. [Google Scholar] [CrossRef]
- Wang, H.; Wang, A.; Wang, X.; Zeng, X.; Xing, H. AMPK/PPAR-γ/NF-ΚB Axis Participates in ROS-Mediated Apoptosis and Autophagy Caused by Cadmium in Pig Liver. Environ. Pollut. 2022, 294, 118659. [Google Scholar] [CrossRef] [PubMed]
- Poliandri, A.H.B.; Esquifino, A.I.; Cano, P.; Jiménez, V.; Lafuente, A.; Cardinali, D.P.; Duvilanski, B.H. In Vivo Protective Effect of Melatonin on Cadmium-Induced Changes in Redox Balance and Gene Expression in Rat Hypothalamus and Anterior Pituitary. J. Pineal Res. 2006, 41, 238–246. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A. Mechanism of Cadmium Induced Apoptosis in the Immunocyte. Toxicol. Lett. 2008, 177, 83–89. [Google Scholar] [CrossRef]
- El-Kott, A.F.; Alshehri, A.S.; Khalifa, H.S.; Abd-Lateif, A.E.; Karim, M.; Alshehri, M.A.; El-Maksoud, M.M.A.; Eid, R.A.; Bin-Meferij, M.M. Cadmium Chloride Induces Memory Deficits and Hippocampal Damage by Activating the JNK/P66Shc/NADPH Oxidase Axis. Int. J. Toxicol. 2020, 39, 477–490. [Google Scholar] [CrossRef]
- Mohammadi-Bardbori, A.; Rannug, A. Arsenic, Cadmium, Mercury and Nickel Stimulate Cell Growth via NADPH Oxidase Activation. Chem. Biol. Interact. 2014, 224, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Souza, V.; Escobar, M.d.C.; Bucio, L.; Hernández, E.; Gómez-Quiroz, L.E.; Gutiérrez Ruiz, M.C. NADPH Oxidase and ERK1/2 Are Involved in Cadmium Induced-STAT3 Activation in HepG2 Cells. Toxicol. Lett. 2009, 187, 180–186. [Google Scholar] [CrossRef]
- Gupta, D.K.; Pena, L.B.; Romero-Puertas, M.C.; Hernández, A.; Inouhe, M.; Sandalio, L.M. NADPH Oxidases Differentially Regulate ROS Metabolism and Nutrient Uptake under Cadmium Toxicity. Plant Cell Environ. 2016, 40, 509–526. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312. [Google Scholar] [CrossRef]
- Andrews, G.K. Regulation of Metallothionein Gene Expression by Oxidative Stress and Metal Ions. Biochem. Pharmacol. 2000, 59, 95–104. [Google Scholar] [CrossRef]
- Hauck, A.K.; Bernlohr, D.A. Oxidative Stress and Lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium Stress: An Oxidative Challenge. BioMetals 2010, 23, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qu, W.; Kadiiska, M.B. Role of Oxidative Stress in Cadmium Toxicity and Carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 209–214. [Google Scholar] [CrossRef]
- Liu, L.; Tao, R.; Huang, J.; He, X.; Qu, L.; Jin, Y.; Zhang, S.; Fu, Z. Hepatic Oxidative Stress and Inflammatory Responses with Cadmium Exposure in Male Mice. Environ. Toxicol. Pharmacol. 2015, 39, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Ma, H.; Liu, G.; Fan, S.; Guo, Z. Mechanism of Cadmium Exposure Induced Hepatotoxicity in the Mud Crab (Scylla Paramamosain): Activation of Oxidative Stress and Nrf2 Signaling Pathway. Antioxidants 2022, 11, 978. [Google Scholar] [CrossRef]
- Wimmer, U.; Wang, Y.; Georgiev, O.; Schaffner, W. Two Major Branches of Anti-Cadmium Defense in the Mouse: MTF-1/Metallothioneins and Glutathione. Nucleic Acids Res. 2005, 33, 5715–5727. [Google Scholar] [CrossRef]
- Wang, Y.; Mandal, A.K.; Son, Y.O.K.; Pratheeshkumar, P.; Wise, J.T.F.; Wang, L.; Zhang, Z.; Shi, X.; Chen, Z. Roles of ROS, Nrf2, and Autophagy in Cadmium-Carcinogenesis and Its Prevention by Sulforaphane. Toxicol. Appl. Pharmacol. 2018, 353, 23–30. [Google Scholar] [CrossRef]
- Montes, S.; Juárez-Rebollar, D.; Nava-Ruíz, C.; Sánchez-García, A.; Heras-Romero, Y.; Rios, C.; Méndez-Armenta, M. Immunohistochemical Study of Nrf2-Antioxidant Response Element as Indicator of Oxidative Stress Induced by Cadmium in Developing Rats. Oxidative Med. Cell. Longev. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Son, Y.O.; Pratheeshkumar, P.; Roy, R.V.; Hitron, J.A.; Wang, L.; Zhang, Z.; Shi, X. Nrf2/P62 Signaling in Apoptosis Resistance and Its Role in Cadmium-Induced Carcinogenesis. J. Biol. Chem. 2014, 289, 28660–28675. [Google Scholar] [CrossRef]
- Saed-Moucheshi, A.; Sohrabi, F.; Fasihfar, E.; Baniasadi, F.; Riasat, M.; Mozafari, A.A. Superoxide Dismutase (SOD) as a Selection Criterion for Triticale Grain Yield under Drought Stress: A Comprehensive Study on Genomics and Expression Profiling, Bioinformatics, Heritability, and Phenotypic Variability. BMC Plant Biol. 2021, 21, 148. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Candas, D.; Li, J.J. MnSOD in Oxidative Stress Response-Potential Regulation via Mitochondrial Protein Influx. Antioxid. Redox Signal. 2014, 20, 1599–1617. [Google Scholar] [CrossRef]
- Bresciani, G.; da Cruz, I.; González-Gallego, J. Manganese Superoxide Dismutase and Oxidative Stress Modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar] [CrossRef] [PubMed]
- López, E.; Arce, C.; Oset-Gasque, M.; Cañadas, S.; González, M. Cadmium Induces Reactive Oxygen Species Generation and Lipid Peroxidation in Cortical Neurons in Culture. Free Radic. Biol. Med. 2006, 40, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Jurczuk, M.; Brzóska, M.M.; Moniuszko-Jakoniuk, J.; Gałażyn-Sidorczuk, M.; Kulikowska-Karpińska, E. Antioxidant Enzymes Activity and Lipid Peroxidation in Liver and Kidney of Rats Exposed to Cadmium and Ethanol. Food Chem. Toxicol. 2004, 42, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Gungor, H.; Kara, H. Effects of Selenium, Zinc, Insulin and Metallothionein on Cadmium-Induced Oxidative Stress and Metallothionein Gene Expression Levels in Diabetic Rats. J. Basic Clin. Physiol. Pharmacol. 2020, 31, 2. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Shih, C.M.; Huang, C.J.; Lin, C.M.; Chou, C.M.; Tsai, M.L.; Liu, T.P.; Chiu, J.F.; Chen, C.T. Effects of Cadmium on Structure and Enzymatic Activity of Cu,Zn-SOD and Oxidative Status in Neural Cells. J. Cell. Biochem. 2006, 98, 577–589. [Google Scholar] [CrossRef]
- Jihen, E.H.; Sonia, S.; Fatima, H.; Mohamed Tahar, S.; Abdelhamid, K. Interrelationships between Cadmium, Zinc and Antioxidants in the Liver of the Rat Exposed Orally to Relatively High Doses of Cadmium and Zinc. Ecotoxicol. Environ. Saf. 2011, 74, 2099–2104. [Google Scholar] [CrossRef]
- Glorieux, C.; Zamocky, M.; Sandoval, J.; Verrax, J.; Calderon, P. Regulation of Catalase Expression in Healthy and Cancerous Cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef]
- Schrader, M.; Fahimi, H. Mammalian Peroxisomes and Reactive Oxygen Species. Histochem. Cell Biol. 2004, 122, 383–393. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the Basics of Redox Biology to Medical and Graduate Students: Oxidants, Antioxidants and Disease Mechanisms. Redox Biol. 2013, 1, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Svensson, B.E. Abilities of Peroxidases to Catalyse Peroxidase-Oxidase Oxidation of Thiols. Biochem. J. 1988, 256, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P. Catalase, a Remarkable Enzyme: Targeting the Oldest Antioxidant Enzyme to Find a New Cancer Treatment Approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M. Effects of Antioxidant Enzymes in the Molecular Control of Reactive Oxygen Species Toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Panday, S.; Talreja, R.; Kavdia, M. The Role of Glutathione and Glutathione Peroxidase in Regulating Cellular Level of Reactive Oxygen and Nitrogen Species. Microvasc. Res. 2020, 131, 104010. [Google Scholar] [CrossRef]
- Vairavamurthy, M.A.; Goldenberg, W.S.; Ouyang, S.; Khalid, S. The Interaction of Hydrophilic Thiols with Cadmium: Investigation with a Simple Model, 3-Mercaptopropionic Acid. Mar. Chem. 2000, 70, 181–189. [Google Scholar] [CrossRef]
- Mah, V.; Jalilehvand, F. Cadmium(II) Complex Formation with Glutathione. J. Biol. Inorg. Chem. 2010, 15, 441–458. [Google Scholar] [CrossRef]
- Leverrier, P.; Montigny, C.; Garrigos, M.; Champeil, P. Metal Binding to Ligands: Cadmium Complexes with Glutathione Revisited. Anal. Biochem. 2007, 371, 215–228. [Google Scholar] [CrossRef]
- Delalande, O.; Desvaux, H.; Godat, E.; Valleix, A.; Junot, C.; Labarre, J.; Boulard, Y. Cadmium—Glutathione Solution Structures Provide New Insights into Heavy Metal Detoxification. FEBS J. 2010, 277, 5086–5096. [Google Scholar] [CrossRef]
- Adamis, P.D.B.; Gomes, D.S.; Pinto, M.L.C.C.; Panek, A.D.; Eleutherio, E.C.A. The Role of Glutathione Transferases in Cadmium Stress. Toxicol. Lett. 2004, 154, 81–88. [Google Scholar] [CrossRef]
- Sandbichler, A.M.; Höckner, M. Cadmium Protection Strategies—A Hidden Trade-Off? Int. J. Mol. Sci. 2016, 17, 139. [Google Scholar] [CrossRef] [PubMed]
- Henkel, G.; Krebs, B. Metallothioneins: Zinc, Cadmium, Mercury, and Copper Thiolates and Selenolates Mimicking Protein Active Site Features—Structural Aspects and Biological Implications. Chem. Rev. 2004, 104, 801–824. [Google Scholar] [CrossRef] [PubMed]
- Sabolić, I.; Breljak, D.; Škarica, M.; Herak-Kramberger, C.M. Role of Metallothionein in Cadmium Traffic and Toxicity in Kidneys and Other Mammalian Organs. BioMetals 2010, 23, 897–926. [Google Scholar] [CrossRef]
- Lei, L.; Jin, T.; YF, Z. Insulin Expression in Rats Exposed to Cadmium—PubMed. Biomed. Environ. Sci. 2007, 20, 295–301. [Google Scholar] [PubMed]
- Lei, L.J.; Jin, T.Y.; Zhou, Y.F. Effects of Cadmium on Levels of Insulin in Rats. Wei Sheng Yan Jiu 2005, 34, 394–396. [Google Scholar] [PubMed]
- El Muayed, M.; Raja, M.R.; Zhang, X.; MacRenaris, K.W.; Bhatt, S.; Chen, X.; Urbanek, M.; O’Halloran, T.V.; Lowe, W.L. Accumulation of Cadmium in Insulin-Producing β Cells. Islets 2012, 4, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.V. Zinc and Insulin in Pancreatic Beta-Cells. Endocrine 2014, 45, 178–189. [Google Scholar] [CrossRef]
- Dodson, G.; Steiner, D. The Role of Assembly in Insulin’s Biosynthesis. Curr. Opin. Struct. Biol. 1998, 8, 189–194. [Google Scholar] [CrossRef]
- Moulis, J.-M. Cellular Mechanisms of Cadmium Toxicity Related to the Homeostasis of Essential Metals. BioMetals 2010, 23, 877–896. [Google Scholar] [CrossRef]
- Sarmiento-Ortega, V.E.; Treviño, S.; Flores-Hernández, J.Á.; Aguilar-Alonso, P.; Moroni-González, D.; Aburto-Luna, V.; Diaz, A.; Brambila, E. Changes on Serum and Hepatic Lipidome after a Chronic Cadmium Exposure in Wistar Rats. Arch. Biochem. Biophys. 2017, 635, 52–59. [Google Scholar] [CrossRef]
- Treviño, S.; Waalkes, M.P.; Flores Hernández, J.A.; León-Chavez, B.A.; Aguilar-Alonso, P.; Brambila, E. Chronic Cadmium Exposure in Rats Produces Pancreatic Impairment and Insulin Resistance in Multiple Peripheral Tissues. Arch. Biochem. Biophys. 2015, 583, 27–35. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Pathogenesis of Type 2 Diabetes Mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- Paschen, M.; Moede, T.; Valladolid-Acebes, I.; Leibiger, B.; Moruzzi, N.; Jacob, S.; García-Prieto, C.F.; Brismar, K.; Leibiger, I.B.; Berggren, P.-O. Diet-Induced β-Cell Insulin Resistance Results in Reversible Loss of Functional β-Cell Mass. FASEB J. 2019, 33, 204–218. [Google Scholar] [CrossRef]
- Shanik, J.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is Hyperinsulinemia the Cart or the Horse? Diabetes Care 2008, 31 (Suppl. S2), S262–S268. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Ferrannini, E.; Miyazaki, Y.; Matsuda, M.; DeFronzo, R.A. San Antonio metabolism study Beta-Cell Dysfunction and Glucose Intolerance: Results from the San Antonio Metabolism (SAM) Study. Diabetologia 2004, 47, 31–39. [Google Scholar] [CrossRef]
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef]
- Swisa, A.; Glaser, B.; Dor, Y. Metabolic Stress and Compromised Identity of Pancreatic Beta Cells. Front. Genet. 2017, 08, 21. [Google Scholar] [CrossRef]
- Kahn, S.E. The Relative Contributions of Insulin Resistance and Beta-Cell Dysfunction to the Pathophysiology of Type 2 Diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef]
- Corkey, B.E. Metabolic Regulation of Insulin Secretion. In Pancreatic Beta Cell in Health and Disease; Springer: Japan, Tokyo, 2007; pp. 53–74. [Google Scholar]
- Ali, I.; Damdimopoulou, P.; Stenius, U.; Halldin, K. Cadmium at Nanomolar Concentrations Activates Raf–MEK–ERK1/2 MAPKs Signaling via EGFR in Human Cancer Cell Lines. Chem. Biol. Interact. 2015, 231, 44–52. [Google Scholar] [CrossRef]
- Thévenod, F.; Friedmann, J.; Katsen, A.; Hauser, I. Up-Regulation of Multidrug Resistance P-Glycoprotein via Nuclear Factor-KappaB Activation Protects Kidney Proximal Tubule Cells from Cadmium- and Reactive Oxygen Species-Induced Apoptosis. J. Biol. Chem. 2000, 275, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-K.; Thévenod, F. Cell Organelles as Targets of Mammalian Cadmium Toxicity. Arch. Toxicol. 2020, 94, 1017–1049. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Chong, W.L.; Hu, J.; Hinault, C.; Michael, M.D.; Krützfeldt, J.; Yin, C.; Holzenberger, M.; Stoffel, M.; Kulkarni, R.N. Insulin Receptors in β-Cells Are Critical for Islet Compensatory Growth Response to Insulin Resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 8977–8982. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, B.R.; Brun, T.; Sarret, E.J.; Ishihara, H.; Schaad, O.; Descombes, P.; Wollheim, C.B. Oligonucleotide Microarray Analysis Reveals PDX1 as an Essential Regulator of Mitochondrial Metabolism in Rat Islets. J. Biol. Chem. 2004, 279, 31121–31130. [Google Scholar] [CrossRef]
- Poitout, V.; Hagman, D.; Stein, R.; Artner, I.; Robertson, R.P.; Harmon, J.S. Regulation of the Insulin Gene by Glucose and Fatty Acids. J. Nutr. 2006, 136, 873–876. [Google Scholar] [CrossRef]
- Sarkar, A.; Zhang, M.; Liu, S.; Sarkar, S.; Brunicardi, F.C.; Berger, D.H.; Belaguli, N.S. Serum Response Factor Expression Is Enriched in Pancreatic β Cells and Regulates Insulin Gene Expression. FASEB J. 2011, 25, 2592–2603. [Google Scholar] [CrossRef]
- Treisman, R. The Serum Response Element. Trends Biochem. Sci. 1992, 17, 423–426. [Google Scholar] [CrossRef]
- Vivas, Y.; Martínez-García, C.; Izquierdo, A.; Garcia-Garcia, F.; Callejas, S.; Velasco, I.; Campbell, M.; Ros, M.; Dopazo, A.; Dopazo, J.; et al. Early Peroxisome Proliferator-Activated Receptor Gamma Regulated Genes Involved in Expansion of Pancreatic Beta Cell Mass. BMC Med. Genom. 2011, 4, 86. [Google Scholar] [CrossRef]
- Kitamura, T.; Ido Kitamura, Y. Role of FoxO Proteins in Pancreatic β Cells. Endocr. J. 2007, 54, 507–515. [Google Scholar] [CrossRef]
- Kitamura, T. The Role of FOXO1 in β-Cell Failure and Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2013, 9, 615–623. [Google Scholar] [CrossRef]
- Lv, L.; Chen, H.; Sun, J.; Lu, D.; Chen, C.; Liu, D. PRMT1 Promotes Glucose Toxicity-Induced β Cell Dysfunction by Regulating the Nucleo-Cytoplasmic Trafficking of PDX-1 in a FOXO1-Dependent Manner in INS-1 Cells. Endocrine 2015, 49, 669–682. [Google Scholar] [CrossRef]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.; Wright, C.; White, M.; Arden, K.; Accili, D. The Forkhead Transcription Factor Foxo1 Links Insulin Signaling to Pdx1 Regulation of Pancreatic Beta Cell Growth. J. Clin. Investig. 2002, 110, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The Role of Macrophages in Obesity-Associated Islet Inflammation and β-Cell Abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Nagai, R. Islet Inflammation in Type 2 Diabetes and Physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting β Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474.e5. [Google Scholar] [CrossRef] [PubMed]
- El Alwani, M.; Wu, B.X.; Obeid, L.M.; Hannun, Y.A. Bioactive Sphingolipids in the Modulation of the Inflammatory Response. Pharmacol. Ther. 2006, 112, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Xu, Y.; Xu, J.; Zhang, J.; Xi, Y.; Pi, H.; Yang, L.; Yu, Z.; Wu, Q.; Meng, Z.; et al. Cadmium Exposure Impairs Pancreatic β-Cell Function and Exaggerates Diabetes by Disrupting Lipid Metabolism. Environ. Int. 2021, 149, 106406. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef]
- Ježek, P.; Holendová, B.; Jabůrek, M.; Tauber, J.; Dlasková, A.; Plecitá-Hlavatá, L. The Pancreatic β-Cell: The Perfect Redox System. Antioxidants 2021, 10, 197. [Google Scholar] [CrossRef]
- Chabosseau, P.; Rutter, G.A. Zinc and Diabetes. Arch. Biochem. Biophys. 2016, 611, 79–85. [Google Scholar] [CrossRef]
- Bellomo, E.A.; Meur, G.; Rutter, G.A. Glucose Regulates Free Cytosolic Zn2+ Concentration, Slc39 (ZiP), and Metallothionein Gene Expression in Primary Pancreatic Islet β-Cells. J. Biol. Chem. 2011, 286, 25778–25789. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Laybutt, D.R.; Jonas, J.-C. Emerging Roles of Metallothioneins in Beta Cell Pathophysiology: Beyond and Above Metal Homeostasis and Antioxidant Response. Biology 2021, 10, 176. [Google Scholar] [CrossRef] [PubMed]
- Günther, V.; Lindert, U.; Schaffner, W. The Taste of Heavy Metals: Gene Regulation by MTF-1. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 1416–1425. [Google Scholar] [CrossRef]
- Zimny, S.; Gogolin, F.; Abel, J.; Gleichmann, H. Metallothionein in Isolated Pancreatic Islets of Mice: Induction by Zinc and Streptozotocin, a Naturally Occurring Diabetogen. Arch. Toxicol. 1993, 67, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Ohly, P.; Gleichmann, H. Metallothionein: In Vitro Induction With Zinc and Streptozotocin in Pancreatic Islets of Mice. Exp. Clin. Endocrinol. Diabetes 1995, 103, 79–82. [Google Scholar] [CrossRef]
- Ohly, P.; Dohle, C.; Abel, J.; Seissler, J.; Gleichmann, H. Zinc Sulphate Induces Metallothionein in Pancreatic Islets of Mice and Protects against Diabetes Induced by Multiple Low Doses of Streptozotocin. Diabetologia 2000, 43, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Duprez, J.; Roma, L.; Close, A.; Jonas, J. Protective Antioxidant and Antiapoptotic Effects of ZnCl2 in Rat Pancreatic Islets Cultured in Low and High Glucose Concentrations. PLoS ONE 2012, 7, e46831. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.C.; Bensellam, M.; Duprez, J.; Elouil, H.; Guiot, Y.; Pascal, S.M.A. Glucose Regulation of Islet Stress Responses and β-Cell Failure in Type 2 Diabetes. Diabetes Obes. Metab. 2009, 11, 65–81. [Google Scholar] [CrossRef]
- Ortis, F.; Naamane, N.; Flamez, D.; Ladrière, L.; Moore, F.; Cunha, D.; Colli, M.; Thykjaer, T.; Thorsen, K.; Orntoft, T.; et al. Cytokines Interleukin-1beta and Tumor Necrosis Factor-Alpha Regulate Different Transcriptional and Alternative Splicing Networks in Primary Beta-Cells. Diabetes 2010, 59, 358–374. [Google Scholar] [CrossRef]
- Cardoso, S.; Seiça, R.; Moreira, P.I. Diabesity and Brain Energy Metabolism: The Case of Alzheimer’s Disease. In Advances in Neurobiology; Springer: New York, NY, USA, 2017; Volume 19, pp. 117–150. [Google Scholar]
- Boyd, S.D.; Ullrich, M.S.; Skopp, A.; Winkler, D.D. Copper Sources for Sod1 Activation. Antioxidants 2020, 9, 500. [Google Scholar] [CrossRef]
- Miura, T.; Muraoka, S.; Ogiso, T. Antioxidant Activity of Metallothionein Compared with Reduced Glutathione. Life Sci. 1997, 60, 301–309. [Google Scholar] [CrossRef]
- Langston, W.; Circu, M.L.; Aw, T.Y. Insulin Stimulation of γ-Glutamylcysteine Ligase Catalytic Subunit Expression Increases Endothelial GSH during Oxidative Stress: Influence of Low Glucose. Free Radic. Biol. Med. 2008, 45, 1591. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, Function, and Post-Translational Regulation of the Catalytic and Modifier Subunits of Glutamate Cysteine Ligase. Mol. Asp. Med. 2009, 30, 86. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.; Tran, M.; Stathopoulos, M.; Wiede, F.; Fam, B.; Dodd, G.; Clarke, I.; Watt, M.; Andrikopoulos, S.; Tiganis, T. High-Fat-Fed Obese Glutathione Peroxidase 1-Deficient Mice Exhibit Defective Insulin Secretion but Protection from Hepatic Steatosis and Liver Damage. Antioxid. Redox Signal. 2014, 20, 2114–2129. [Google Scholar] [CrossRef] [PubMed]
- Alnahdi, A.; John, A.; Raza, H. N-Acetyl Cysteine Attenuates Oxidative Stress and Glutathione-Dependent Redox Imbalance Caused by High Glucose/High Palmitic Acid Treatment in Pancreatic Rin-5F Cells. PLoS ONE 2019, 14, e0226696. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. Oxidative Stress: The Vulnerable β-Cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef]
- Grankvist, K.; Marklund, S.L.; Taljedal, I.B. CuZn-Superoxide Dismutase, Mn-Superoxide Dismutase, Catalase and Glutathione Peroxidase in Pancreatic Islets and Other Tissues in the Mouse. Biochem. J. 1981, 199, 393–398. [Google Scholar] [CrossRef]
- Aronis, A.; Madar, Z.; Tirosh, O. Mechanism Underlying Oxidative Stress-Mediated Lipotoxicity: Exposure of J774.2 Macrophages to Triacylglycerols Facilitates Mitochondrial Reactive Oxygen Species Production and Cellular Necrosis. Free Radic. Biol. Med. 2005, 38, 1221–1230. [Google Scholar] [CrossRef]
- Mulder, H.; Ling, C. Mitochondrial Dysfunction in Pancreatic β-Cells in Type 2 Diabetes. Mol. Cell. Endocrinol. 2009, 297, 34–40. [Google Scholar] [CrossRef]
- Jacquet, A.; Cottet-Rousselle, C.; Arnaud, J.; Julien Saint Amand, K.; Ben Messaoud, R.; Lénon, M.; Demeilliers, C.; Moulis, J.-M. Mitochondrial Morphology and Function of the Pancreatic β-Cells INS-1 Model upon Chronic Exposure to Sub-Lethal Cadmium Doses. Toxics 2018, 6, 20. [Google Scholar] [CrossRef]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Pall, M.L.; Levine, S. Nrf2, a Master Regulator of Detoxification and Also Antioxidant, Anti-Inflammatory and Other Cytoprotective Mechanisms, Is Raised by Health Promoting Factors. Sheng Li Xue Bao 2015, 67, 1–18. [Google Scholar]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative Stress Pathways in Pancreatic β-Cells and Insulin-Sensitive Cells and Tissues: Importance to Cell Metabolism, Function, and Dysfunction. Am. J. Physiol. Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Fukutomi, T.; Sugawara, A.; Kawamura, H.; Takahashi, T.; Pi, J.; Uruno, A.; Yamamoto, M. Nrf2 Protects Pancreatic β-Cells From Oxidative and Nitrosative Stress in Diabetic Model Mice. Diabetes 2014, 63, 605–618. [Google Scholar] [CrossRef] [PubMed]
- El-Mansy, A.A.; Mazroa, S.A.; Hamed, W.S.; Yaseen, A.H.; El-Mohandes, E.A. Histological and Immunohistochemical Effects of Curcuma Longa on Activation of Rat Hepatic Stellate Cells after Cadmium Induced Hepatotoxicity. Biotech. Histochem. 2016, 91, 170–181. [Google Scholar] [CrossRef] [PubMed]
- El-Sokkary, G.H.; Nafady, A.A.; Shabash, E.H. Melatonin Administration Ameliorates Cadmium-Induced Oxidative Stress and Morphological Changes in the Liver of Rat. Ecotoxicol. Environ. Saf. 2010, 73, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Ortega, V.E.; Moroni-González, D.; Díaz, A.; Eduardo, B.; Samuel, T. Oral Subacute Exposure to Cadmium LOAEL Dose Induces Insulin Resistance and Impairment of the Hormonal and Metabolic Liver-Adipose Axis in Wistar Rats. Biol. Trace Elem. Res. 2021, 200, 4370–4384. [Google Scholar] [CrossRef]
- Harstad, E.B.; Klaassen, C.D. Gadolinium Chloride Pretreatment Prevents Cadmium Chloride-Induced Liver Damage in Both Wild-Type and MT-Null Mice. Toxicol. Appl. Pharmacol. 2002, 180, 178–185. [Google Scholar] [CrossRef]
- Souza, V.; Escobar, M.D.C.; Gómez-Quiroz, L.; Bucio, L.; Hernández, E.; Cossio, E.C.; Gutiérrez-Ruiz, M.C. Acute Cadmium Exposure Enhances AP-1 DNA Binding and Induces Cytokines Expression and Heat Shock Protein 70 in HepG2 Cells. Toxicology 2004, 197, 213–228. [Google Scholar] [CrossRef]
- Matović, V.; Buha, A.; Dukić-Ćosić, D.; Bulat, Z. Insight into the Oxidative Stress Induced by Lead and/or Cadmium in Blood, Liver and Kidneys. Food Chem. Toxicol. 2015, 78, 130–140. [Google Scholar] [CrossRef]
- Dai, S.; Yin, Z.; Yuan, G.; Lu, H.; Jia, R.; Xu, J.; Song, X.; Li, L.; Shu, Y.; Liang, X.; et al. Quantification of Metallothionein on the Liver and Kidney of Rats by Subchronic Lead and Cadmium in Combination. Environ. Toxicol. Pharmacol. 2013, 36, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Xia, M.Z.; Wang, H.; Liu, X.J.; Hu, Y.F.; Chen, Y.H.; Zhang, C.; Xu, D.X. Cadmium Selectively Induces MIP-2 and COX-2 through PTEN-Mediated Akt Activation in RAW264.7 Cells. Toxicol. Sci. 2014, 138, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Okoye, C.N.; MacDonald-Jay, N.; Kamunde, C. Effects of Bioenergetics, Temperature and Cadmium on Liver Mitochondria Reactive Oxygen Species Production and Consumption. Aquat. Toxicol. 2019, 214, 105264. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R. Prevention of Cadmium-Induced Toxicity in Liver-Derived Cells by the Combination Preparation Hepeel®. Environ. Toxicol. Pharmacol. 2009, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, J.; Zhang, K.; Liu, X.; Li, J. Effects of Chronic Cadmium Poisoning on Zn, Cu, Fe, Ca, and Metallothionein in Liver and Kidney of Rats. Biol. Trace Elem. Res. 2012, 149, 57–63. [Google Scholar] [CrossRef]
- Beyrami, M.; Karimi, E.; Oskoueian, E. Synthesized Chrysin-Loaded Nanoliposomes Improves Cadmium-Induced Toxicity in Mice. Environ. Sci. Pollut. Res. 2020, 27, 40643–40651. [Google Scholar] [CrossRef]
- Pal, M.; Febbraio, M.A.; Lancaster, G.I. The Roles of C-Jun NH2-Terminal Kinases (JNKs) in Obesity and Insulin Resistance. J. Physiol. 2016, 594, 267–279. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the Crossroad of Obesity, Insulin Resistance, and Cell Stress Response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C.; Laybutt, D.R.; Burchfield, J.G.; Gurisik, E.; Narasimhan, S.; Mitchell, C.J.; Pedersen, D.J.; Braun, U.; Cooney, G.J.; Leitges, M.; et al. Inhibition of PKCε Improves Glucose-Stimulated Insulin Secretion and Reduces Insulin Clearance. Cell Metab. 2007, 6, 320–328. [Google Scholar] [CrossRef]
- Chapatwala, K.D.; Rajanna, E.; Desaiah, D. Cadmium Induced Changes in Gluconeogenic Enzymes in Rat Kidney and Liver. Drug Chem. Toxicol. 1980, 3, 407–420. [Google Scholar] [CrossRef]
- Chapatwala, K.D.; Hobson, M.; Desaiah, D.; Rajanna, B. Effect of Cadmium on Hepatic and Renal Gluconeogenic Enzymes in Female Rats. Toxicol. Lett. 1982, 12, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Bashir, N.; Shagirtha, K.; Manoharan, V.; Miltonprabu, S. The Molecular and Biochemical Insight View of Grape Seed Proanthocyanidins in Ameliorating Cadmium-Induced Testes-Toxicity in Rat Model: Implication of PI3K/Akt/Nrf-2 Signaling. Biosci. Rep. 2019, 39, BSR20180515. [Google Scholar] [CrossRef] [PubMed]
- Shati, A.A.; Alfaifi, M.Y. Trans-Resveratrol Inhibits Tau Phosphorylation in the Brains of Control and Cadmium Chloride-Treated Rats by Activating PP2A and PI3K/Akt Induced-Inhibition of GSK3β. Neurochem. Res. 2019, 44, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Zhang, Y.; Xing, H.; Xu, S. Ameliorative Effect of Selenomethionine on Cadmium-Induced Hepatocyte Apoptosis via Regulating PI3K/AKT Pathway in Chickens. Biol. Trace Elem. Res. 2019, 195, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Guangliang, S.; Qing, Z.; Qingqing, L.; Hang, Y.; Yiming, Z.; Shu, L. Astilbin Protects Chicken Peripheral Blood Lymphocytes from Cadmium-Induced Necroptosis via Oxidative Stress and the PI3K/Akt Pathway. Ecotoxicol. Environ. Saf. 2020, 190, 110064. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and Inactivation of Glycogen Synthase Kinase 3 by Protein Kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Sarmiento-Ortega, V.E.; Moroni-González, D.; Díaz, A.; Morán, C.; Brambila, E.; Treviño, S. Sodium Metavanadate Treatment Improves Glycogen Levels in Multiple Tissues in a Model of Metabolic Syndrome Caused by Chronic Cadmium Exposure in Wistar Rats. BioMetals 2021, 34, 245–258. [Google Scholar] [CrossRef]
- Sarmiento-Ortega, V.; Brambila, E.; Flores-Hernández, J.; Díaz, A.; Peña-Rosas, U.; Moroni-González, D.; Aburto-Luna, V.; Treviño, S. The NOAEL Metformin Dose Is Ineffective against Metabolic Disruption Induced by Chronic Cadmium Exposure in Wistar Rats. Toxics 2018, 6, 55. [Google Scholar] [CrossRef]
- Treviño, S.; Diaz, A. Vanadium and Insulin: Partners in Metabolic Regulation. J. Inorg. Biochem. 2020, 208, 111094. [Google Scholar] [CrossRef]
- Treviño, S.; Sánchez-Lara, E.; Sarmiento-Ortega, V.E.; Sánchez-Lombardo, I.; Flores-Hernández, J.Á.; Pérez-Benítez, A.; Brambila-Colombres, E.; González-Vergara, E. Hypoglycemic, Lipid-Lowering and Metabolic Regulation Activities of Metforminium Decavanadate (H2Metf)3 [V10O28]·8H2O Using Hypercaloric-Induced Carbohydrate and Lipid Deregulation in Wistar Rats as b. J. Inorg. Biochem. 2015, 147, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Santamaria-Juarez, C.; Atonal-Flores, F.; Diaz, A.; Sarmiento-Ortega, V.E.; Garcia-Gonzalez, M.; Aguilar-Alonso, P.; Lopez-Lopez, G.; Brambila, E.; Treviño, S. Aortic Dysfunction by Chronic Cadmium Exposure Is Linked to Multiple Metabolic Risk Factors That Converge in Anion Superoxide Production. Arch. Physiol. Biochem. 2020, 128, 748–756. [Google Scholar] [CrossRef]
- Hirano, T. Pathophysiology of Diabetic Dyslipidemia. J. Atheroscler. Thromb. 2018, 25, 771–782. [Google Scholar] [CrossRef]
- Adiels, M.; Olofsson, S.O.; Taskinen, M.R.; Borén, J. Diabetic Dyslipidaemia. Curr. Opin. Lipidol. 2006, 17, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Packard, C.J. Triacylglycerol-Rich Lipoproteins and the Generation of Small, Dense Low-Density Lipoprotein. Biochem. Soc. Trans. 2003, 31, 1066–1069. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Häkkinen, A.; Olofsson, S.O.; Yki-Järvinen, H.; et al. Overproduction of Large VLDL Particles Is Driven by Increased Liver Fat Content in Man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.; Wroblewski, J.M.; Webb, N.R.; Van Der Westhuyzen, D.R. Impact of Phospholipid Transfer Protein on Nascent High-Density Lipoprotein Formation and Remodeling. Arter. Thromb. Vasc. Biol. 2014, 34, 1910–1916. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Elangovan, P.; Pari, L. Protective Role of Tetrahydrocurcumin: An Active Polyphenolic Curcuminoid on Cadmium-InducedOxidative Damage in Rats. Appl. Biochem. Biotechnol. 2017, 183, 51–69. [Google Scholar] [CrossRef]
- Camont, L.; Lhomme, M.; Rached, F.; Le Goff, W.; Nègre-Salvayre, A.; Salvayre, R.; Calzada, C.; Lagarde, M.; Chapman, M.J.; Kontush, A. Small, Dense High-Density Lipoprotein-3 Particles Are Enriched in Negatively Charged Phospholipids: Relevance to Cellular Cholesterol Efflux, Antioxidative, Antithrombotic, Anti-Inflammatory, and Antiapoptotic Functionalities. Arter. Thromb. Vasc. Biol. 2013, 33, 2715–2723. [Google Scholar] [CrossRef]
- Jaeschke, H.; Gores, G.J.; Cederbaum, A.I.; Hinson, J.A.; Pessayre, D.; Lemasters, J.J. Mechanisms of Hepatotoxicity. Toxicol. Sci. 2002, 65, 166–176. [Google Scholar] [CrossRef]
- Martelli, A.; Rousselet, E.; Dycke, C.; Bouron, A.; Moulis, J.M. Cadmium Toxicity in Animal Cells by Interference with Essential Metals. Biochimie 2006, 88, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Souza, V.; Bucio, L.; Gutiérrez-Ruiz, M.C. Cadmium Uptake by a Human Hepatic Cell Line (WRL-68 Cells). Toxicology 1997, 120, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Min, K.S.; Ueda, H.; Tanaka, K. Involvement of Intestinal Calcium Transporter 1 and Metallothionein in Cadmium Accumulation in the Liver and Kidney of Mice Fed a Low-Calcium Diet. Toxicol. Lett. 2008, 176, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, H.; Okugaki, S.; Kubota, K.; Fujiyama, T.; Miyataka, H.; Himeno, S. The Role of ZIP8 Down-Regulation in Cadmium-Resistant Metallothionein-Null Cells. J. Appl. Toxicol. 2009, 29, 367–373. [Google Scholar] [CrossRef]
- DelRaso, N.J.; Foy, B.D.; Gearhart, J.M.; Frazier, J.M. Cadmium Uptake Kinetics in Rat Hepatocytes: Correction for Albumin Binding. Toxicol. Sci. 2003, 72, 19–30. [Google Scholar] [CrossRef]
- Nordberg, G.F.; Piscator, M.; Lind, B. Distribution of Cadmium among Protein Fractions of Mouse Liver. Acta Pharmacol. Toxicol. 1971, 29, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Goering, P.L.; Klaassen, C.D. Tolerance to Cadmium-Induced Toxicity Depends on Presynthesized Metallothionein in Liver. J. Toxicol. Environ. Health 1984, 14, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Qi, K.; Zhang, L.; Bai, Z.; Ren, C.; Xu, X.; Zhang, Z.; Li, X. Glutathione Might Attenuate Cadmium-Induced Liver Oxidative Stress and Hepatic Stellate Cell Activation. Biol. Trace Elem. Res. 2019, 191, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.V.S.; Verma, S. Protective Effects of GSH, Vitamin E, and Selenium on Lipid Peroxidation in Cadmium-Fed Rats. Biol. Trace Elem. Res. 1996, 51, 161–168. [Google Scholar] [CrossRef]
- Renugadevi, J.; Prabu, S.M. Cadmium-Induced Hepatotoxicity in Rats and the Protective Effect of Naringenin. Exp. Toxicol. Pathol. 2010, 62, 171–181. [Google Scholar] [CrossRef]
- Prabu, S.M.; Shagirtha, K.; Renugadevi, J. Amelioration of Cadmium-Induced Oxidative Stress, Impairment in Lipids and Plasma Lipoproteins by the Combined Treatment with Quercetin and α-Tocopherol in Rats. J. Food Sci. 2010, 75, T132–T140. [Google Scholar] [CrossRef]
- Haouem, S.; El Hani, A. Effect of Cadmium on Lipid Peroxidation and on Some Antioxidants in the Liver, Kidneys and Testes of Rats Given Diet Containing Cadmium-Polluted Radish Bulbs. J. Toxicol. Pathol. 2013, 26, 359. [Google Scholar] [CrossRef] [PubMed]
- Zwolak, I. The Role of Selenium in Arsenic and Cadmium Toxicity: An Updated Review of Scientific Literature. Biol. Trace Elem. Res. 2020, 193, 44. [Google Scholar] [CrossRef] [PubMed]
- Unsal, V.; Dalkiran, T.; Çiçek, M.; Kölükçü, E. The Role of Natural Antioxidants Against Reactive Oxygen Species Produced by Cadmium Toxicity: A Review. Adv. Pharm. Bull. 2020, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Newairy, A.A.; El-Sharaky, A.S.; Badreldeen, M.M.; Eweda, S.M.; Sheweita, S.A. The Hepatoprotective Effects of Selenium against Cadmium Toxicity in Rats. Toxicology 2007, 242, 23–30. [Google Scholar] [CrossRef]
- Casalino, E.; Calzaretti, G.; Sblano, C.; Landriscina, C. Molecular Inhibitory Mechanisms of Antioxidant Enzymes in Rat Liver and Kidney by Cadmium. Toxicology 2002, 179, 37–50. [Google Scholar] [CrossRef]
- Yalin, S.; Comelekoglu, U.; Bagis, S.; Sahin, N.O.; Ogenler, O.; Hatungil, R. Acute Effect of Single-Dose Cadmium Treatment on Lipid Peroxidation and Antioxidant Enzymes in Ovariectomized Rats. Ecotoxicol. Environ. Saf. 2006, 65, 140–144. [Google Scholar] [CrossRef]
- Lazarus, M.; Orct, T.; Aladrović, J.; Ljubić, B.B.; Jurasović, J.; Blanuša, M. Effect of Selenium Pre-Treatment on Antioxidative Enzymes and Lipid Peroxidation in Cd-Exposed Suckling Rats. Biol. Trace Elem. Res. 2011, 142, 611–622. [Google Scholar] [CrossRef]
- Gong, P.; Chen, F.X.; Ma, G.F.; Feng, Y.; Zhao, Q.Y.; Wang, R. Endomorphin 1 Effectively Protects Cadmium Chloride-Induced Hepatic Damage in Mice. Toxicology 2008, 251, 35–44. [Google Scholar] [CrossRef]
- Tandon, S.K.; Singh, S.; Prasad, S.; Khandekar, K.; Dwivedi, V.K.; Chatterjee, M.; Mathur, N. Reversal of Cadmium Induced Oxidative Stress by Chelating Agent, Antioxidant or Their Combination in Rat. Toxicol. Lett. 2003, 145, 211–217. [Google Scholar] [CrossRef]
- Dey, A.; Lakshmanan, J. The Role of Antioxidants and Other Agents in Alleviating Hyperglycemia Mediated Oxidative Stress and Injury in Liver. Food Funct. 2013, 4, 1148–1184. [Google Scholar] [CrossRef]
- Tang, W.; Jiang, Y.F.; Ponnusamy, M.; Diallo, M. Role of Nrf2 in Chronic Liver Disease. World J. Gastroenterol. 2014, 20, 13079–13087. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Reisman, S.A. Nrf2 the Rescue: Effects of the Antioxidative/Electrophilic Response on the Liver. Toxicol. Appl. Pharmacol. 2010, 244, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, J.; Ge, J.; Wang, L.L.; Li, N.; Sun, X.T.; Cao, H.B.; Li, J.L. Selenium Triggers Nrf2-Mediated Protection against Cadmium-Induced Chicken Hepatocyte Autophagy and Apoptosis. Toxicol. Vitr. 2017, 44, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.G.; Wang, X.Y.; Wang, J.H.; Fan, R.F.; Wang, L. Trehalose Prevents Cadmium-Induced Hepatotoxicity by Blocking Nrf2 Pathway, Restoring Autophagy and Inhibiting Apoptosis. J. Inorg. Biochem. 2019, 192, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, Y.; Lu, Z.; Guo, W.; Tumen, B.; He, Y.; Chen, C.; Hu, S.; Xu, K.; Wang, Y.; et al. Cadmium Induces Acute Liver Injury by Inhibiting Nrf2 and the Role of NF-ΚB, NLRP3, and MAPKS Signaling Pathway. Int. J. Environ. Res. Public Health 2020, 17, 138. [Google Scholar] [CrossRef]
- Lawal, A.O.; Ellis, E.M. Nrf2-Mediated Adaptive Response to Cadmium-Induced Toxicity Involves Protein Kinase C Delta in Human 1321N1 Astrocytoma Cells. Environ. Toxicol. Pharmacol. 2011, 32, 54–62. [Google Scholar] [CrossRef]
- Wang, C.C.; Si, L.F.; Guo, S.N.; Zheng, J.L. Negative Effects of Acute Cadmium on Stress Defense, Immunity, and Metal Homeostasis in Liver of Zebrafish: The Protective Role of Environmental Zinc Dpre-Exposure. Chemosphere 2019, 222, 91–97. [Google Scholar] [CrossRef]
- Shinkai, Y.; Kimura, T.; Itagaki, A.; Yamamoto, C.; Taguchi, K.; Yamamoto, M.; Kumagai, Y.; Kaji, T. Partial Contribution of the Keap1-Nrf2 System to Cadmium-Mediated Metallothionein Expression in Vascular Endothelial Cells. Toxicol. Appl. Pharmacol. 2016, 295, 37–46. [Google Scholar] [CrossRef]
- Buha, A.; Baralić, K.; Djukic-Cosic, D.; Bulat, Z.; Tinkov, A.; Panieri, E.; Saso, L. The Role of Toxic Metals and Metalloids in Nrf2 Signaling. Antioxidants 2021, 10, 630. [Google Scholar] [CrossRef]
- He, X.; Chen, M.G.; Ma, Q. Activation of Nrf2 in Defense against Cadmium-Induced Oxidative Stress. Chem. Res. Toxicol. 2008, 21, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Liu, M. Adipose Tissue in Control of Metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Lazar, M.A. Physiology. The Health Risk of Obesity—Better Metrics Imperative. Science 2013, 341, 856–858. [Google Scholar] [CrossRef]
- Akingbemi, B.T. Adiponectin Receptors in Energy Homeostasis and Obesity Pathogenesis. Prog. Mol. Biol. Transl. Sci. 2013, 114, 317–342. [Google Scholar] [CrossRef] [PubMed]
- Martínez-García, M.Á.; Moncayo, S.; Insenser, M.; Álvarez-Blasco, F.; Luque-Ramírez, M.; Escobar-Morreale, H.F. Postprandial Responses of Circulating Energy Homeostasis Mediators to Single Macronutrient Challenges: Influence of Obesity and Sex Hormones. Food Funct. 2021, 12, 1051–1062. [Google Scholar] [CrossRef]
- Treviño, S.; Cortezano-Esteban, S.; Hernández-Fragoso, H.; Díaz, A.; Vázquez-Roque, R.; Enrique Sarmiento-Ortega, V.; Moroni-González, D.; Pelayo, R.; Brambila, E. Clinical Monitored in Subjects Metabolically Healthy and Unhealthy before and during a SARS-CoV-2 Infection- A Cross-Sectional Study in Mexican Population. Cytokine 2022, 153, 155868. [Google Scholar] [CrossRef]
- Jaganathan, R.; Ravindran, R.; Dhanasekaran, S. Emerging Role of Adipocytokines in Type 2 Diabetes as Mediators of Insulin Resistance and Cardiovascular Disease. Can. J. Diabetes 2018, 42, 446–456.e1. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of Adipose Tissue: An Endocrine Organ. Arch. Med. Sci. 2013, 9, 191. [Google Scholar] [CrossRef]
- Qatanani, M.; Lazar, M.A. Mechanisms of Obesity-Associated Insulin Resistance: Many Choices on the Menu. Genes Dev. 2007, 21, 1443–1455. [Google Scholar] [CrossRef]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine Dysregulation and Adipose Tissue Inflammation in Human Obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef]
- Kawakami, T.; Sugimoto, H.; Furuichi, R.; Kadota, Y.; Inoue, M.; Setsu, K.; Suzuki, S.; Sato, M. Cadmium Reduces Adipocyte Size and Expression Levels of Adiponectin and Peg1/Mest in Adipose Tissue. Toxicology 2010, 267, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Nishiyama, K.; Kadota, Y.; Sato, M.; Inoue, M.; Suzuki, S. Cadmium Modulates Adipocyte Functions in Metallothionein-Null Mice. Toxicol. Appl. Pharmacol. 2013, 272, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, R.; Ribeiro, M.; Kajdacsy-Balla, A. The Toxic Effect of Environmental Cadmium on Visceral Adipose Tissue. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Lee, E.J.; Moon, J.Y.; Yoo, B.S. Cadmium Inhibits the Differentiation of 3T3-L1 Preadipocyte through the C/EBPα and PPARγ Pathways. Drug Chem. Toxicol. 2012, 35, 225–231. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Gustafson, B.; Gogg, S.; Hedjazifar, S.; Jenndahl, L.; Hammarstedt, A.; Smith, U. Inflammation and Impaired Adipogenesis in Hypertrophic Obesity in Man. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E999–E1003. [Google Scholar] [CrossRef]
- Zhu, S.; Sun, F.; Li, W.; Cao, Y.; Wang, C.; Wang, Y.; Liang, D.; Zhang, R.; Zhang, S.; Wang, H.; et al. Apelin Stimulates Glucose Uptake through the PI3K/Akt Pathway and Improves Insulin Resistance in 3T3-L1 Adipocytes. Mol. Cell. Biochem. 2011, 353, 305–313. [Google Scholar] [CrossRef]
- Tsuchiya, A.; Kanno, T.; Nishizaki, T. PI3 Kinase Directly Phosphorylates Akt1/2 at Ser473/474 in the Insulin Signal Transduction Pathway. J. Endocrinol. 2014, 220, 49–59. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Simental-Mendía, L.E.; Barreto, G.E.; Sahebkar, A. Metabolic Effects of Antidiabetic Drugs on Adipocytes and Adipokine Expression. J. Cell. Physiol. 2019, 234, 16987–16997. [Google Scholar] [CrossRef]
- Summers, S.A.; Whiteman, E.L.; Birnbaum, M.J. Insulin Signaling in the Adipocyte. Int. J. Obes. 2000, 24, S67–S70. [Google Scholar] [CrossRef]
- Han, J.C.; Park, S.Y.; Hah, B.G.; Choi, G.H.; Kim, Y.K.; Kwon, T.H.; Kim, E.K.; Lachaal, M.; Jung, C.Y.; Lee, W. Cadmium Induces Impaired Glucose Tolerance in Rat by Down-Regulating GLUT4 Expression in Adipocytes. Arch. Biochem. Biophys. 2003, 413, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Ficková, M.; Eybl, V.; Kotyzová, D.; Mičková, V.; Möstbök, S.; Brtko, J. Long Lasting Cadmium Intake Is Associated with Reduction of Insulin Receptors in Rat Adipocytes. Biometals 2003, 16, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Saltiel, A.R. Adapting to Obesity with Adipose Tissue Inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Laurencikiene, J.; Van Harmelen, V.; Nordström, E.A.; Dicker, A.; Blomqvist, L.; Näslund, E.; Langin, D.; Arner, P.; Rydén, M. NF-ΚB Is Important for TNF-α-Induced Lipolysis in Human Adipocytes. J. Lipid Res. 2007, 48, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.G.; Araujo, T.G.; Carvalho, B.M.; Guadagnini, D.; Rocha, G.Z.; Bagarolli, R.A.; Carvalheira, J.B.C.; Saad, M.J.A. Acute Exercise Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization in Diet-Induced Obese Rats. Obesity 2013, 21, 2545–2556. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory Links between Obesity and Metabolic Disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Zhang, J.; Qi, X.; Cui, Y.; Yin, K.; Lin, H. Cadmium Induced Inflammation and Apoptosis of Porcine Epididymis via Activating RAF1/MEK/ERK and NF-ΚB Pathways. Toxicol. Appl. Pharmacol. 2021, 415, 115449. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Llorens, S.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Talebi, M.; Shakibaei, M.; Samarghandian, S. An Overview of the Role of Adipokines in Cardiometabolic Diseases. Molecules 2020, 25, 5218. [Google Scholar] [CrossRef]
- Park, S.E.; Park, C.Y.; Sweeney, G. Biomarkers of Insulin Sensitivity and Insulin Resistance: Past, Present and Future. Crit. Rev. Clin. Lab. Sci. 2015, 52, 180–190. [Google Scholar] [CrossRef]
- Fasshauer, M.; Blüher, M. Adipokines in Health and Disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Graziano, M.; Sciacca, L.; Baratta, R.; Frittitta, L. Adipose Tissue, Obesity and Adiponectin: Role in Endocrine Cancer Risk. Int. J. Mol. Sci. 2019, 20, 2863. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sánchez, N. There and Back Again: Leptin Actions in White Adipose Tissue. Int. J. Mol. Sci. 2020, 21, 6039. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased Oxidative Stress in Obesity and Its Impact on Metabolic Syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Wonisch, W.; Falk, A.; Sundl, I.; Winklhofer-Roob, B.M.; Lindschinger, M. Oxidative Stress Increases Continuously with BMI and Age with Unfavourable Profiles in Males. Aging Male 2012, 15, 159–165. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Khemka, V.K.; Chatterjee, G.; Ganguly, A.; Mukhopadhyay, S.; Chakrabarti, S. Enhanced ROS Production and Oxidative Damage in Subcutaneous White Adipose Tissue Mitochondria in Obese and Type 2 Diabetes Subjects. Mol. Cell. Biochem. 2015, 399, 95–103. [Google Scholar] [CrossRef]
- Boyer, F.; Diotel, N.; Girard, D.; Rondeau, P.; Essop, M.F.; Bourdon, E. Enhanced Oxidative Stress in Adipose Tissue from Diabetic Mice, Possible Contribution of Glycated Albumin. Biochem. Biophys. Res. Commun. 2016, 473, 154–160. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef]
- Hauck, A.K.; Huang, Y.; Hertzel, A.V.; Bernlohr, D.A. Adipose Oxidative Stress and Protein Carbonylation. J. Biol. Chem. 2019, 294, 1083–1088. [Google Scholar] [CrossRef]
- Curtis, J.M.; Grimsrud, P.A.; Wright, W.S.; Xu, X.; Foncea, R.E.; Graham, D.W.; Brestoff, J.R.; Wiczer, B.M.; Ilkayeva, O.; Cianflone, K.; et al. Downregulation of Adipose Glutathione S-Transferase A4 Leads to Increased Protein Carbonylation, Oxidative Stress, and Mitochondrial Dysfunction. Diabetes 2010, 59, 1132–1142. [Google Scholar] [CrossRef]
- Roškarić, P.; Šperanda, M.; Mašek, T.; Verbanac, D.; Starčević, K. Low Dietary N6/N3 Ratio Attenuates Changes in the NRF 2 Gene Expression, Lipid Peroxidation, and Inflammatory Markers Induced by Fructose Overconsumption in the Rat Abdominal Adipose Tissue. Antioxidants 2021, 10, 2005. [Google Scholar] [CrossRef]
- Den Hartigh, L.J.; Omer, M.; Goodspeed, L.; Wang, S.; Wietecha, T.; O’Brien, K.D.; Han, C.Y. Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity. Arter. Thromb. Vasc. Biol. 2017, 37, 466–475. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- De Pauw, A.; Tejerina, S.; Raes, M.; Keijer, J.; Arnould, T. Mitochondrial (Dys)Function in Adipocyte (de)Differentiation and Systemic Metabolic Alterations. Am. J. Pathol. 2009, 175, 927–939. [Google Scholar] [CrossRef]
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.-i.; et al. Increased Oxidative Stress Precedes the Onset of High-Fat Diet-Induced Insulin Resistance and Obesity. Metabolism 2008, 57, 1071–1077. [Google Scholar] [CrossRef]
- Paglialunga, S.; Ludzki, A.; Root-McCaig, J.; Holloway, G.P. In Adipose Tissue, Increased Mitochondrial Emission of Reactive Oxygen Species Is Important for Short-Term High-Fat Diet-Induced Insulin Resistance in Mice. Diabetologia 2015, 58, 1071–1080. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stöckli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial Oxidative Stress Causes Insulin Resistance without Disrupting Oxidative Phosphorylation. J. Biol. Chem. 2018, 293, 7315–7328. [Google Scholar] [CrossRef]
- Greenstein, A.S.; Khavandi, K.; Withers, S.B.; Sonoyama, K.; Clancy, O.; Jeziorska, M.; Laing, I.; Yates, A.P.; Pemberton, P.W.; Malik, R.A.; et al. Local Inflammation and Hypoxia Abolish the Protective Anticontractile Properties of Perivascular Fat in Obese Patients. Circulation 2009, 119, 1661–1670. [Google Scholar] [CrossRef]
- Sun, J.; Jiao, Z.; Zhu, W.; Li, X.; Wang, P.; Wang, J.; Tai, T.; Wang, Y.; Wang, H.; Shi, G. Astilbin Attenuates Cadmium-Induced Adipose Tissue Damage by Inhibiting NF-ΚB Pathways and Regulating the Expression of HSPs in Chicken. Biol. Trace Elem. Res. 2022, 2022, 1–12. [Google Scholar] [CrossRef]
- Huh, J.Y.; Kim, Y.; Jeong, J.; Park, J.; Kim, I.; Huh, K.H.; Kim, Y.S.; Woo, H.A.; Rhee, S.G.; Lee, K.J.; et al. Peroxiredoxin 3 Is a Key Molecule Regulating Adipocyte Oxidative Stress, Mitochondrial Biogenesis, and Adipokine Expression. Antioxid. Redox Signal. 2012, 16, 229–243. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J.Y.; Kim, J.H.; Lee, H.S.; Huh, J.W.; Lee, D.S. Peroxiredoxin 2 Deficiency Reduces White Adipogenesis Due to the Excessive ROS Generation. Cell Biol. Int. 2020, 44, 2086–2093. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Fukuhara, A.; Hashimoto, E.; Kobayashi, H.; Kobayashi, S.; Otsuki, M.; Shimomura, I. Oxidative Stress Inhibits Healthy Adipose Expansion Through Suppression of SREBF1-Mediated Lipogenic Pathway. Diabetes 2018, 67, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Hosick, P.A.; Weeks, M.F.; Hankins, M.W.; Moore, K.H.; Stec, D.E. Sex-Dependent Effects of HO-1 Deletion from Adipocytes in Mice. Int. J. Mol. Sci. 2017, 18, 611. [Google Scholar] [CrossRef]
- Cao, J.; Peterson, S.J.; Sodhi, K.; Vanella, L.; Barbagallo, I.; Rodella, L.F.; Schwartzman, M.L.; Abraham, N.G.; Kappas, A. Heme Oxygenase Gene Targeting to Adipocytes Attenuates Adiposity and Vascular Dysfunction in Mice Fed a High-Fat Diet. Hypertension 2012, 60, 467–475. [Google Scholar] [CrossRef]
- Kim, R.; Hu, H.; Rotnitzky, A.; Bellinger, D.; Needleman, H. A Longitudinal Study of Chronic Lead Exposure and Physical Growth in Boston Children. Environ. Health Perspect. 1995, 103, 952–957. [Google Scholar] [CrossRef]
- Wang, N.; Chen, C.; Nie, X.; Han, B.; Li, Q.; Chen, Y.; Zhu, C.; Chen, Y.; Xia, F.; Cang, Z.; et al. Blood Lead Level and Its Association with Body Mass Index and Obesity in China—Results from SPECT-China Study. Sci. Rep. 2015, 5, 18299. [Google Scholar] [CrossRef]
- Hildebrand, J.; Thakar, S.; Watts, T.L.; Banfield, L.; Thabane, L.; Macri, J.; Hill, S.; Samaan, M.C. The Impact of Environmental Cadmium Exposure on Type 2 Diabetes Risk: A Protocol for an Overview of Systematic Reviews. Syst. Rev. 2019, 8, 309. [Google Scholar] [CrossRef]
- Tian, L.L.; Zhao, Y.C.; Wang, X.C.; Gu, J.L.; Sun, Z.J.; Zhang, Y.L.; Wang, J.X. Effects of Gestational Cadmium Exposure on Pregnancy Outcome and Development in the Offspring at Age 4.5 Years. Biol. Trace Elem. Res. 2009, 132, 51–59. [Google Scholar] [CrossRef]
- Vidal, A.C.; Semenova, V.; Darrah, T.; Vengosh, A.; Huang, Z.; King, K.; Nye, M.D.; Fry, R.; Skaar, D.; Maguire, R.; et al. Maternal Cadmium, Iron and Zinc Levels, DNA Methylation and Birth Weight. BMC Pharmacol. Toxicol. 2015, 16, 20. [Google Scholar] [CrossRef]
- Lin, C.M.; Doyle, P.; Wang, D.; Hwang, Y.H.; Chen, P.C. Does Prenatal Cadmium Exposure Affect Fetal and Child Growth? Occup. Environ. Med. 2011, 68, 641–646. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Eriksson, J.G.; Forsén, T.; Osmond, C. Fetal Origins of Adult Disease: Strength of Effects and Biological Basis. Int. J. Epidemiol. 2002, 31, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Tinkov, A.A.; Filippini, T.; Ajsuvakova, O.P.; Skalnaya, M.G.; Aaseth, J.; Bjørklund, G.; Gatiatulina, E.R.; Popova, E.V.; Nemereshina, O.N.; Huang, P.-T.; et al. Cadmium and Atherosclerosis: A Review of Toxicological Mechanisms and a Meta-Analysis of Epidemiologic Studies. Environ. Res. 2018, 162, 240–260. [Google Scholar] [CrossRef] [PubMed]
- Haswell-Elkins, M.; McGrath, V.; Moore, M.; Satarug, S.; Walmby, M.; Ng, J. Exploring Potential Dietary Contributions Including Traditional Seafood and Other Determinants of Urinary Cadmium Levels among Indigenous Women of a Torres Strait Island (Australia). J. Expo. Sci. Environ. Epidemiol. 2007, 17, 298–306. [Google Scholar] [CrossRef]
- Skalnaya, M.G.; Tinkov, A.A.; Demidov, V.A.; Serebryansky, E.P.; Nikonorov, A.A.; Skalny, A.V. Hair Toxic Element Content in Adult Men and Women in Relation to Body Mass Index. Biol. Trace Elem. Res. 2014, 161, 13–19. [Google Scholar] [CrossRef]
- Filippini, T.; Michalke, B.; Malagoli, C.; Grill, P.; Bottecchi, I.; Malavolti, M.; Vescovi, L.; Sieri, S.; Krogh, V.; Cherubini, A.; et al. Determinants of Serum Cadmium Levels in a Northern Italy Community: A Cross-Sectional Study. Environ. Res. 2016, 150, 219–226. [Google Scholar] [CrossRef]
- Wang, X.; Mukherjee, B.; Park, S.K. Associations of Cumulative Exposure to Heavy Metal Mixtures with Obesity and Its Comorbidities among U.S. Adults in NHANES 2003–2014. Environ. Int. 2018, 121, 683–694. [Google Scholar] [CrossRef]
- Nie, X.; Wang, N.; Chen, Y.; Chen, C.; Han, B.; Zhu, C.; Chen, Y.; Xia, F.; Cang, Z.; Lu, M.; et al. Blood Cadmium in Chinese Adults and Its Relationships with Diabetes and Obesity. Environ. Sci. Pollut. Res. Int. 2016, 23, 18714–18723. [Google Scholar] [CrossRef]
- MERALI, Z.; SINGHAL, R.L. Diabetogenic Effects of Chronic Oral Cadmium Adminstration to Neonatal Rats. Br. J. Pharmacol. 1980, 69, 151–157. [Google Scholar] [CrossRef]
- Ba, Q.; Li, M.; Chen, P.; Huang, C.; Duan, X.; Lu, L.; Li, J.; Chu, R.; Xie, D.; Song, H.; et al. Sex-Dependent Effects of Cadmium Exposure in Early Life on Gut Microbiota and Fat Accumulation in Mice. Environ. Health Perspect. 2017, 125, 437–446. [Google Scholar] [CrossRef]
- Yu, Y.; Ma, R.; Yu, L.; Cai, Z.; Li, H.; Zuo, Y.; Wang, Z.; Li, H. Combined Effects of Cadmium and Tetrabromobisphenol a (TBBPA) on Development, Antioxidant Enzymes Activity and Thyroid Hormones in Female Rats. Chem. Biol. Interact. 2018, 289, 23–31. [Google Scholar] [CrossRef]
- Edwards, J.; Ackerman, C. A Review of Diabetes Mellitus and Exposure to the Environmental Toxicant Cadmium with an Emphasis on Likely Mechanisms of Action. Curr. Diabetes Rev. 2016, 12, 252–258. [Google Scholar] [CrossRef]
- Bimonte, V.M.; Besharat, Z.M.; Antonioni, A.; Cella, V.; Lenzi, A.; Ferretti, E.; Migliaccio, S. The Endocrine Disruptor Cadmium: A New Player in the Pathophysiology of Metabolic Diseases. J. Endocrinol. Investig. 2021, 44, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.; Olsen, A.; Nguyen, J.; Wong, W.; Muayed, M.E.; Edwards, J. Pancreatic Islets Accumulate Cadmium in a Rodent Model of Cadmium-Induced Hyperglycemia. Int. J. Mol. Sci. 2021, 22, 360. [Google Scholar] [CrossRef] [PubMed]
- Fatima, G.; Raza, A.M.; Hadi, N.; Nigam, N.; Mahdi, A.A. Cadmium in Human Diseases: It’s More than Just a Mere Metal. Indian J. Clin. Biochem. 2019, 34, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Barregard, L.; Bergström, G.; Fagerberg, B. Cadmium Exposure in Relation to Insulin Production, Insulin Sensitivity and Type 2 Diabetes: A Cross-Sectional and Prospective Study in Women. Environ. Res. 2013, 121, 104–109. [Google Scholar] [CrossRef]
- Borné, Y.; Fagerberg, B.; Persson, M.; Sallsten, G.; Forsgard, N.; Hedblad, B.; Barregard, L.; Engström, G. Cadmium Exposure and Incidence of Diabetes Mellitus—Results from the Malmö Diet and Cancer Study. PLoS ONE 2014, 9, e112277. [Google Scholar] [CrossRef]
- Filippini, T.; Wise, L.A.; Vinceti, M. Cadmium Exposure and Risk of Diabetes and Prediabetes: A Systematic Review and Dose-Response Meta-Analysis. Environ. Int. 2022, 158, 106920. [Google Scholar] [CrossRef]
- Tinkov, A.A.; Filippini, T.; Ajsuvakova, O.P.; Aaseth, J.; Gluhcheva, Y.G.; Ivanova, J.M.; Bjørklund, G.; Skalnaya, M.G.; Gatiatulina, E.R.; Popova, E.V.; et al. The Role of Cadmium in Obesity and Diabetes. Sci. Total Environ. 2017, 601–602, 741–755. [Google Scholar] [CrossRef]
- Swaddiwudhipong, W.; Limpatanachote, P.; Mahasakpan, P.; Krintratun, S.; Punta, B.; Funkhiew, T. Progress in Cadmium-Related Health Effects in Persons with High Environmental Exposure in Northwestern Thailand: A Five-Year Follow-Up. Environ. Res. 2012, 112, 194–198. [Google Scholar] [CrossRef]
- Zhang, W.L.; Du, Y.; Zhai, M.M.; Shang, Q. Cadmium Exposure and Its Health Effects: A 19-Year Follow-up Study of a Polluted Area in China. Sci. Total Environ. 2014, 470–471, 224–228. [Google Scholar] [CrossRef]
- Madrigal, J.M.; Ricardo, A.C.; Persky, V.; Turyk, M. Associations between Blood Cadmium Concentration and Kidney Function in the U.S. Population: Impact of Sex, Diabetes and Hypertension. Environ. Res. 2019, 169, 180. [Google Scholar] [CrossRef]
- Wu, M.; Song, J.; Zhu, C.; Wang, Y.; Yin, X.; Huang, G.; Zhao, K.; Zhu, J.; Duan, Z.; Su, L. Association between Cadmium Exposure and Diabetes Mellitus Risk: A Prisma-Compliant Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 113129. [Google Scholar] [CrossRef]
- Wallia, A.; Allen, N.B.; Badon, S.; El Muayed, M. Association between Urinary Cadmium Levels and Prediabetes in the NHANES 2005-2010 Population. Int. J. Hyg. Environ. Health 2014, 217, 854–860. [Google Scholar] [CrossRef]
- Menke, A.; Guallar, E.; Cowie, C.C. Metals in Urine and Diabetes in U.S. Adults. Diabetes 2016, 65, 164–171. [Google Scholar] [CrossRef]
- Swaddiwudhipong, W.; Mahasakpan, P.; Limpatanachote, P.; Krintratun, S. Correlations of Urinary Cadmium with Hypertension and Diabetes in Persons Living in Cadmium-Contaminated Villages in Northwestern Thailand: A Population Study. Environ. Res. 2010, 110, 612–616. [Google Scholar] [CrossRef]
- Kuo, C.C.; Moon, K.; Thayer, K.A.; Navas-Acien, A. Environmental Chemicals and Type 2 Diabetes: An Updated Systematic Review of the Epidemiologic Evidence. Curr. Diabetes Rep. 2013, 13, 831–849. [Google Scholar] [CrossRef]
- Moon, S.S. Association of Lead, Mercury and Cadmium with Diabetes in the Korean Population: The Korea National Health and Nutrition Examination Survey (KNHANES) 2009–2010. Diabet. Med. 2013, 30, e143–e148. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moroni-González, D.; Sarmiento-Ortega, V.E.; Diaz, A.; Brambila, E.; Treviño, S. Pancreas–Liver–Adipose Axis: Target of Environmental Cadmium Exposure Linked to Metabolic Diseases. Toxics 2023, 11, 223. https://doi.org/10.3390/toxics11030223
Moroni-González D, Sarmiento-Ortega VE, Diaz A, Brambila E, Treviño S. Pancreas–Liver–Adipose Axis: Target of Environmental Cadmium Exposure Linked to Metabolic Diseases. Toxics. 2023; 11(3):223. https://doi.org/10.3390/toxics11030223
Chicago/Turabian StyleMoroni-González, Diana, Victor Enrique Sarmiento-Ortega, Alfonso Diaz, Eduardo Brambila, and Samuel Treviño. 2023. "Pancreas–Liver–Adipose Axis: Target of Environmental Cadmium Exposure Linked to Metabolic Diseases" Toxics 11, no. 3: 223. https://doi.org/10.3390/toxics11030223
APA StyleMoroni-González, D., Sarmiento-Ortega, V. E., Diaz, A., Brambila, E., & Treviño, S. (2023). Pancreas–Liver–Adipose Axis: Target of Environmental Cadmium Exposure Linked to Metabolic Diseases. Toxics, 11(3), 223. https://doi.org/10.3390/toxics11030223