
The Measurement of Atmospheric Black Carbon: A Review



Abstract
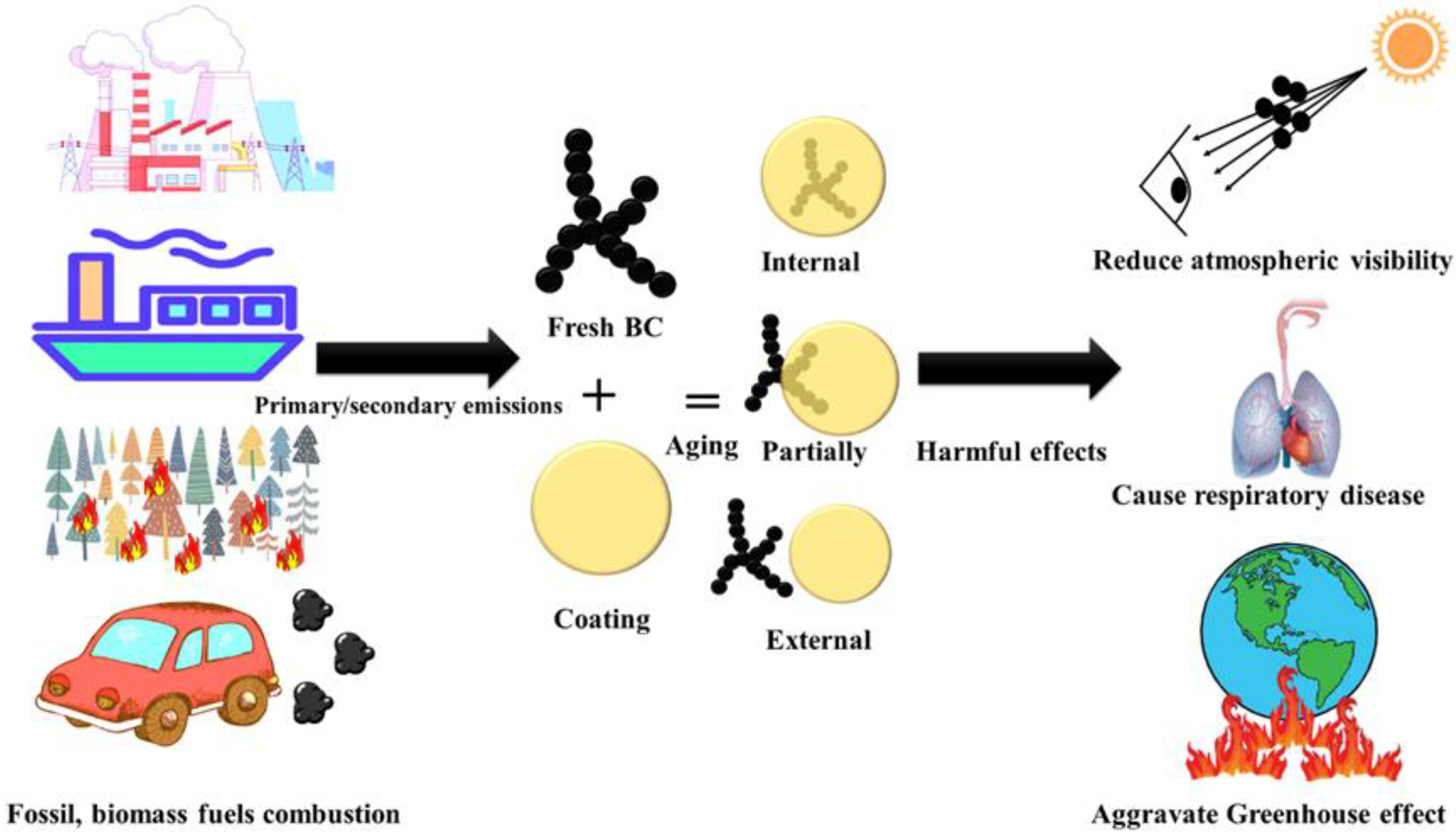
1. Introduction
2. Elemental Carbon
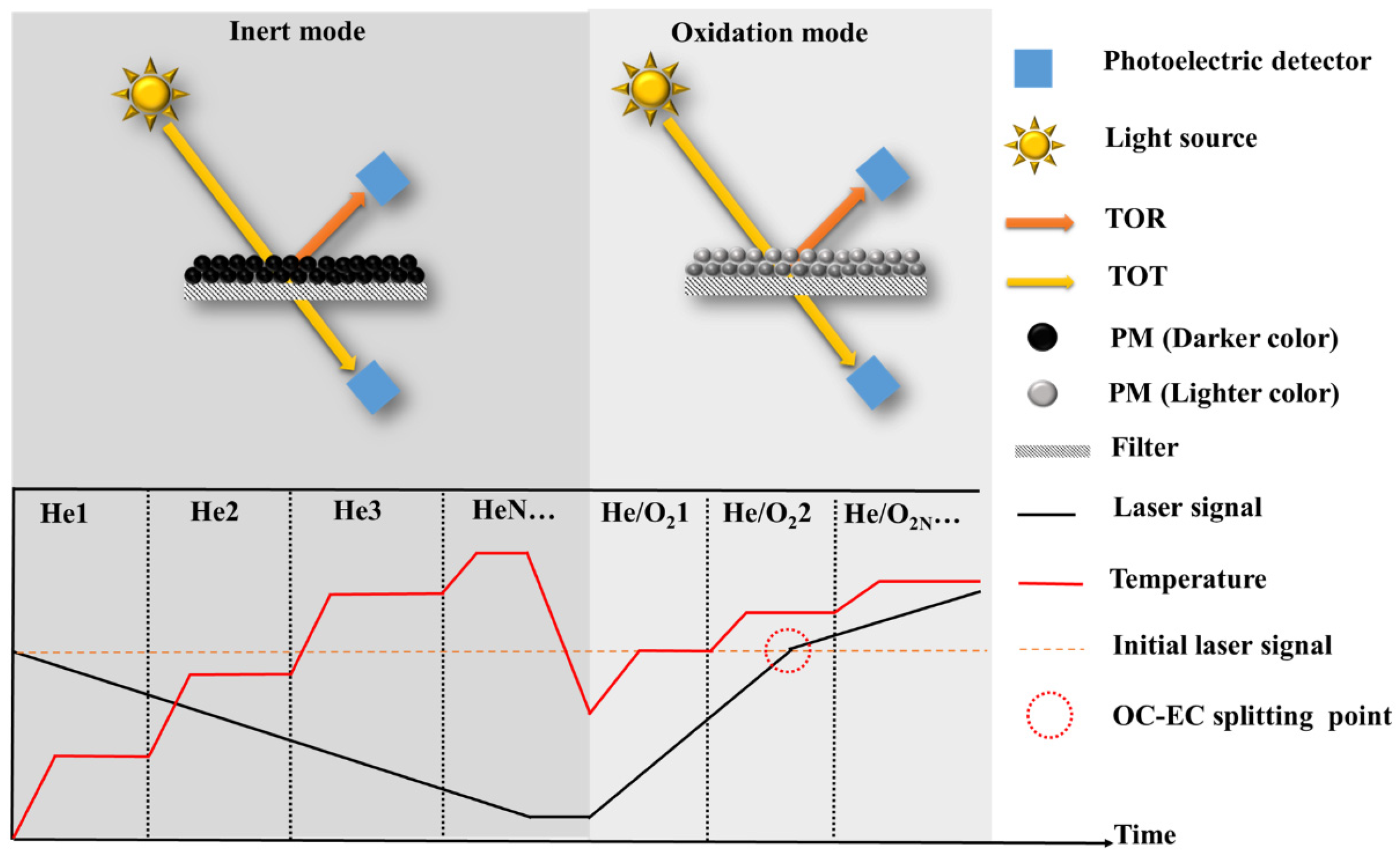
2.1. The Effect of Thermo-Optical Protocol on Elemental Carbon Quantification
2.2. The Effect of Chemical Composition on Elemental Carbon Quantification
2.3. Solvent Extraction Method
3. Equivalent Black Carbon
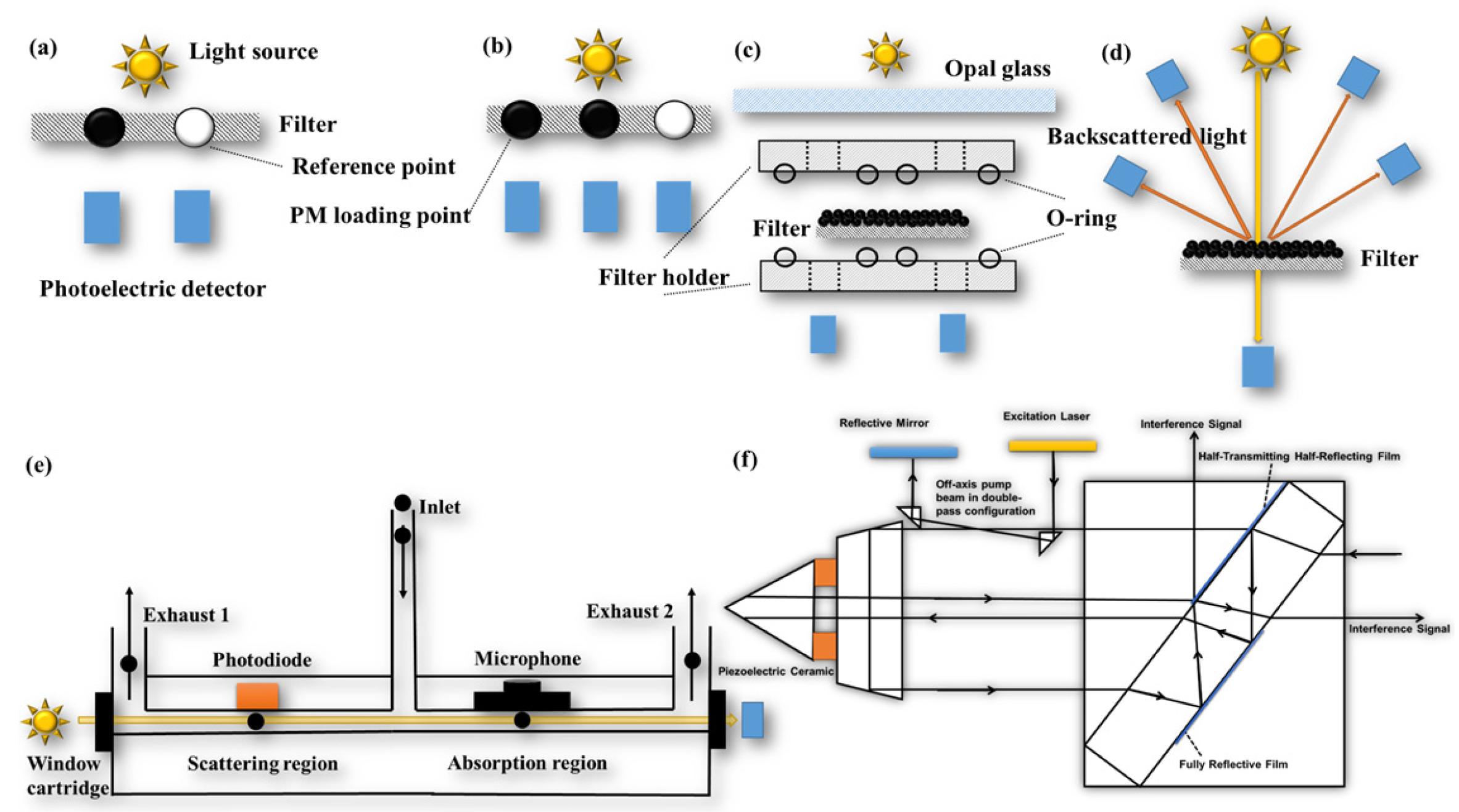
3.1. Filter-Based Technique
3.2. In Situ Technique
3.3. Comparison of babs Measurement Results of Different Optical Instruments
3.4. Sample Heating Pretreatment Method
4. Refractory Black Carbon
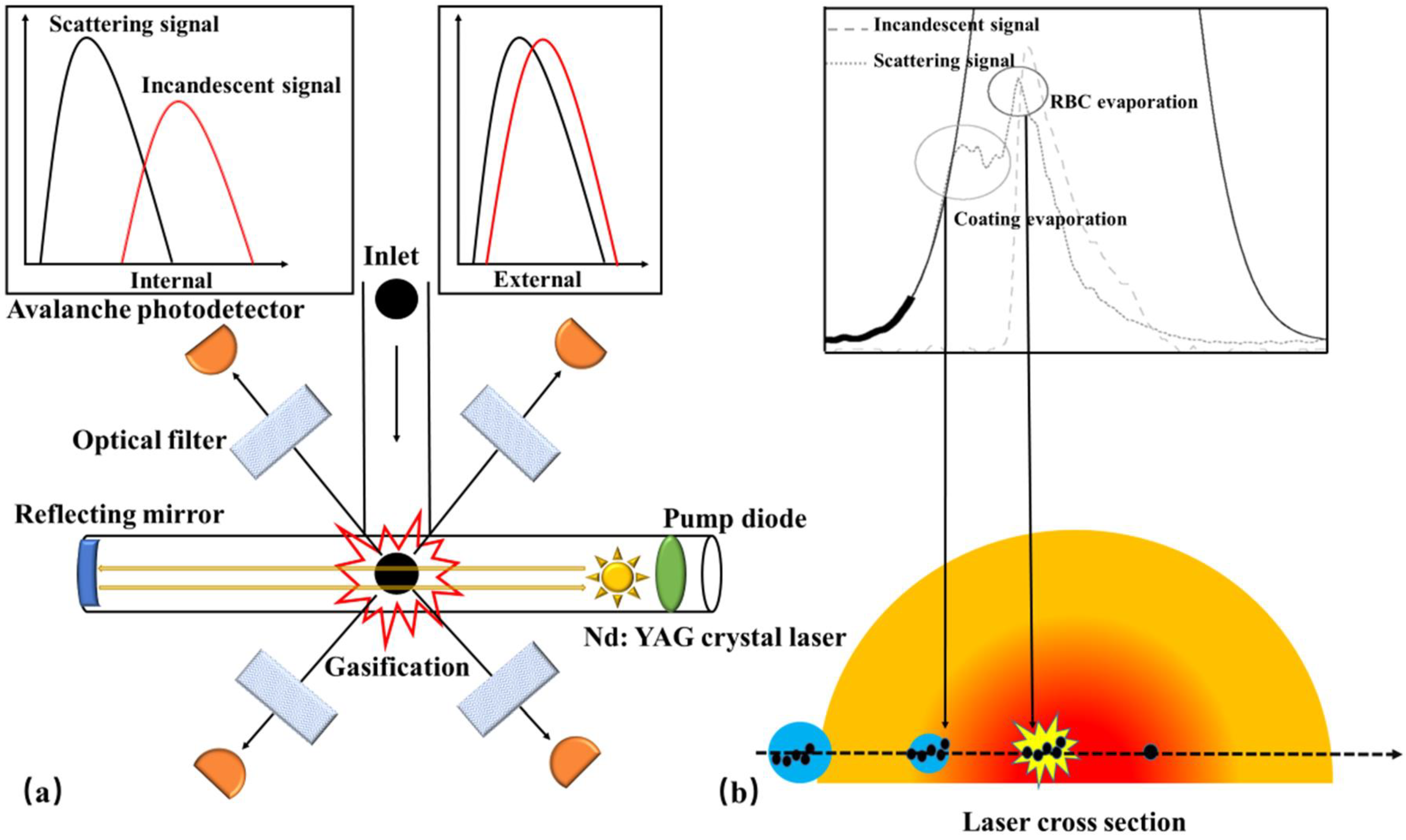
4.1. Single Particle Soot Photometer
4.2. Soot Particle–Aerosol Mass Spectrometer
4.3. Correction Scheme for Refractory Black Carbon Mass Loss
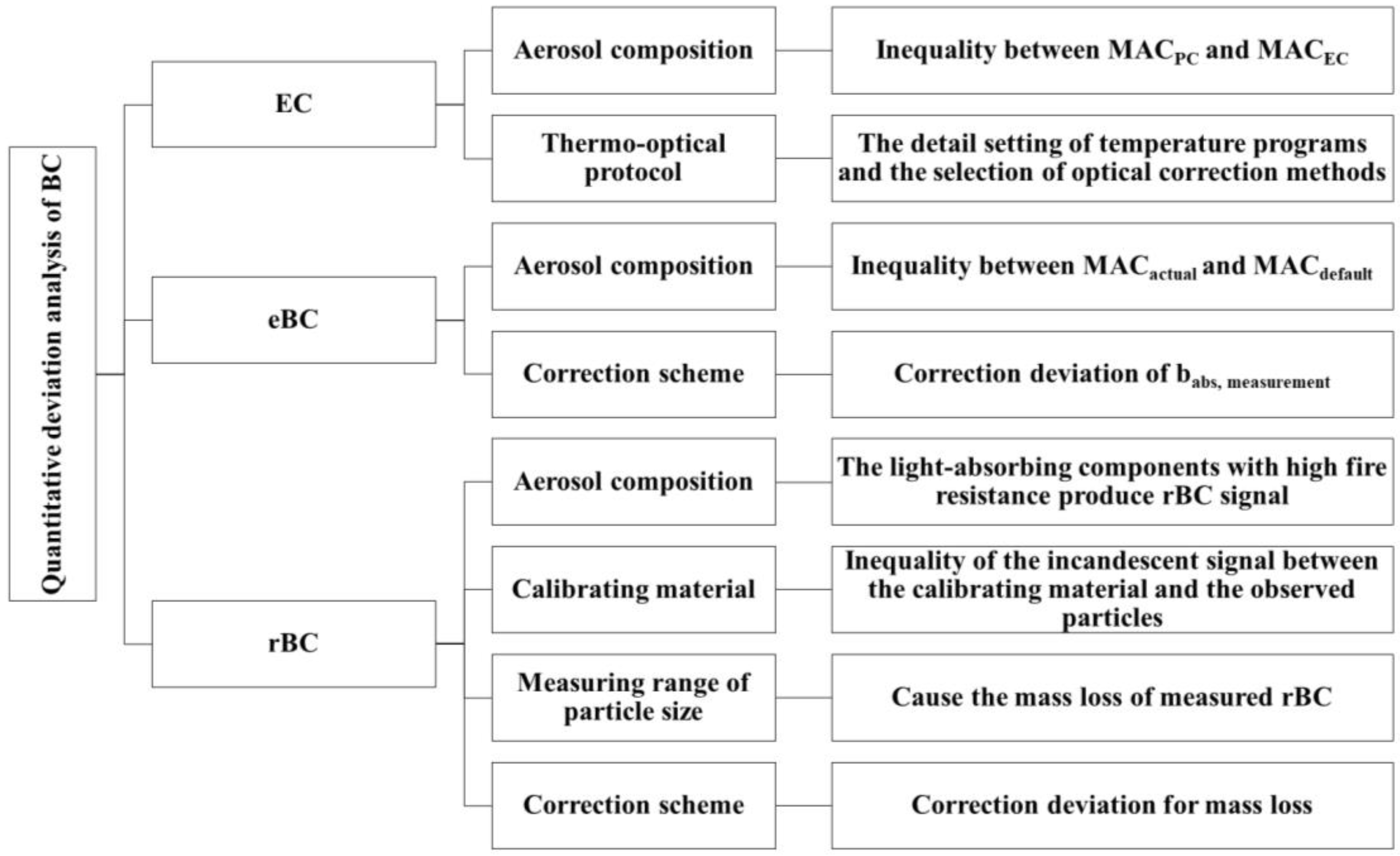
5. Inter-Comparison of Black Carbon Quantification between Techniques
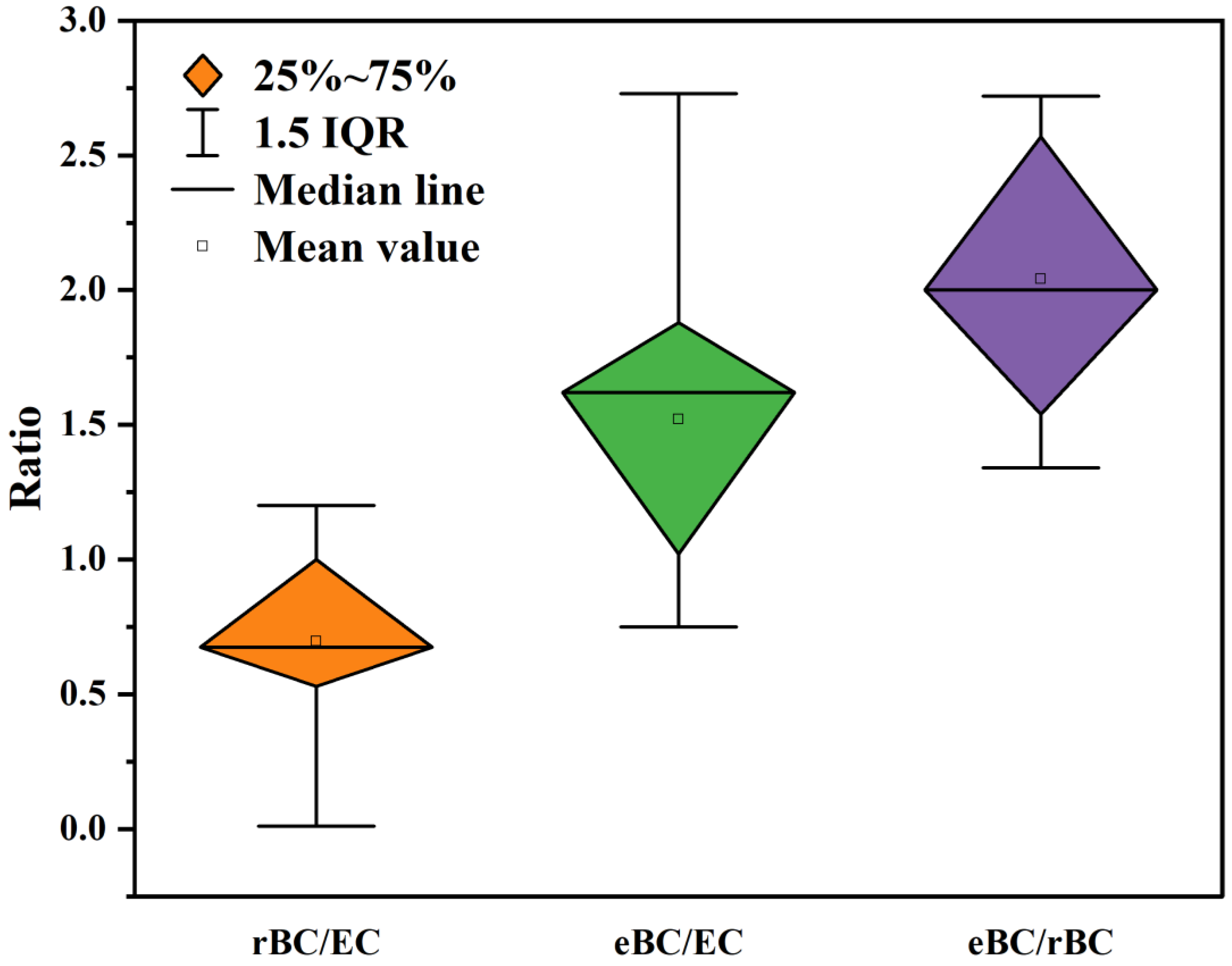
5.1. Refractory Black Carbon vs. Elemental Carbon
5.2. Equivalent Black Carbon vs. Elemental Carbon
5.3. Equivalent Black Carbon vs. Refractory Black Carbon
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bond, T.C.; Doherty, S.J.; Fahey, D.W.; Forster, P.M.; Berntsen, T.; DeAngelo, B.J.; Flanner, M.G.; Ghan, S.; Kärcher, B.; Koch, D.; et al. Bounding the Role of Black Carbon in the Climate System: A Scientific Assessment. J. Geophys. Res. Atmos. 2013, 118, 5380–5552. [Google Scholar] [CrossRef]
- Li, J.; Carlson, B.E.; Yung, Y.L.; Lv, D.; Hansen, J.; Penner, J.E.; Liao, H.; Ramaswamy, V.; Kahn, R.A.; Zhang, P.; et al. Scattering and Absorbing Aerosols in the Climate System. Nat. Rev. Earth Environ. 2022, 3, 363–379. [Google Scholar] [CrossRef]
- Moteki, N. Climate-Relevant Properties of Black Carbon Aerosols Revealed by in Situ Measurements: A Review. Prog. Earth Planet. Sci. 2023, 10, 12. [Google Scholar] [CrossRef]
- Petzold, A.; Ogren, J.A.; Fiebig, M.; Laj, P.; Li, S.M.; Baltensperger, U.; Holzer-Popp, T.; Kinne, S.; Pappalardo, G.; Sugimoto, N. Recommendations for Reporting “Black Carbon” Measurements. Atmos. Chem. Phys. 2013, 13, 8365–8379. [Google Scholar] [CrossRef]
- Barker, J.D.; Kaspari, S.; Gabrielli, P.; Wegner, A.; Beaudon, E.; Sierra-Hernández, M.R.; Thompson, L. Drought-Induced Biomass Burning as a Source of Black Carbon to the Central Himalaya since 1781 CE as Reconstructed from the Dasuopu Ice Core. Atmos. Chem. Phys. 2021, 21, 5615–5633. [Google Scholar] [CrossRef]
- Blanco-Donado, E.P.; Schneider, I.L.; Artaxo, P.; Lozano-Osorio, J.; Portz, L.; Oliveira, M.L.S. Source Identification and Global Implications of Black Carbon. Geosci. Front. 2022, 13, 101149. [Google Scholar] [CrossRef]
- Deng, J.; Guo, H.; Zhang, H.; Zhu, J.; Wang, X.; Fu, P. Source Apportionment of Black Carbon Aerosols from Light Absorption Observation and Source-Oriented Modeling: An Implication in a Coastal City in China. Atmos. Chem. Phys. 2020, 20, 14419–14435. [Google Scholar] [CrossRef]
- Fang, W.; Evangeliou, N.; Eckhardt, S.; Xing, J.; Zhang, H.; Xiao, H.; Zhao, M.; Kim, S.-W. Increased Contribution of Biomass Burning to Haze Events in Shanghai since China’s Clean Air Actions. Commun. Earth Environ. 2023, 4, 310. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, J.; Gagné, S.; Chan, T.W.; Thomson, K.; Fofie, E.; Cary, R.A.; Rutherford, D.; Comer, B.; Swanson, J.; et al. Sources of Variance in BC Mass Measurements from a Small Marine Engine: Influence of the Instruments, Fuels and Loads. Atmos. Environ. 2018, 182, 128–137. [Google Scholar] [CrossRef]
- Jing, A.; Zhu, B.; Wang, H.; Yu, X.; An, J.; Kang, H. Source Apportionment of Black Carbon in Different Seasons in the Northern Suburb of Nanjing, China. Atmos. Environ. 2019, 201, 190–200. [Google Scholar] [CrossRef]
- Sedlacek, A.J.; Onasch, T.B.; Nichman, L.; Lewis, E.R.; Davidovits, P.; Freedman, A.; Williams, L. Formation of Refractory Black Carbon by SP2-Induced Charring of Organic Aerosol. Aerosol Sci. Technol. 2018, 52, 1345–1350. [Google Scholar] [CrossRef]
- Yan, C.; Zheng, M.; Shen, G.; Cheng, Y.; Ma, S.; Sun, J.; Cui, M.; Zhang, F.; Han, Y.; Chen, Y. Characterization of Carbon Fractions in Carbonaceous Aerosols from Typical Fossil Fuel Combustion Sources. Fuel 2019, 254, 115620. [Google Scholar] [CrossRef]
- Cappa, C.D.; Zhang, X.; Russell, L.M.; Collier, S.; Lee, A.K.Y.; Chen, C.-L.; Betha, R.; Chen, S.; Liu, J.; Price, D.J.; et al. Light Absorption by Ambient Black and Brown Carbon and Its Dependence on Black Carbon Coating State for Two California, USA, Cities in Winter and Summer. J. Geophys. Res. Atmos. 2019, 124, 1550–1577. [Google Scholar] [CrossRef]
- Ivančič, M.; Rigler, M.; Alföldy, B.; Lavrič, G.; Ježek Brecelj, I.; Gregorič, A. Highly Time-Resolved Apportionment of Carbonaceous Aerosols from Wildfire Using the TC–BC Method: Camp Fire 2018 Case Study. Toxics 2023, 11, 497. [Google Scholar] [CrossRef] [PubMed]
- Krasowsky, T.S.; McMeeking, G.R.; Wang, D.; Sioutas, C.; Ban-Weiss, G.A. Measurements of the Impact of Atmospheric Aging on Physical and Optical Properties of Ambient Black Carbon Particles in Los Angeles. Atmos. Environ. 2016, 142, 496–504. [Google Scholar] [CrossRef]
- Sun, J.Y.; Wu, C.; Wu, D.; Cheng, C.; Li, M.; Li, L.; Deng, T.; Yu, J.Z.; Li, Y.J.; Zhou, Q.; et al. Amplification of Black Carbon Light Absorption Induced by Atmospheric Aging: Temporal Variation at Seasonal and Diel Scales in Urban Guangzhou. Atmos. Chem. Phys. 2020, 20, 2445–2470. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, N.; Wang, J.; Huang, X.; Wang, Z.; Liu, T.; Geng, G.; Qi, X.; Nie, W.; Chi, X.; et al. Strong Haze-Black Carbon-Climate Connections Observed Across Northern and Eastern China. J. Geophys. Res. Atmos. 2023, 128, e2023JD038505. [Google Scholar] [CrossRef]
- Hu, W.; Hu, M.; Hu, W.; Jimenez, J.L.; Yuan, B.; Chen, W.; Wang, M.; Wu, Y.; Chen, C.; Wang, Z.; et al. Chemical Composition, Sources, and Aging Process of Submicron Aerosols in Beijing: Contrast between Summer and Winter. J. Geophys. Res. Atmos. 2016, 121, 1955–1977. [Google Scholar] [CrossRef]
- Feng, X.; Wang, J.; Teng, S.; Xu, X.; Zhu, B.; Wang, J.; Zhu, X.; Yurkin, M.A.; Liu, C. Can Light Absorption of Black Carbon Still Be Enhanced by Mixing with Absorbing Materials? Atmos. Environ. 2021, 253, 118358. [Google Scholar] [CrossRef]
- Bove, H.; Bongaerts, E.; Slenders, E.; Bijnens, E.M.; Saenen, N.D.; Gyselaers, W.; Van Eyken, P.; Plusquin, M.; Roeffaers, M.B.J.; Ameloot, M.; et al. Ambient Black Carbon Particles Reach the Fetal Side of Human Placenta. Nat. Commun. 2019, 10, 3866. [Google Scholar] [CrossRef]
- Li, B.; Gasser, T.; Ciais, P.; Piao, S.; Tao, S.; Balkanski, Y.; Hauglustaine, D.; Boisier, J.P.; Chen, Z.; Huang, M.; et al. The Contribution of China’s Emissions to Global Climate Forcing. Nature 2016, 531, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Shindell, D.; Kuylenstierna, J.C.I.; Vignati, E.; van Dingenen, R.; Amann, M.; Klimont, Z.; Anenberg, S.C.; Muller, N.; Janssens-Maenhout, G.; Raes, F.; et al. Simultaneously Mitigating Near-Term Climate Change and Improving Human Health and Food Security. Science 2012, 335, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Cai, R.; Liu, C.; Jiang, J.; Nie, W.; Wang, J.; Moteki, N.; Zaveri, R.A.; Huang, X.; et al. Unified Theoretical Framework for Black Carbon Mixing State Allows Greater Accuracy of Climate Effect Estimation. Nat. Commun. 2023, 14, 2703. [Google Scholar] [CrossRef] [PubMed]
- Rönkkö, T.; Saarikoski, S.; Kuittinen, N.; Karjalainen, P.; Keskinen, H.; Järvinen, A.; Mylläri, F.; Aakko-Saksa, P.; Timonen, H. Review of Black Carbon Emission Factors from Different Anthropogenic Sources. Environ. Res. Lett. 2023, 18, 033004. [Google Scholar] [CrossRef]
- Zhai, J.; Yang, X.; Li, L.; Bai, B.; Liu, P.; Huang, Y.; Fu, T.-M.; Zhu, L.; Zeng, Z.; Tao, S.; et al. Absorption Enhancement of Black Carbon Aerosols Constrained by Mixing-State Heterogeneity. Environ. Sci. Technol. 2022, 56, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Kalbermatter, D.M.; Močnik, G.; Drinovec, L.; Visser, B.; Röhrbein, J.; Oscity, M.; Weingartner, E.; Hyvärinen, A.-P.; Vasilatou, K. Comparing Black-Carbon- and Aerosol-Absorption-Measuring Instruments—A New System Using Lab-Generated Soot Coated with Controlled Amounts of Secondary Organic Matter. Atmos. Meas. Tech. 2022, 15, 561–572. [Google Scholar] [CrossRef]
- Lack, D.A.; Moosmüller, H.; McMeeking, G.R.; Chakrabarty, R.K.; Baumgardner, D. Characterizing Elemental, Equivalent Black, and Refractory Black Carbon Aerosol Particles: A Review of Techniques, Their Limitations and Uncertainties. Anal. Bioanal. Chem. 2014, 406, 99–122. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, M.; Liu, Y.; Jin, Y.; Liu, J.; Zhang, B.; Yang, X.; Wu, Y.; Zhang, T.; Xiang, Y.; et al. Intercomparison of Equivalent Black Carbon (eBC) and Elemental Carbon (EC) Concentrations with Three-Year Continuous Measurement in Beijing, China. Environ. Res. 2022, 209, 112791. [Google Scholar] [CrossRef]
- Schwarz, J.P.; Gao, R.S.; Spackman, J.R.; Watts, L.A.; Thomson, D.S.; Fahey, D.W.; Ryerson, T.B.; Peischl, J.; Holloway, J.S.; Trainer, M.; et al. Measurement of the Mixing State, Mass, and Optical Size of Individual Black Carbon Particles in Urban and Biomass Burning Emissions. Geophys. Res. Lett. 2008, 35, L13810. [Google Scholar] [CrossRef]
- Sedlacek, A.J.; Lewis, E.R.; Kleinman, L.; Xu, J.; Zhang, Q. Determination of and Evidence for Non-Core-Shell Structure of Particles Containing Black Carbon Using the Single-Particle Soot Photometer (SP2). Geophys. Res. Lett. 2012, 39, L06802. [Google Scholar] [CrossRef]
- Pileci, R.E.; Modini, R.L.; Bertò, M.; Yuan, J.; Corbin, J.C.; Marinoni, A.; Henzing, B.; Moerman, M.M.; Putaud, J.P.; Spindler, G.; et al. Comparison of Co-Located Refractory Black Carbon (rBC) and Elemental Carbon (EC) Mass Concentration Measurements during Field Campaigns at Several European Sites. Atmos. Meas. Tech. 2021, 14, 1379–1403. [Google Scholar] [CrossRef]
- Aakko-Saksa, P.; Kuittinen, N.; Murtonen, T.; Koponen, P.; Aurela, M.; Järvinen, A.; Teinilä, K.; Saarikoski, S.; Barreira, L.M.F.; Salo, L.; et al. Suitability of Different Methods for Measuring Black Carbon Emissions from Marine Engines. Atmosphere 2022, 13, 31. [Google Scholar] [CrossRef]
- Malik, A.; Aggarwal, S.G.; Ohata, S.; Mori, T.; Kondo, Y.; Sinha, P.R.; Patel, P.; Kumar, B.; Singh, K.; Soni, D.; et al. Measurement of Black Carbon in Delhi: Evidences of Regional Transport, Meteorology and Local Sources for Pollution Episodes. Aerosol Air Qual. Res. 2022, 22, 220128. [Google Scholar] [CrossRef]
- Asmi, E.; Backman, J.; Servomaa, H.; Virkkula, A.; Gini, M.I.; Eleftheriadis, K.; Müller, T.; Ohata, S.; Kondo, Y.; Hyvärinen, A. Absorption Instruments Inter-Comparison Campaign at the Arctic Pallas Station. Atmos. Meas. Tech. 2021, 14, 5397–5413. [Google Scholar] [CrossRef]
- Chan, T.W.; Huang, L.; Banwait, K.; Zhang, W.; Ernst, D.; Wang, X.; Watson, J.G.; Chow, J.C.; Green, M.; Czimczik, C.I.; et al. Inter-Comparison of Elemental and Organic Carbon Mass Measurements from Three North American National Long-Term Monitoring Networks at a Co-Located Site. Atmos. Meas. Tech. 2019, 12, 4543–4560. [Google Scholar] [CrossRef]
- Li, H.; Lamb, K.D.; Schwarz, J.P.; Selimovic, V.; Yokelson, R.J.; McMeeking, G.R.; May, A.A. Inter-Comparison of Black Carbon Measurement Methods for Simulated Open Biomass Burning Emissions. Atmos. Environ. 2019, 206, 156–169. [Google Scholar] [CrossRef]
- Onasch, T.B.; Trimborn, A.; Fortner, E.C.; Jayne, J.T.; Kok, G.L.; Williams, L.R.; Davidovits, P.; Worsnop, D.R. Soot Particle Aerosol Mass Spectrometer: Development, Validation, and Initial Application. Aerosol Sci. Technol. 2012, 46, 804–817. [Google Scholar] [CrossRef]
- Cheng, Y.; He, K.B.; Duan, F.K.; Zheng, M.; Ma, Y.L.; Tan, J.H.; Du, Z.Y. Improved Measurement of Carbonaceous Aerosol: Evaluation of the Sampling Artifacts and Inter-Comparison of the Thermal–optical Analysis Methods. Atmos. Chem. Phys. 2010, 10, 8533–8548. [Google Scholar] [CrossRef]
- Watson, J.G.; Chow, J.C.; Chen, L.W.A.; Frank, N.H. Methods to Assess Carbonaceous Aerosol Sampling Artifacts for IMPROVE and Other Long-Term Networks. J. Air Waste Manag. Assoc. 2009, 59, 898–911. [Google Scholar] [CrossRef]
- Haller, T.; Sommer, E.; Steinkogler, T.; Rentenberger, C.; Wonaschuetz, A.; Kasper-Giebl, A.; Grothe, H.; Hitzenberger, R. Investigation of Structural Changes of Atmospheric Aerosol Samples during Two Thermal–Optical Measurement Procedures (EUSAAR2, NIOSH870). Atmos. Meas. Tech. 2021, 14, 3721–3735. [Google Scholar] [CrossRef]
- Haller, T.; Rentenberger, C.; Meyer, J.C.; Felgitsch, L.; Grothe, H.; Hitzenberger, R. Structural Changes of CAST Soot during a Thermal–Optical Measurement Protocol. Atmos. Meas. Tech. 2019, 12, 3503–3519. [Google Scholar] [CrossRef]
- Chow, J.C.; Yu, J.Z.; Watson, J.G.; Hang Ho, S.S.; Bohannan, T.L.; Hays, M.D.; Fung, K.K. The Application of Thermal Methods for Determining Chemical Composition of Carbonaceous Aerosols: A Review. J. Environ. Sci. Health Part A Toxic/Hazard. Subst. Environ. Eng. 2007, 42, 1521–1541. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.C.; Watson, J.G.; Robles, J.; Wang, X.; Chen, L.-W.A.; Trimble, D.L.; Kohl, S.D.; Tropp, R.J.; Fung, K.K. Quality Assurance and Quality Control for Thermal/Optical Analysis of Aerosol Samples for Organic and Elemental Carbon. Anal. Bioanal. Chem. 2011, 401, 3141–3152. [Google Scholar] [CrossRef]
- Wu, C.; Yu, J.Z. Evaluation of Linear Regression Techniques for Atmospheric Applications: The Importance of Appropriate Weighting. Atmos. Meas. Tech. 2018, 11, 1233–1250. [Google Scholar] [CrossRef]
- Blanchard, C.L.; Hidy, G.M.; Tanenbaum, S.; Edgerton, E.S.; Hartsell, B.E. The Southeastern Aerosol Research and Characterization (SEARCH) Study: Spatial Variations and Chemical Climatology, 1999–2010. J. Air Waste Manag. Assoc. 2013, 63, 260–275. [Google Scholar] [CrossRef]
- Cavalli, F.; Viana, M.; Yttri, K.E.; Genberg, J.; Putaud, J.-P. Toward a Standardised Thermal–optical Protocol for Measuring Atmospheric Organic and Elemental Carbon: The EUSAAR Protocol. Atmos. Meas. Tech. 2010, 3, 79–89. [Google Scholar] [CrossRef]
- Chow, J.C.; Chen, L.-W.A.; Watson, J.G.; Lowenthal, D.H.; Magliano, K.A.; Turkiewicz, K.; Lehrman, D.E. PM2.5 Chemical Com-1364 Position and Spatiotemporal Variability during the California Regional PM10/PM2.5 Air Quality Study (CRPAQS). J. Geophys. Res. Atmos. 2006, 111, 2005JD006457. [Google Scholar] [CrossRef]
- Dabek-Zlotorzynska, E.; Dann, T.F.; Kalyani Martinelango, P.; Celo, V.; Brook, J.R.; Mathieu, D.; Ding, L.; Austin, C.C. Canadian National Air Pollution Surveillance (NAPS) PM2.5 Speciation Program: Methodology and PM2.5 Chemical Composition for the Years 2003–2008. Atmos. Environ. 2011, 45, 673–686. [Google Scholar] [CrossRef]
- Flanagan, J.B.; Jayanty, R.K.M.; Rickman, E.E., Jr.; Peterson, M.R. PM2.5 Speciation Trends Network: Evaluation of Whole-System Uncertainties Using Data from Sites with Collocated Samplers. J. Air Waste Manag. Assoc. 2006, 56, 492–499. [Google Scholar] [CrossRef][Green Version]
- Malm, W.C.; Hand, J.L. An Examination of the Physical and Optical Properties of Aerosols Collected in the IMPROVE Program. Atmos. Environ. 2007, 41, 3407–3427. [Google Scholar] [CrossRef]
- Schauer, J.J.; Mader, B.T.; Deminter, J.T.; Heidemann, G.; Bae, M.S.; Seinfeld, J.H.; Flagan, R.C.; Cary, R.A.; Smith, D.; Huebert, B.J.; et al. ACE-Asia Intercomparison of a Thermal–optical Method for the Determination of Particle-Phase Organic and Elemental Carbon. Environ. Sci. Technol. 2003, 37, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.C.; Watson, J.G.; Pritchett, L.C.; Pierson, W.R.; Frazier, C.A.; Purcell, R.G. The Dri Thermal/Optical Reflectance Carbon Analysis System: Description, Evaluation and Applications in U.S. Air Quality Studies. Atmos. Environ. Part A 1993, 27, 1185–1201. [Google Scholar] [CrossRef]
- Wu, C.; Ng, W.M.; Huang, J.; Wu, D.; Yu, J.Z. Determination of Elemental and Organic Carbon in PM2.5 in the Pearl River Delta Region: Inter-Instrument (Sunset vs. DRI Model 2001 Thermal/Optical Carbon Analyzer) and Inter-Protocol Comparisons (IMPROVE vs. ACE-Asia Protocol). Aerosol Sci. Technol. 2012, 46, 610–621. [Google Scholar] [CrossRef]
- Zhang, X.; Trzepla, K.; White, W.; Raffuse, S.; Hyslop, N.P. Intercomparison of Thermal–Optical Carbon Measurements by Sunset and Desert Research Institute (DRI) Analyzers Using the IMPROVE_A Protocol. Atmos. Meas. Tech. 2021, 14, 3217–3231. [Google Scholar] [CrossRef]
- Bauer, J.J.; Yu, X.-Y.; Cary, R.; Laulainen, N.; Berkowitz, C. Characterization of the Sunset Semi-Continuous Carbon Aerosol Analyzer. J. Air Waste Manag. Assoc. 2009, 59, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Ammerlaan, B.A.J.; Jedynska, A.D.; Henzing, J.S.; Holzinger, R. On a Possible Bias in Elemental Carbon Measurements with the Sunset Thermal/Optical Carbon Analyser Caused by Unstable Laser Signal. Atmos. Environ. 2015, 122, 571–576. [Google Scholar] [CrossRef]
- Yang, H.; Yu, J.Z. Uncertainties in Charring Correction in the Analysis of Elemental and Organic Carbon in Atmospheric Particles by Thermal/Optical Methods. Environ. Sci. Technol. 2002, 36, 5199–5204. [Google Scholar] [CrossRef]
- Yu, J.Z.; Xu, J.; Yang, H. Charring Characteristics of Atmospheric Organic Particulate Matter in Thermal Analysis. Environ. Sci. Technol. 2002, 36, 754–761. [Google Scholar] [CrossRef]
- Huntzicker, J.J.; Johnson, R.L.; Shah, J.J.; Cary, R.A. Analysis of Organic and Elemental Carbon in Ambient Aerosols by a Thermal–optical Method. In Particulate Carbon: Atmospheric Life Cycle; Wolff, G.T., Klimisch, R.L., Eds.; Springer: Boston, MA, USA, 1982; pp. 79–88. ISBN 978-1-4684-4154-3. [Google Scholar]
- Birch, M.E.; Cary, R.A. Elemental Carbon-Based Method for Monitoring Occupational Exposures to Particulate Diesel Exhaust. Aerosol Sci. Technol. 1996, 25, 221–241. [Google Scholar] [CrossRef]
- Chow, J.C.; Watson, J.G.; Chen, L.-W.A.; Arnott, W.P.; Moosmüller, H.; Fung, K. Equivalence of Elemental Carbon by Thermal/Optical Reflectance and Transmittance with Different Temperature Protocols. Environ. Sci. Technol. 2004, 38, 4414–4422. [Google Scholar] [CrossRef]
- Subramanian, R.; Khlystov, A.Y.; Robinson, A.L. Effect of Peak Inert-Mode Temperature on Elemental Carbon Measured Using Thermal–optical Analysis. Aerosol Sci. Technol. 2006, 40, 763–780. [Google Scholar] [CrossRef]
- Chow, J.C.; Watson, J.G.; Zhang, H.; Yu, J.Z. Refining Temperature Measures in Thermal/Optical Carbon Analysis. Atmos. Chem. Phys. 2005, 5, 2961–2972. [Google Scholar] [CrossRef]
- Karanasiou, A.; Minguillón, M.C.; Viana, M.; Alastuey, A.; Putaud, J.-P.; Maenhaut, W.; Panteliadis, P.; Močnik, G.; Favez, O.; Kuhlbusch, T.A.J. Thermal–optical Analysis for the Measurement of Elemental Carbon (EC) and Organic Carbon (OC) in Ambient Air a Literature Review. Atmos. Meas. Tech. Discuss. 2015, 8, 9649–9712. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, Y.; Guo, H.; Zhi, G.; Xiong, S.; Li, J.; Sheng, G.; Fu, J. Characteristics of Organic and Elemental Carbon in PM2.5 Samples in Shanghai, China. Atmos. Res. 2009, 92, 434–442. [Google Scholar] [CrossRef]
- Cheng, Y.; He, K.-B.; Duan, F.-K.; Du, Z.-Y.; Zheng, M.; Ma, Y.-L. Ambient Organic Carbon to Elemental Carbon Ratios: Influence of the Thermal–Optical Temperature Protocol and Implications. Sci. Total Environ. 2014, 468–469, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.; Hays, M.D.; Geron, C.; Jetter, J. Differences in the OC/EC Ratios That Characterize Ambient and Source Aerosols Due to Thermal–optical Analysis. Aerosol Sci. Technol. 2012, 46, 127–137. [Google Scholar] [CrossRef]
- Piazzalunga, A.; Belis, C.; Bernardoni, V.; Cazzuli, O.; Fermo, P.; Valli, G.; Vecchi, R. Estimates of Wood Burning Contribution to PM by the Macro-Tracer Method Using Tailored Emission Factors. Atmos. Environ. 2011, 45, 6642–6649. [Google Scholar] [CrossRef]
- Wu, C.; Huang, X.H.H.; Ng, W.M.; Griffith, S.M.; Yu, J.Z. Inter-Comparison of NIOSH and IMPROVE Protocols for OC and EC Determination: Implications for Inter-Protocol Data Conversion. Atmos. Meas. Tech. 2016, 9, 4547–4560. [Google Scholar] [CrossRef]
- Watson, J.G.; Chow, J.C.; Chen, L.W.A. Summary of Organic and Elemental Carbon/Black Carbon Analysis Methods and Intercomparisons. Aerosol Air Qual. Res. 2005, 5, 65–102. [Google Scholar] [CrossRef]
- Chow, J.C.; Watson, J.G.; Crow, D.; Lowenthal, D.H.; Merrifield, T. Comparison of IMPROVE and NIOSH Carbon Measurements. Aerosol Sci. Technol. 2001, 34, 23–34. [Google Scholar] [CrossRef]
- Cheng, Y.; Duan, F.-K.; He, K.-B.; Zheng, M.; Du, Z.-Y.; Ma, Y.-L.; Tan, J.-H. Intercomparison of Thermal–Optical Methods for the Determination of Organic and Elemental Carbon: Influences of Aerosol Composition and Implications. Environ. Sci. Technol. 2011, 45, 10117–10123. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-W.A.; Chow, J.C.; Watson, J.G.; Moosmüller, H.; Arnott, W.P. Modeling Reflectance and Transmittance of Quartz-Fiber Filter Samples Containing Elemental Carbon Particles: Implications for Thermal/Optical Analysis. J. Aerosol Sci. 2004, 35, 765–780. [Google Scholar] [CrossRef]
- Chiappini, L.; Verlhac, S.; Aujay, R.; Maenhaut, W.; Putaud, J.P.; Sciare, J.; Jaffrezo, J.L.; Liousse, C.; Galy-Lacaux, C.; Alleman, L.Y.; et al. Clues for a Standardised Thermal–optical Protocol for the Assessment of Organic and Elemental Carbon within Ambient Air Particulate Matter. Atmos. Meas. Tech. 2014, 7, 1649–1661. [Google Scholar] [CrossRef]
- Brown, R.J.C.; Beccaceci, S.; Butterfield, D.M.; Quincey, P.G.; Harris, P.M.; Maggos, T.; Panteliadis, P.; John, A.; Jedynska, A.; Kuhlbusch, T.A.J.; et al. Standardisation of a European Measurement Method for Organic Carbon and Elemental Carbon in Ambient Air: Results of the Field Trial Campaign and the Determination of a Measurement Uncertainty and Working Range. Environ. Sci. Process. Impacts 2017, 19, 1249–1259. [Google Scholar] [CrossRef]
- Chow, J.C.; Watson, J.G.; Louie, P.K.K.; Chen, L.W.A.; Sin, D. Comparison of PM2.5 Carbon Measurement Methods in Hong Kong, China. Environ. Pollut. 2005, 137, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Zhi, G.; Chen, Y.; Sun, J.; Chen, L.; Tian, W.; Duan, J.; Zhang, G.; Chai, F.; Sheng, G.; Fu, J. Harmonizing Aerosol Carbon Measurements between Two Conventional Thermal/Optical Analysis Methods. Environ. Sci. Technol. 2011, 45, 2902–2908. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, M.; Calzolai, G.; Chiari, M.; Cincinelli, A.; Lucarelli, F.; Martellini, T.; Nava, S. A Comparison between Thermal–optical Transmittance Elemental Carbon Measured by Different Protocols in PM2.5 Samples. Sci. Total Environ. 2016, 571, 195–205. [Google Scholar] [CrossRef]
- Hu, Z.; Kang, S.; Xu, J.; Zhang, C.; Li, X.; Yan, F.; Zhang, Y.; Chen, P.; Li, C. Significant Overestimation of Black Carbon Concentration Caused by High Organic Carbon in Aerosols of the Tibetan Plateau. Atmos. Environ. 2023, 294, 119486. [Google Scholar] [CrossRef]
- Karanasiou, A.; Diapouli, E.; Cavalli, F.; Eleftheriadis, K.; Viana, M.; Alastuey, A.; Querol, X.; Reche, C. On the Quantification of Atmospheric Carbonate Carbon by Thermal/Optical Analysis Protocols. Atmos. Meas. Tech. 2011, 4, 2409–2419. [Google Scholar] [CrossRef]
- Zhang, Q.; Shen, Z.; Zhang, L.; Zeng, Y.; Ning, Z.; Zhang, T.; Lei, Y.; Wang, Q.; Li, G.; Sun, J.; et al. Investigation of Primary and Secondary Particulate Brown Carbon in Two Chinese Cities of Xi’an and Hong Kong in Wintertime. Environ. Sci. Technol. 2020, 54, 3803–3813. [Google Scholar] [CrossRef]
- Han, Y.M.; Chen, L.W.A.; Huang, R.J.; Chow, J.C.; Watson, J.G.; Ni, H.Y.; Liu, S.X.; Fung, K.K.; Shen, Z.X.; Wei, C.; et al. Carbonaceous Aerosols in Megacity Xi’an, China: Implications of Thermal/Optical Protocols Comparison. Atmos. Environ. 2016, 132, 58–68. [Google Scholar] [CrossRef]
- Fung, K.; Chow, J.C.; Watson, J.G. Evaluation of OC/EC Speciation by Thermal Manganese Dioxide Oxidation and the IMPROVE Method. J. Air Waste Manag. Assoc. 2002, 52, 1333–1341. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.; Chung, A.; Paulson, S.E. The Effect of Metal Salts on Quantification of Elemental and Organic Carbon in Diesel Exhaust Particles Using Thermal–optical Evolved Gas Analysis. Atmos. Chem. Phys. 2010, 10, 11447–11457. [Google Scholar] [CrossRef]
- Novakov, T.; Corrigan, C.E. Thermal Characterization of Biomass Smoke Particles. Microchim. Acta 1995, 119, 157–166. [Google Scholar] [CrossRef]
- Liu, J.M.; Du, Z.Y.; Liang, L.L.; Yu, Q.Q.; Shen, G.F.; Ma, Y.L.; Zheng, M.; Cheng, Y.; He, K.B. Uncertainties in Thermal–optical Measurements of Black Carbon: Insights from Source and Ambient Samples. Sci. Total Environ. 2019, 656, 239–249. [Google Scholar] [CrossRef]
- Kang, H.; Shang, X.; Abdumutallip, M.; Chen, Y.; Li, L.; Wang, X.; Li, C.; Ouyang, H.; Tang, X.; Wang, L.; et al. Accurate Observation of Black and Brown Carbon in Atmospheric Fine Particles via a Versatile Aerosol Concentration Enrichment System (VACES). Sci. Total Environ. 2022, 837, 155817. [Google Scholar] [CrossRef]
- Rathod, T.D.; Sahu, S.K. Measurements of Optical Properties of Black and Brown Carbon Using Multi-Wavelength Absorption Technique at Mumbai, India. J. Earth Syst. Sci. 2022, 131, 32. [Google Scholar] [CrossRef]
- Soni, A.; Gupta, T. Alternative Approach for the In Situ Measurement of Absorption Enhancement of Atmospheric Black Carbon Due to Atmospheric Mixing. ACS Earth Space Chem. 2022, 6, 261–267. [Google Scholar] [CrossRef]
- Reisinger, P.; Wonaschütz, A.; Hitzenberger, R.; Petzold, A.; Bauer, H.; Jankowski, N.; Puxbaum, H.; Chi, X.; Maenhaut, W. Intercomparison of Measurement Techniques for Black or Elemental Carbon Under Urban Background Conditions in Wintertime: Influence of Biomass Combustion. Environ. Sci. Technol. 2008, 42, 884–889. [Google Scholar] [CrossRef]
- Cheng, Y.; Zheng, M.; He, K.-B.; Chen, Y.; Yan, B.; Russell, A.G.; Shi, W.; Jiao, Z.; Sheng, G.; Fu, J.; et al. Comparison of Two Thermal–optical Methods for the Determination of Organic Carbon and Elemental Carbon: Results from the Southeastern United States. Atmos. Environ. 2011, 45, 1913–1918. [Google Scholar] [CrossRef]
- Bao, M.; Zhang, Y.-L.; Cao, F.; Lin, Y.-C.; Wang, Y.; Liu, X.; Zhang, W.; Fan, M.; Xie, F.; Cary, R.; et al. Highly Time-Resolved Characterization of Carbonaceous Aerosols Using a Two-Wavelength Sunset Thermal–Optical Carbon Analyzer. Atmos. Meas. Tech. 2021, 14, 4053–4068. [Google Scholar] [CrossRef]
- Chen, L.-W.A.; Chow, J.C.; Wang, X.L.; Robles, J.A.; Sumlin, B.J.; Lowenthal, D.H.; Zimmermann, R.; Watson, J.G. Multi-Wavelength Optical Measurement to Enhance Thermal/Optical Analysis for Carbonaceous Aerosol. Atmos. Meas. Tech. 2015, 8, 451–461. [Google Scholar] [CrossRef]
- Sillanpää, M.; Frey, A.; Hillamo, R.; Pennanen, A.S.; Salonen, R.O. Organic, Elemental and Inorganic Carbon in Particulate Matter of Six Urban Environments in Europe. Atmos. Chem. Phys. 2005, 5, 2869–2879. [Google Scholar] [CrossRef]
- Jankowski, N.; Schmidl, C.; Marr, I.L.; Bauer, H.; Puxbaum, H. Comparison of Methods for the Quantification of Carbonate Carbon in Atmospheric PM10 Aerosol Samples. Atmos. Environ. 2008, 42, 8055–8064. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Perron, N.; Ciobanu, V.G.; Zotter, P.; Minguillón, M.C.; Wacker, L.; Prévôt, A.S.H.; Baltensperger, U.; Szidat, S. On the Isolation of OC and EC and the Optimal Strategy of Radiocarbon-Based Source Apportionment of Carbonaceous Aerosols. Atmos. Chem. Phys. 2012, 12, 10841–10856. [Google Scholar] [CrossRef]
- Lappi, M.K.; Ristimäki, J.M. Evaluation of Thermal Optical Analysis Method of Elemental Carbon for Marine Fuel Exhaust. J. Air Waste Manag. Assoc. 2017, 67, 1298–1318. [Google Scholar] [CrossRef]
- Cui, M.; Chen, Y.; Tian, C.; Zhang, F.; Yan, C.; Zheng, M. Chemical Composition of PM2.5 from Two Tunnels with Different Vehicular Fleet Characteristics. Sci. Total Environ. 2016, 550, 123–132. [Google Scholar] [CrossRef]
- Yttri, K.E.; Dye, C.; Braathen, O.-A.; Simpson, D.; Steinnes, E. Carbonaceous Aerosols in Norwegian Urban Areas. Atmos. Chem. Phys. 2009, 9, 2007–2020. [Google Scholar] [CrossRef]
- Piazzalunga, A.; Bernardoni, V.; Fermo, P.; Valli, G.; Vecchi, R. Technical Note: On the Effect of Water-Soluble Compounds Removal on EC Quantification by TOT Analysis in Urban Aerosol Samples. Atmos. Chem. Phys. 2011, 11, 10193–10203. [Google Scholar] [CrossRef]
- Srinivas, B.; Sarin, M.M. Light Absorbing Organic Aerosols (Brown Carbon) over the Tropical Indian Ocean: Impact of Biomass Burning Emissions. Environ. Res. Lett. 2013, 8, 044042. [Google Scholar] [CrossRef]
- Cong, Z.; Kang, S.; Kawamura, K.; Liu, B.; Wan, X.; Wang, Z.; Gao, S.; Fu, P. Carbonaceous Aerosols on the South Edge of the Tibetan Plateau: Concentrations, Seasonality and Sources. Atmos. Chem. Phys. 2015, 15, 1573–1584. [Google Scholar] [CrossRef]
- Bai, Z.; Cui, X.; Wang, X.; Xie, H.; Chen, B. Light Absorption of Black Carbon Is Doubled at Mt. Tai and Typical Urban Area in North China. Sci. Total Environ. 2018, 635, 1144–1151. [Google Scholar] [CrossRef]
- Chen, Y.; Bond, T.C. Light Absorption by Organic Carbon from Wood Combustion. Atmos. Chem. Phys. 2010, 10, 1773–1787. [Google Scholar] [CrossRef]
- Huang, R.-J.; Yang, L.; Cao, J.; Chen, Y.; Chen, Q.; Li, Y.; Duan, J.; Zhu, C.; Dai, W.; Wang, K.; et al. Brown Carbon Aerosol in Urban Xi’an, Northwest China: The Composition and Light Absorption Properties. Environ. Sci. Technol. 2018, 52, 6825–6833. [Google Scholar] [CrossRef]
- Yan, F.; Kang, S.; Sillanpaa, M.; Hu, Z.; Gao, S.; Chen, P.; Gautam, S.; Reinikainen, S.P.; Li, C. A New Method for Extraction of Methanol-Soluble Brown Carbon: Implications for Investigation of Its Light Absorption Ability. Environ. Pollut. 2020, 262, 114300. [Google Scholar] [CrossRef]
- Aakko-Saksa, P.; Koponen, P.; Aurela, M.; Vesala, H.; Piimäkorpi, P.; Murtonen, T.; Sippula, O.; Koponen, H.; Karjalainen, P.; Kuittinen, N.; et al. Considerations in Analysing Elemental Carbon from Marine Engine Exhaust Using Residual, Distillate and Biofuels. J. Aerosol Sci. 2018, 126, 191–204. [Google Scholar] [CrossRef]
- Hecobian, A.; Zhang, X.; Zheng, M.; Frank, N.; Edgerton, E.S.; Weber, R.J. Water-Soluble Organic Aerosol Material and the Light-Absorption Characteristics of Aqueous Extracts Measured over the Southeastern United States. Atmos. Chem. Phys. 2010, 10, 5965–5977. [Google Scholar] [CrossRef]
- Cheng, Y.; He, K.; Du, Z.; Engling, G.; Liu, J.; Ma, Y.; Zheng, M.; Weber, R.J. The Characteristics of Brown Carbon Aerosol during Winter in Beijing. Atmos. Environ. 2016, 127, 355–364. [Google Scholar] [CrossRef]
- Cheng, Y.; He, K.B.; Engling, G.; Weber, R.; Liu, J.M.; Du, Z.Y.; Dong, S.P. Brown and Black Carbon in Beijing Aerosol: Implications for the Effects of Brown Coating on Light Absorption by Black Carbon. Sci. Total Environ. 2017, 599–600, 1047–1055. [Google Scholar] [CrossRef]
- Wallén, A.; Lidén, G.; Hansson, H.-C. Measured Elemental Carbon by Thermo-Optical Transmittance Analysis in Water-Soluble Extracts from Diesel Exhaust, Woodsmoke, and Ambient Particulate Samples. J. Occup. Environ. Hyg. 2009, 7, 35–45. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Yan, C.; Xiao, J.; Ye, J.; Guo, L.; Zheng, M. Metrological Traceability of Black Carbon Measurement Based on Optical Methods and Its Challenges in China: A Review. Atmos. Res. 2023, 292, 106854. [Google Scholar] [CrossRef]
- Nakayama, T.; Suzuki, H.; Kagamitani, S.; Ikeda, Y.; Uchiyama, A.; Matsumi, Y. Characterization of a Three Wavelength Photoacoustic Soot Spectrometer (PASS-3) and a Photoacoustic Extinctiometer (PAX). J. Meteorol. Soc. Jpn. 2015, 93, 285–308. [Google Scholar] [CrossRef]
- Grondin, D.; Geara, S.; Breuil, P.; Viricelle, J.P.; Vernoux, P. Influence of Electrodes Polarization on the Response of Resistive Soot Sensor. Procedia Eng. 2016, 168, 31–34. [Google Scholar] [CrossRef]
- Ammerlaan, B.A.J.; Holzinger, R.; Jedynska, A.D.; Henzing, J.S. Technical Note: Aerosol Light Absorption Measurements with a Carbon Analyser—Calibration and Precision Estimates. Atmos. Environ. 2017, 164, 1–7. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Ming, J. Aerosol Optical Properties Measured Using a PAX in Central Asia from 2016 to 2019 and the Climatic and Environmental Outlooks. ACS Earth Space Chem. 2021, 5, 95–105. [Google Scholar] [CrossRef]
- Yu, Z.; Cheng, Z.; Magoon, G.R.; Hajj, O.E.; Saleh, R. Characterization of Light-Absorbing Aerosols from a Laboratory Combustion Source with Two Different Photoacoustic Techniques. Aerosol Sci. Technol. 2021, 55, 387–397. [Google Scholar] [CrossRef]
- Masey, N.; Ezani, E.; Gillespie, J.; Sutherland, F.; Lin, C.; Hamilton, S.; Heal, M.R.; Beverland, I.J. Consistency of Urban Background Black Carbon Concentration Measurements by Portable AE51 and Reference AE22 Aethalometers: Effect of Corrections for Filter Loading. Aerosol Air Qual. Res. 2020, 20, 329–340. [Google Scholar] [CrossRef]
- Virkkula, A. Correction of the Calibration of the 3-Wavelength Particle Soot Absorption Photometer (3 PSAP). Aerosol Sci. Technol. 2010, 44, 706–712. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Kondo, Y.; Sahu, L.K.; Imaru, J.; Fukushima, N.; Kano, M. Performance of a Newly Designed Continuous Soot Monitoring System (COSMOS). J. Environ. Monit. 2008, 10, 1195–1201. [Google Scholar] [CrossRef]
- Ogren, J.A.; Wendell, J.; Andrews, E.; Sheridan, P.J. Continuous Light Absorption Photometer for Long-Term Studies. Atmos. Meas. Tech. 2017, 10, 4805–4818. [Google Scholar] [CrossRef]
- Cai, J.; Yan, B.; Ross, J.; Zhang, D.; Kinney, P.L.; Perzanowski, M.S.; Jung, K.; Miller, R.; Chillrud, S.N. Validation of MicroAeth® as a Black Carbon Monitor for Fixed-Site Measurement and Optimization for Personal Exposure Characterization. Aerosol Air Qual. Res. 2014, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cappa, C.D.; Lim, C.Y.; Hagan, D.H.; Coggon, M.; Koss, A.; Sekimoto, K.; de Gouw, J.; Onasch, T.B.; Warneke, C.; Kroll, J.H. Biomass-Burning-Derived Particles from a Wide Variety of Fuels—Part 2: Effects of Photochemical Aging on Particle Optical and Chemical Properties. Atmos. Chem. Phys. 2020, 20, 8511–8532. [Google Scholar] [CrossRef]
- Knox, A.; Evans, G.J.; Brook, J.R.; Yao, X.; Jeong, C.H.; Godri, K.J.; Sabaliauskas, K.; Slowik, J.G. Mass Absorption Cross-Section of Ambient Black Carbon Aerosol in Relation to Chemical Age. Aerosol Sci. Technol. 2009, 43, 522–532. [Google Scholar] [CrossRef]
- Kondo, Y.; Sahu, L.; Kuwata, M.; Miyazaki, Y.; Takegawa, N.; Moteki, N.; Imaru, J.; Han, S.; Nakayama, T.; Oanh, N.T.K.; et al. Stabilization of the Mass Absorption Cross Section of Black Carbon for Filter-Based Absorption Photometry by the Use of a Heated Inlet. Aerosol Sci. Technol. 2009, 43, 741–756. [Google Scholar] [CrossRef]
- Sedlacek, A.; Lee, J. Photothermal Interferometric Aerosol Absorption Spectrometry. Aerosol Sci. Technol. 2007, 41, 1089–1101. [Google Scholar] [CrossRef]
- Davies, N.W.; Fox, C.; Szpek, K.; Cotterell, M.I.; Taylor, J.W.; Allan, J.D.; Williams, P.I.; Trembath, J.; Haywood, J.M.; Langridge, J.M. Evaluating Biases in Filter-Based Aerosol Absorption Measurements Using Photoacoustic Spectroscopy. Atmos. Meas. Tech. 2019, 12, 3417–3434. [Google Scholar] [CrossRef]
- Rosen, H.; Hansen, A.D.A.; Gundel, L.; Novakov, T. Identification of the Optically Absorbing Component in Urban Aerosols. Appl. Opt. 1978, 17, 3859. [Google Scholar] [CrossRef]
- Moosmüller, H.; Chakrabarty, R.K.; Arnott, W.P. Aerosol Light Absorption and Its Measurement: A Review. J. Quant. Spectrosc. Radiat. Transf. 2009, 110, 844–878. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, S.-W.; Ogren, J.A.; Sheridan, P.J.; Yoon, S.-C.; Sharma, S.; Lin, N.-H. Multiple Scattering Correction Factor Estimation for Aethalometer Aerosol Absorption Coefficient Measurement. Aerosol Sci. Technol. 2018, 53, 160–171. [Google Scholar] [CrossRef]
- Drinovec, L.; Močnik, G.; Zotter, P.; Prévôt, A.S.H.; Ruckstuhl, C.; Coz, E.; Rupakheti, M.; Sciare, J.; Müller, T.; Wiedensohler, A.; et al. The “Dual-Spot” Aethalometer: An Improved Measurement of Aerosol Black Carbon with Real-Time Loading Compensation. Atmos. Meas. Tech. 2015, 8, 1965–1979. [Google Scholar] [CrossRef]
- Bond, T.C.; Anderson, T.L.; Campbell, D. Calibration and Intercomparison of Filter-Based Measurements of Visible Light Absorption by Aerosols. Aerosol Sci. Technol. 1999, 30, 582–600. [Google Scholar] [CrossRef]
- Arnott, W.P.; Hamasha, K.; Moosmüller, H.; Sheridan, P.J.; Ogren, J.A. Towards Aerosol Light-Absorption Measurements with a 7-Wavelength Aethalometer: Evaluation with a Photoacoustic Instrument and 3-Wavelength Nephelometer. Aerosol Sci. Technol. 2005, 39, 17–29. [Google Scholar] [CrossRef]
- Collaud Coen, M.; Weingartner, E.; Apituley, A.; Ceburnis, D.; Fierz-Schmidhauser, R.; Flentje, H.; Henzing, J.S.; Jennings, S.G.; Moerman, M.; Petzold, A.; et al. Minimizing Light Absorption Measurement Artifacts of the Aethalometer: Evaluation of Five Correction Algorithms. Atmos. Meas. Tech. 2010, 3, 457–474. [Google Scholar] [CrossRef]
- Schmid, O.; Artaxo, P.; Arnott, W.P.; Chand, D.; Gatti, L.V.; Frank, G.P.; Hoffer, A.; Schnaiter, M.; Andreae, M.O. Spectral Light Absorption by Ambient Aerosols Influenced by Biomass Burning in the Amazon Basin. I: Comparison and Field Calibration of Absorption Measurement Techniques. Atmos. Chem. Phys. 2006, 6, 3443–3462. [Google Scholar] [CrossRef]
- Virkkula, A.; Makela, T.; Hillamo, R.; Yli-Tuomi, T.; Hirsikko, A.; Hameri, K.; Koponen, I.K. A Simple Procedure for Correcting Loading Effects of Aethalometer Data. J. Air Waste Manag. Assoc. 2007, 57, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Weingartner, E.; Saathoff, H.; Schnaiter, M.; Streit, N.; Bitnar, B.; Baltensperger, U. Absorption of Light by Soot Particles: Determination of the Absorption Coefficient by Means of Aethalometers. J. Aerosol Sci. 2003, 34, 1445–1463. [Google Scholar] [CrossRef]
- Ogren, J.A. Comment on “Calibration and Intercomparison of Filter-Based Measurements of Visible Light Absorption by Aerosols”. Aerosol Sci. Technol. 2010, 44, 589–591. [Google Scholar] [CrossRef]
- Petzold, A.; Schloesser, H.; Sheridan, P.J.; Arnott, W.P.; Ogren, J.A.; Virkkula, A. Evaluation of Multiangle Absorption Photometry for Measuring Aerosol Light Absorption. Aerosol Sci. Technol. 2005, 39, 40–51. [Google Scholar] [CrossRef]
- Petzold, A.; Schonlinner, M. Multi-Angle Absorption Photometry—A New Method for the Measurement of Aerosol Light Absorption and Atmospheric Black Carbon. J. Aerosol Sci. 2004, 35, 421–441. [Google Scholar] [CrossRef]
- Bond, T.C.; Bergstrom, R.W. Light Absorption by Carbonaceous Particles: An Investigative Review. Aerosol Sci. Technol. 2006, 40, 27–67. [Google Scholar] [CrossRef]
- Moosmuller, H.; Arnott, W.P.; Rogers, C.F. Methods for Real-Time, In Situ Measurement of Aerosol Light Absorption. J. Air Waste Manag. Assoc. 1997, 47, 157–166. [Google Scholar] [CrossRef]
- Modini, R.L.; Corbin, J.C.; Brem, B.T.; Irwin, M.; Bertò, M.; Pileci, R.E.; Fetfatzis, P.; Eleftheriadis, K.; Henzing, B.; Moerman, M.M.; et al. Detailed Characterization of the CAPS Single-Scattering Albedo Monitor (CAPS PMssa) as a Field-Deployable Instrument for Measuring Aerosol Light Absorption with the Extinction-Minus-Scattering Method. Atmos. Meas. Tech. 2021, 14, 819–851. [Google Scholar] [CrossRef]
- Abu-Rahmah, A.; Arnott, W.P.; Moosmüller, H. Integrating Nephelometer with a Low Truncation Angle and an Extended Calibration Scheme. Meas. Sci. Technol. 2006, 17, 1723–1732. [Google Scholar] [CrossRef]
- Saathoff, H.; Naumann, K.H.; Schnaiter, M.; Schock, W.; Weingartner, E.; Baltensperger, U.; Kramer, L.; Bozoki, Z.; Poschl, U.; Niessner, R.; et al. Carbon Mass Determinations during the AIDA Soot Aerosol Campaign 1999. J. Aerosol Sci. 2003, 34, 1399–1420. [Google Scholar] [CrossRef]
- Schnaiter, M.; Horvath, H.; Mohler, O.; Naumann, K.H.; Saathoff, H.; Schock, O.W. UV-VIS-NIR Spectral Optical Properties of Soot and Soot-Containing Aerosols. J. Aerosol Sci. 2003, 34, 1421–1444. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, W.; Zhang, Q.; Wang, S.; Fang, B.; Chen, W.; Venables, D.S.; Wang, X.; Pu, W.; Wang, X.; et al. Optical Properties of Atmospheric Fine Particles near Beijing during the HOPE-J3A Campaign. Atmos. Chem. Phys. 2016, 16, 6421–6439. [Google Scholar] [CrossRef]
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Allan, J.D.; Coe, H.; Ulbrich, I.; Alfarra, M.R.; Takami, A.; Middlebrook, A.M.; Sun, Y.L.; et al. Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes. Geophys. Res. Lett. 2007, 34, L13801. [Google Scholar] [CrossRef]
- Lack, D.A.; Cappa, C.D.; Covert, D.S.; Baynard, T.; Massoli, P.; Sierau, B.; Bates, T.S.; Quinn, P.K.; Lovejoy, E.R.; Ravishankara, A.R. Bias in Filter-Based Aerosol Light Absorption Measurements Due to Organic Aerosol Loading: Evidence from Ambient Measurements. Aerosol Sci. Technol. 2008, 42, 1033–1041. [Google Scholar] [CrossRef]
- Subramanian, R.; Roden, C.A.; Boparai, P.; Bond, T.C. Yellow Beads and Missing Particles: Trouble Ahead for Filter-Based Absorption Measurements. Aerosol Sci. Technol. 2007, 41, 630–637. [Google Scholar] [CrossRef]
- Arnott, W.P.; Moosmuller, H.; Sheridan, P.J.; Ogren, J.A.; Raspet, R.; Slaton, W.V.; Hand, J.L.; Kreidenweis, S.M.; Collett, J.L. Photoacoustic and Filter-Based Ambient Aerosol Light Absorption Measurements: Instrument Comparisons and the Role of Relative Humidity. J. Geophys. Res. Atmos. 2003, 108, AAC 15-1–AAC 15-11. [Google Scholar] [CrossRef]
- Tasoglou, A.; Subramanian, R.; Pandis, S.N. An Inter-Comparison of Black-Carbon-Related Instruments in a Laboratory Study of Biomass Burning Aerosol. Aerosol Sci. Technol. 2018, 52, 1320–1331. [Google Scholar] [CrossRef]
- Holder, A.L.; Hagler, G.S.W.; Aurell, J.; Hays, M.D.; Gullett, B.K. Particulate Matter and Black Carbon Optical Properties and Emission Factors from Prescribed Fires in the Southeastern United States. J. Geophys. Res. Atmos. 2016, 121, 3465–3483. [Google Scholar] [CrossRef]
- Laing, J.R.; Jaffe, D.A.; Sedlacek, I.I.I.A.J. Comparison of Filter-Based Absorption Measurements of Biomass Burning Aerosol and Background Aerosol at the Mt. Bachelor Observatory. Aerosol Air Qual. Res. 2020, 20, 663–678. [Google Scholar] [CrossRef]
- Backman, J.; Virkkula, A.; Vakkari, V.; Beukes, J.P.; Van Zyl, P.G.; Josipovic, M.; Piketh, S.; Tiitta, P.; Chiloane, K.; Petäjä, T.; et al. Differences in Aerosol Absorption Ångström Exponents between Correction Algorithms for a Particle Soot Absorption Photometer Measured on the South African Highveld. Atmos. Meas. Tech. 2014, 7, 4285–4298. [Google Scholar] [CrossRef]
- Kondo, Y.; Sahu, L.; Moteki, N.; Khan, F.; Takegawa, N.; Liu, X.; Koike, M.; Miyakawa, T. Consistency and Traceability of Black Carbon Measurements Made by Laser-Induced Incandescence, Thermal–optical Transmittance, and Filter-Based Photo-Absorption Techniques. Aerosol Sci. Technol. 2011, 45, 295–312. [Google Scholar] [CrossRef]
- Kanaya, Y.; Komazaki, Y.; Pochanart, P.; Liu, Y.; Akimoto, H.; Gao, J.; Wang, T.; Wang, Z. Mass Concentrations of Black Carbon Measured by Four Instruments in the Middle of Central East China in June 2006. Atmos. Chem. Phys. 2008, 8, 7637–7649. [Google Scholar] [CrossRef]
- Yuan, R.; Lobo, P.; Smallwood, G.J.; Johnson, M.P.; Parker, M.C.; Butcher, D.; Spencer, A. Measurement of Black Carbon Emissions from Multiple Engine and Source Types Using Laser-Induced Incandescence: Sensitivity to Laser Fluence. Atmos. Meas. Tech. 2022, 15, 241–259. [Google Scholar] [CrossRef]
- Cross, E.S.; Onasch, T.B.; Ahern, A.; Wrobel, W.; Slowik, J.G.; Olfert, J.; Lack, D.A.; Massoli, P.; Cappa, C.D.; Schwarz, J.P.; et al. Soot Particle Studies—Instrument Inter-Comparison—Project Overview. Aerosol Sci. Technol. 2010, 44, 592–611. [Google Scholar] [CrossRef]
- Rademacher, H.P. Airborne and Laboratory Performance Characterization of a New Aerosol and BC Instrument, The SP2-XR; University of Wyoming: Laramie, WY, USA, 2022; ISBN 9798363522567. [Google Scholar]
- Chakrabarty, R.K.; Moosmüller, H.; Garro, M.A.; Arnott, W.P.; Walker, J.; Susott, R.A.; Babbitt, R.E.; Wold, C.E.; Lincoln, E.N.; Hao, W.M. Emissions from the Laboratory Combustion of Wildland Fuels: Particle Morphology and Size. J. Geophys. Res. Atmos. 2006, 111, 2005JD006659. [Google Scholar] [CrossRef]
- Chakrabarty, R.; Moosmüller, H.; Arnott, W.; Garro, M.; Slowik, J.; Cross, E.; Han, J.; Davidovits, P.; Onasch, T.; Worsnop, D. Light Scattering and Absorption by Fractal-like Carbonaceous Chain Aggregates:: Comparison of Theories and Experiment. Appl. Opt. 2007, 46, 6990–7006. [Google Scholar] [CrossRef]
- Pósfai, M.; Simonics, R.; Li, J.; Hobbs, P.V.; Buseck, P.R. Individual Aerosol Particles from Biomass Burning in Southern Africa: 1. Compositions and Size Distributions of Carbonaceous Particles. J. Geophys. Res. Atmos. 2003, 108, 2002JD002291. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S.; Wang, J.; Hua, Y.; Liu, C.; Cai, J.; Xu, Q.; Xu, X.; Jiang, S.; Zheng, G.; et al. Significant Contribution of Coarse Black Carbon Particles to Light Absorption in North China Plain. Environ. Sci. Technol. Lett. 2022, 9, 134–139. [Google Scholar] [CrossRef]
- Laborde, M.; Schnaiter, M.; Linke, C.; Saathoff, H.; Naumann, K.H.; Möhler, O.; Berlenz, S.; Wagner, U.; Taylor, J.W.; Liu, D.; et al. Single Particle Soot Photometer Intercomparison at the AIDA Chamber. Atmos. Meas. Tech. 2012, 5, 3077–3097. [Google Scholar] [CrossRef]
- Moteki, N.; Kondo, Y. Effects of Mixing State on Black Carbon Measurements by Laser-Induced Incandescence. Aerosol Sci. Technol. 2007, 41, 398–417. [Google Scholar] [CrossRef]
- Raatikainen, T.; Brus, D.; Hyvarinen, A.P.; Svensson, J.; Asmi, E.; Lihavainen, H. Black Carbon Concentrations and Mixing State in the Finnish Arctic. Atmos. Chem. Phys. 2015, 15, 10057–10070. [Google Scholar] [CrossRef]
- Schwarz, J.P.; Gao, R.S.; Fahey, D.W.; Thomson, D.S.; Watts, L.A.; Wilson, J.C.; Reeves, J.M.; Darbeheshti, M.; Baumgardner, D.G.; Kok, G.L.; et al. Single-Particle Measurements of Midlatitude Black Carbon and Light-Scattering Aerosols from the Boundary Layer to the Lower Stratosphere. J. Geophys. Res. Atmos. 2006, 111, D16207. [Google Scholar] [CrossRef]
- Stephens, M.; Turner, N.; Sandberg, J. Particle Identification by Laser-Induced Incandescence in a Solid-State Laser Cavity. Appl. Opt. 2003, 42, 3726–3736. [Google Scholar] [CrossRef] [PubMed]
- Baumgardner, D.; Popovicheva, O.; Allan, J.; Bernardoni, V.; Cao, J.; Cavalli, F.; Cozic, J.; Diapouli, E.; Eleftheriadis, K.; Genberg, P.J.; et al. Soot Reference Materials for Instrument Calibration and Intercomparisons: A Workshop Summary with Recommendations. Atmos. Meas. Tech. 2012, 5, 1869–1887. [Google Scholar] [CrossRef]
- Laborde, M.; Mertes, P.; Zieger, P.; Dommen, J.; Baltensperger, U.; Gysel, M. Sensitivity of the Single Particle Soot Photometer to Different Black Carbon Types. Atmos. Meas. Tech. 2012, 5, 1031–1043. [Google Scholar] [CrossRef]
- Moteki, N.; Kondo, Y. Dependence of Laser-Induced Incandescence on Physical Properties of Black Carbon Aerosols: Measurements and Theoretical Interpretation. Aerosol Sci. Technol. 2010, 44, 663–675. [Google Scholar] [CrossRef]
- Miyakawa, T.; Kanaya, Y.; Komazaki, Y.; Taketani, F.; Pan, X.; Irwin, M.; Symonds, J. Intercomparison between a Single Particle Soot Photometer and Evolved Gas Analysis in an Industrial Area in Japan: Implications for the Consistency of Soot Aerosol Mass Concentration Measurements. Atmos. Environ. 2016, 127, 14–21. [Google Scholar] [CrossRef]
- Schwarz, J.P.; Spackman, J.R.; Gao, R.S.; Perring, A.E.; Cross, E.; Onasch, T.B.; Ahern, A.; Wrobel, W.; Davidovits, P.; Olfert, J.; et al. The Detection Efficiency of the Single Particle Soot Photometer. Aerosol Sci. Technol. 2010, 44, 612–628. [Google Scholar] [CrossRef]
- Shiraiwa, M.; Kondo, Y.; Moteki, N.; Takegawa, N.; Sahu, L.K.; Takami, A.; Hatakeyama, S.; Yonemura, S.; Blake, D.R. Radiative Impact of Mixing State of Black Carbon Aerosol in Asian Outflow. J. Geophys. Res. Atmos. 2008, 113, D24210. [Google Scholar] [CrossRef]
- Kupiszewski, P.; Zanatta, M.; Mertes, S.; Vochezer, P.; Lloyd, G.; Schneider, J.; Schenk, L.; Schnaiter, M.; Baltensperger, U.; Weingartner, E.; et al. Ice Residual Properties in Mixed-Phase Clouds at the High-Alpine Jungfraujoch Site. J. Geophys. Res. Atmos. 2016, 121, 12343–12362. [Google Scholar] [CrossRef] [PubMed]
- Moteki, N.; Adachi, K.; Ohata, S.; Yoshida, A.; Harigaya, T.; Koike, M.; Kondo, Y. Anthropogenic Iron Oxide Aerosols Enhance Atmospheric Heating. Nat. Commun. 2017, 8, 15329. [Google Scholar] [CrossRef] [PubMed]
- Canagaratna, M.R.; Jayne, J.T.; Jimenez, J.L.; Allan, J.D.; Alfarra, M.R.; Zhang, Q.; Onasch, T.B.; Drewnick, F.; Coe, H.; Middlebrook, A.; et al. Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer. Mass Spectrom. Rev. 2007, 26, 185–222. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S.; et al. Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Rivellini, L.-H.; Cui, Y.; Willis, M.D.; Wilkie, R.; Abbatt, J.P.D.; Canagaratna, M.R.; Wang, J.; Ge, X.; Lee, A.K.Y. Elemental Analysis of Oxygenated Organic Coating on Black Carbon Particles Using a Soot-Particle Aerosol Mass Spectrometer. Atmos. Meas. Tech. 2021, 14, 2799–2812. [Google Scholar] [CrossRef]
- Matthew, B.M.; Middlebrook, A.M.; Onasch, T.B. Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols. Aerosol Sci. Technol. 2008, 42, 884–898. [Google Scholar] [CrossRef]
- Wang, J.; Ge, X.; Chen, Y.; Shen, Y.; Zhang, Q.; Sun, Y.; Xu, J.; Ge, S.; Yu, H.; Chen, M. Highly Time-Resolved Urban Aerosol Characteristics during Springtime in Yangtze River Delta, China: Insights from Soot Particle Aerosol Mass Spectrometry. Atmos. Chem. Phys. 2016, 16, 9109–9127. [Google Scholar] [CrossRef]
- Weimer, S.; Drewnick, F.; Hogrefe, O.; Schwab, J.J.; Rhoads, K.; Orsini, D.; Canagaratna, M.; Worsnop, D.R.; Demerjian, K.L. Size-Selective Nonrefractory Ambient Aerosol Measurements during the Particulate Matter Technology Assessment and Characterization Study–New York 2004 Winter Intensive in New York City. J. Geophys. Res. 2006, 111, D18305. [Google Scholar] [CrossRef]
- Collier, S.; Williams, L.R.; Onasch, T.B.; Cappa, C.D.; Zhang, X.; Russell, L.M.; Chen, C.-L.; Sanchez, K.J.; Worsnop, D.R.; Zhang, Q. Influence of Emissions and Aqueous Processing on Particles Containing Black Carbon in a Polluted Urban Environment: Insights From a Soot Particle-Aerosol Mass Spectrometer. J. Geophys. Res. Atmos. 2018, 123, 6648–6666. [Google Scholar] [CrossRef]
- Massoli, P.; Onasch, T.B.; Cappa, C.D.; Nuamaan, I.; Hakala, J.; Hayden, K.; Li, S.-M.; Sueper, D.T.; Bates, T.S.; Quinn, P.K.; et al. Characterization of Black Carbon-Containing Particles from Soot Particle Aerosol Mass Spectrometer Measurements on the R/V Atlantis during CalNex 2010. J. Geophys. Res. Atmos. 2015, 120, 2575–2593. [Google Scholar] [CrossRef]
- Middlebrook, A.M.; Bahreini, R.; Jimenez, J.L.; Canagaratna, M.R. Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer Using Field Data. Aerosol Sci. Technol. 2012, 46, 258–271. [Google Scholar] [CrossRef]
- Laborde, M.; Crippa, M.; Tritscher, T.; Jurányi, Z.; Decarlo, P.F.; Temime-Roussel, B.; Marchand, N.; Eckhardt, S.; Stohl, A.; Baltensperger, U.; et al. Black Carbon Physical Properties and Mixing State in the European Megacity Paris. Atmos. Chem. Phys. 2013, 13, 5831–5856. [Google Scholar] [CrossRef]
- Dallmann, T.R.; Onasch, T.B.; Kirchstetter, T.W.; Worton, D.R.; Fortner, E.C.; Herndon, S.C.; Wood, E.C.; Franklin, J.P.; Worsnop, D.R.; Goldstein, A.H.; et al. Characterization of Particulate Matter Emissions from On-Road Gasoline and Diesel Vehicles Using a Soot Particle Aerosol Mass Spectrometer. Atmos. Chem. Phys. 2014, 14, 7585–7599. [Google Scholar] [CrossRef]
- Fortner, E.C.; Brooks, W.A.; Onasch, T.B.; Canagaratna, M.R.; Massoli, P.; Jayne, J.T.; Franklin, J.P.; Knighton, W.B.; Wormhoudt, J.; Worsnop, D.R.; et al. Particulate Emissions Measured During the TCEQ Comprehensive Flare Emission Study. Ind. Eng. Chem. Res. 2012, 51, 12586–12592. [Google Scholar] [CrossRef]
- Wang, J.; Ye, J.; Liu, D.; Wu, Y.; Zhao, J.; Xu, W.; Xie, C.; Shen, F.; Zhang, J.; Ohno, P.E.; et al. Characterization of Submicron Organic Particles in Beijing during Summertime: Comparison between SP-AMS and HR-AMS. Atmos. Chem. Phys. 2020, 20, 14091–14102. [Google Scholar] [CrossRef]
- Willis, M.D.; Lee, A.K.Y.; Onasch, T.B.; Fortner, E.C.; Williams, L.R.; Lambe, A.T.; Worsnop, D.R.; Abbatt, J.P.D. Collection Efficiency of the Soot-Particle Aerosol Mass Spectrometer (SP-AMS) for Internally Mixed Particulate Black Carbon. Atmos. Meas. Tech. 2014, 7, 4507–4516. [Google Scholar] [CrossRef]
- Sharma, S.; Leaitch, W.R.; Huang, L.; Veber, D.; Kolonjari, F.; Zhang, W.; Hanna, S.J.; Bertram, A.K.; Ogren, J.A. An Evaluation of Three Methods for Measuring Black Carbon in Alert, Canada. Atmos. Chem. Phys. 2017, 17, 15225–15243. [Google Scholar] [CrossRef]
- Corbin, J.C.; Czech, H.; Massabò, D.; De Mongeot, F.B.; Jakobi, G.; Liu, F.; Lobo, P.; Mennucci, C.; Mensah, A.A.; Orasche, J.; et al. Infrared-Absorbing Carbonaceous Tar Can Dominate Light Absorption by Marine-Engine Exhaust. NPJ Clim. Atmos. Sci. 2019, 2, 12. [Google Scholar] [CrossRef]
- Zhang, X.; Kim, H.; Parworth, C.L.; Young, D.E.; Zhang, Q.; Metcalf, A.R.; Cappa, C.D. Optical Properties of Wintertime Aerosols from Residential Wood Burning in Fresno, CA: Results from DISCOVER-AQ 2013. Environ. Sci. Technol. 2016, 50, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Gysel, M.; Laborde, M.; Olfert, J.S.; Subramanian, R.; Groehn, A.J. Effective Density of Aquadag and Fullerene Soot Black Carbon Reference Materials Used for SP2 Calibration. Atmos. Meas. Tech. 2011, 4, 2851–2858. [Google Scholar] [CrossRef]
- Ram, K.; Sarin, M.M.; Tripathi, S.N. Inter-Comparison of Thermal and Optical Methods for Determination of Atmospheric Black Carbon and Attenuation Coefficient from an Urban Location in Northern India. Atmos. Res. 2010, 97, 335–342. [Google Scholar] [CrossRef]
- Sharma, S.; Brook, J.R.; Cachier, H.; Chow, J.; Gaudenzi, A.; Lu, G. Light Absorption and Thermal Measurements of Black Carbon in Different Regions of Canada. J. Geophys. Res. Atmos. 2002, 107, AAC 11-1–AAC 11-11. [Google Scholar] [CrossRef]
- Sirois, A.; Barrie, L.A. Arctic Lower Tropospheric Aerosol Trends and Composition at Alert, Canada: 1980-1995. J. Geophys. Res. Atmos. 1999, 104, 11599. [Google Scholar] [CrossRef]
- Ahmed, T.; Dutkiewicz, V.A.; Shareef, A.; Tuncel, G.; Tuncel, S.; Husain, L. Measurement of Black Carbon (BC) by an Optical Method and a Thermal–optical Method: Intercomparison for Four Sites. Atmos. Environ. 2009, 43, 6305–6311. [Google Scholar] [CrossRef]
- Jeong, C.-H.; Hopke, P.K.; Kim, E.; Lee, D.-W. The Comparison between Thermal–optical Transmittance Elemental Carbon and Aethalometer Black Carbon Measured at Multiple Monitoring Sites. Atmos. Environ. 2004, 38, 5193–5204. [Google Scholar] [CrossRef]
- Stone, R.S.; Sharma, S.; Herber, A.; Eleftheriadis, K.; Nelson, D.W. A Characterization of Arctic Aerosols on the Basis of Aerosol Optical Depth and Black Carbon Measurements. Elem. Sci. Anthr. 2014, 2, 000027. [Google Scholar] [CrossRef]
- Karanasiou, A.; Panteliadis, P.; Perez, N.; Minguillon, M.C.; Pandolfi, M.; Titos, G.; Viana, M.; Moreno, T.; Querol, X.; Alastuey, A. Evaluation of the Semi-Continuous OCEC Analyzer Performance with the EUSAAR2 Protocol. Sci. Total Environ. 2020, 747, 141266. [Google Scholar] [CrossRef]
- Patterson, E.M.; McMahon, C.K. Absorption Characteristics of Forest Fire Particulate Matter. Atmos. Environ. (1967) 1984, 18, 2541–2551. [Google Scholar] [CrossRef]
- Zhi, G.; Chen, Y.; Xue, Z.; Meng, F.; Cai, J.; Sheng, G.; Fu, J. Comparison of Elemental and Black Carbon Measurements during Normal and Heavy Haze Periods: Implications for Research. Environ. Monit. Assess. 2014, 186, 6097–6106. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Deng, C.; Cao, F.; Cao, C.; Zou, Z.; Liu, S.; Lee, X.; Li, J.; Zhang, G.; Zhang, Y. Assessment of Carbonaceous Aerosols in Shanghai, China—Part 1: Long-Term Evolution, Seasonal Variations, and Meteorological Effects. Atmos. Chem. Phys. 2017, 17, 9945–9964. [Google Scholar] [CrossRef]
- Healy, R.M.; Sofowote, U.; Su, Y.; Debosz, J.; Noble, M.; Jeong, C.-H.; Wang, J.M.; Hilker, N.; Evans, G.J.; Doerksen, G.; et al. Ambient Measurements and Source Apportionment of Fossil Fuel and Biomass Burning Black Carbon in Ontario. Atmos. Environ. 2017, 161, 34–47. [Google Scholar] [CrossRef]
- Rattigan, O.V.; Dirk Felton, H.; Bae, M.-S.; Schwab, J.J.; Demerjian, K.L. Multi-Year Hourly PM2.5 Carbon Measurements in New York: Diurnal, Day of Week and Seasonal Patterns. Atmos. Environ. 2010, 44, 2043–2053. [Google Scholar] [CrossRef]
- Yelverton, T.L.B.; Hays, M.D.; Gullett, B.K.; Linak, W.P. Black Carbon Measurements of Flame-Generated Soot as Determined by Optical, Thermal–optical, Direct Absorption, and Laser Incandescence Methods. Environ. Eng. Sci. 2014, 31, 209–215. [Google Scholar] [CrossRef]
- Slowik, J.G.; Cross, E.S.; Han, J.-H.; Davidovits, P.; Onasch, T.B.; Jayne, J.T.; Williams, L.R.; Canagaratna, M.R.; Worsnop, D.R.; Chakrabarty, R.K.; et al. An Inter-Comparison of Instruments Measuring Black Carbon Content of Soot Particles. Aerosol Sci. Technol. 2007, 41, 295–314. [Google Scholar] [CrossRef]
- Buffaloe, G.M.; Lack, D.A.; Williams, E.J.; Coffman, D.; Hayden, K.L.; Lerner, B.M.; Li, S.M.; Nuaaman, I.; Massoli, P.; Onasch, T.B.; et al. Black Carbon Emissions from In-Use Ships: A California Regional Assessment. Atmos. Chem. Phys. 2014, 14, 1881–1896. [Google Scholar] [CrossRef]
- Holder, A.L.; Hagler, G.S.W.; Yelverton, T.L.B.; Hays, M.D. On-Road Black Carbon Instrument Intercomparison and Aerosol Characteristics by Driving Environment. Atmos. Environ. 2014, 88, 183–191. [Google Scholar] [CrossRef]
- Lack, D.A.; Langridge, J.M. On the Attribution of Black and Brown Carbon Light Absorption Using the Ångström Exponent. Atmos. Chem. Phys. 2013, 13, 10535–10543. [Google Scholar] [CrossRef]
- Cappa, C.D.; Lack, D.A.; Burkholder, J.B.; Ravishankara, A.R. Bias in Filter-Based Aerosol Light Absorption Measurements Due to Organic Aerosol Loading: Evidence from Laboratory Measurements. Aerosol Sci. Technol. 2008, 42, 1022–1032. [Google Scholar] [CrossRef]
- Liu, D.; Whitehead, J.; Alfarra, M.R.; Reyes-Villegas, E.; Spracklen, D.V.; Reddington, C.L.; Kong, S.; Williams, P.I.; Ting, Y.-C.; Haslett, S.; et al. Black-Carbon Absorption Enhancement in the Atmosphere Determined by Particle Mixing State. Nat. Geosci. 2017, 10, 184–188. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, J.; Han, Y.; Tian, J.; Zhu, C.; Zhang, Y.; Zhang, N.; Shen, Z.; Ni, H.; Zhao, S. Sources and Physicochemical Characteristics of Black Carbon Aerosol from the Southeastern Tibetan Plateau: Internal Mixing Enhances Light Absorption. Atmos. Chem. Phys. 2018, 18, 4639–4656. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Abbreviation | Symbol | Unit |
|---|---|---|---|
| Absorption Ångström Exponent | AAE | - | |
| Aerosol Characterization Experiments | ACE | - | - |
| Aerosol Mass Spectrometer-Scanning Mobility Particle Sizer | AMS-SMPS | - | - |
| Aethalometer | AE | - | - |
| Babs produced by PM and filter | - | bpf | m−1 |
| Biomass burning | BB | - | - |
| Black carbon | BC | - | - |
| Brown carbon | BrC | - | - |
| California Regional PM10/PM2.5 Air Quality Study | CRPAQS | - | - |
| Canadian National Air Pollution Surveillance | NAPS | - | - |
| Carbonate carbon | CC | - | - |
| Chemical Speciation Network | CSN | - | - |
| Collection area of the filter | - | A | m2 |
| Collection efficiency | CE | - | % |
| Combustion Aerosol Standard | CAST | - | - |
| Continuous Soot Monitoring System | COSMOS | - | - |
| Correction factor for multiple scattering effects | - | Cref | - |
| Diameter | - | D | μm |
| EC defined by reflected light signal | ECR | - | μg C/cm2 |
| EC defined by transmitted light signal | ECT | - | μg C/cm2 |
| EC quantified by the protocol with an inert peak temperature is 870 °C | EC870 | - | μg C/cm2 |
| Elemental carbon | EC | - | μg C/cm2 |
| Equivalent black carbon | eBC | - | μg/m3 |
| European Supersites for Atmospheric Aerosol Research | EUSAAR | - | - |
| Extinction coefficient | - | bext | Mm−1 |
| Extinction minus scattering | EMS | - | - |
| High resolution-particle time-of-flight aerosol mass spectrometer | HR-ToF-AMS | - | - |
| Humic-like substances | HULIS | - | - |
| Inert peak temperature | Tpeak | - | °C |
| Inert peak temperature is 870 °C | Tpeak-870 °C/He-870 | - | - |
| Insoluble organic carbon | ISOC | - | - |
| Integrating plate | IP | - | - |
| Integrating sandwich | IS | - | - |
| Interagency Monitoring of Protected Visual Environments | IMPROVE | - | - |
| Irradiance | - | I | MW/cm2 |
| Laser-induced incandescence | LII | - | - |
| Light absorbing carbon | LAC | - | - |
| Light absorption coefficient | - | babs | Mm−1 |
| Light intensity | - | I | W/m2 |
| Lower detection limit | LDL | - | nm |
| Mass | - | M | μg |
| Mass absorption cross-section | MAC | - | m2/g |
| Mixing state | - | Dp/DBC | - |
| Multi-Angle Absorption Photometer | MAAP | - | - |
| National Institute for Occupational Safety and Health | NIOSH | - | - |
| Non-refractory particles | NR-PM | - | - |
| Organic aerosol | OA | - | - |
| Organic carbon | OC | - | μg C/cm2 |
| Particle Soot Absorption Photometer | PSAP | - | - |
| Particulate matter | PM | - | μg/m3 |
| Photo thermal interferometry | PTI | - | - |
| Photoacoustic Extinctiometer | PAX | - | - |
| Photoacoustic spectrometer | PAS | - | - |
| Pyrolytic carbon | PC | - | μg C/cm2 |
| Reflected light | R | - | mv (millivolts) |
| Refractory black carbon | rBC | - | μg/m3 |
| Refractory particles | R-PM | - | - |
| Relative humidity | - | RH | % |
| Sampling time | - | ∆t | s |
| Scattering coefficient | - | bsca | Mm−1 |
| Secondary organic aerosol | SOA | - | - |
| Single particle soot photometer | SP2 | - | - |
| Single scattering albedo | SSA | ω | - |
| Soot particle–aerosol mass spectrometer | SP-AMS | - | - |
| Southeastern Aerosol Research and Characterization network | SEARCH | - | - |
| Speciation Trends Network | STN | - | - |
| Stage 4 of the inert mode | He4 | - | - |
| Teflon-coated glass fiber | TFE | - | - |
| Temperature | - | T | °C |
| Thermal optical analysis | TOA | - | - |
| Thermal optical reflectance method | TOR | - | - |
| Thermal optical transmittance method | TOT | - | - |
| Thickness of the filter | - | X | m |
| Total Carbon | TC | - | μg C/cm2 |
| Transmitted light | T | - | mv (millivolts) |
| Transmitted light attenuation | ATN | - | - |
| Tricolor Absorption Photometer | TAP | - | - |
| Upper detection limit | UDL | - | nm |
| Velocity of the gas passing through the filter | - | V | m3/s |
| Volatile organic compounds | VOCs | - | - |
| Water-soluble organic carbon | WSOC | - | - |
| Wavelength | - | λ | nm |
| Technical Classification | Instrument Model | Principle | Deviation Source | Reference |
|---|---|---|---|---|
| Offline | DRI thermal/optical reflectance carbon analyzer | OC and EC are separated sequentially under different temperatures and atmospheres, and optical correction is used to monitor the formation and evolution of PC in real time. | 1. Selection of thermo-optical protocols; 2. Interference of non-EC chemical components in PM | [52] |
| DRI Model 2001 | [53] | |||
| DRI Model 2015 | [54] | |||
| Sunset model 5 L analyzers | ||||
| Semi-continuous | Model RT-4 | [55] |
| IMPROVE a | IMPROVE_A a | NIOSH5040 b | NIOSH870 b | EUSAAR_1 b | EUSAAR_2 b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Step | Gas | Temperature (°C) | Time (s) | Temperature (°C) | Time (s) | Temperature (°C) | Time (s) | Temperature (°C) | Time (s) | Temperature (°C) | Time (s) | Temperature (°C) | Time (s) |
| OC1 | Pure He | 120 | 150–580 | 140 | 150–580 | 250 | 60 | 310 | 80 | 200 | 120 | 200 | 120 |
| OC2 | Pure He | 250 | 150–580 | 280 | 150–580 | 500 | 60 | 475 | 80 | 300 | 150 | 300 | 150 |
| OC3 | Pure He | 450 | 150–580 | 480 | 150–580 | 650 | 60 | 615 | 80 | 450 | 180 | 450 | 180 |
| OC4 | Pure He | 550 | 150–580 | 580 | 150–580 | 850 | 90 | 870 | 110 | 650 | 180 | 650 | 180 |
| EC1 | 2%O2 + 98%He | 550 | 150–580 | 580 | 150–580 | 650 | 30 | 550 | 45 | 550 | 240 | 500 | 120 |
| EC2 | 2%O2 + 98%He | 700 | 150–580 | 740 | 150–580 | 750 | 30 | 625 | 45 | 850 | 150 | 550 | 120 |
| EC3 | 2%O2 + 98%He | 800 | 150–580 | 840 | 150–580 | 825 | 30 | 700 | 45 | n/a | n/a | 700 | 70 |
| EC4 | 2%O2 + 98%He | n/a | n/a | n/a | n/a | 920 | >120 | 775 | 45 | n/a | n/a | 850 | 80 |
| EC5 | 2%O2 + 98%He | n/a | n/a | n/a | n/a | n/a | n/a | 850 | 45 | n/a | n/a | n/a | n/a |
| EC6 | 2%O2 + 98%He | n/a | n/a | n/a | n/a | n/a | n/a | 870 | 110 | n/a | n/a | n/a | n/a |
| Chemical Composition | Specific Classification | Influence Mechanism | Reference |
|---|---|---|---|
| Chemical composition | Calcium carbonate, natural calcite | They form an EC-like carbon signal. | [80] |
| Organic carbon | Brown carbon | 1. It has strong light absorption at short wavelengths and interferes with the laser signal; 2. it forms PC. | [81] |
| Humic-like substances | 1. They have strong thermal stability and can be evolved in the oxidation stage; 2. they form PC. | [82] | |
| Metal | Metallic oxides | The release of O2 in the inert mode causes EC and/or PC to early evolution. | [83] |
| Metal salts | 1. They reduce the oxidation temperature of EC; 2. they can increase the charring degree of OC. | [84] | |
| Inorganic salt | NH4HSO4 | It can change the charring degree of OC. | [58] |
| K+, Na+ | They can change the combustion temperature of carbonaceous components. | [85] | |
| Refractory oxygen-containing surface groups | CO1+, CO2+ | They can catalyze the early evolution of EC. | [86] |
| Solvent Categories | Operation Steps | Sample Types | OC Removal Efficiency | Reference |
|---|---|---|---|---|
| Ultra-pure water | Put the filter on the glass sand core (the area is 1.5 cm2), pour the 100 mL ultra-pure water into the glass sand core. | ambient sample | 28~79% | [99] |
| Ultra-pure water | The liquid soaked in the filter was extracted from the 30 mL bottle with a 5 mL disposable syringe, filtered (0.45 μm PTFE Filter head), and transferred to another 30 mL bottle. | ambient sample | - | [108] |
| Ultra-pure water | The filter is placed on the glass sand core (the diameter is 37 mm), the ultra-pure water is introduced into the glass sand core, and the water consumption is dynamically set according to the TC loading. | ambient sample | - | [100] |
| Ultra-pure water | - | marine aerosol | 50~56% | [101] |
| Ultra-pure water | Ultrasonic extraction of samples with ultra-pure water for 30 min | ambient sample | 53.80% | [102] |
| Ultra-pure water | After cleaning the filter with ultra-pure water (100 mL), the liquid is gradually dripped into the filter sand with a pipette until thoroughly wet and filtered in a vacuum. To prevent the porous diaphragm from fouling, a protective filter is inserted between the sample and the diaphragm. | ambient sample | 28~55% | [78] |
| Ultra-pure water | Same as [98]. | ambient sample | - | [103] |
| Methanol | The filter was immersed in methanol followed by ultrasonic extraction for 1 h. | wood burning smoke | 92~98% | [104] |
| Methanol | Same as [108]. | ambient sample | 85% | [109] |
| Ultra-pure water | 40% | |||
| Methanol | Immerse the filter sample in methanol for 1 h. | ambient sample | 89% | [110] |
| Ultra-pure water | Same as [104]. | ambient sample | 42 ± 18% | [105] |
| Methanol | 76 ± 29% | |||
| Methanol | The sample is placed between the two blank filters and put together on the glass sand core of the vacuum filter. Methanol is added to the filter three times to ensure that the retention time of methanol at the sand core is more than 1 h. | biomass burning sample | 93 ± 3.8% | [106] |
| ambient sample | 79.3 ± 10% | |||
| A mixture of dichloromethane, acetone, and hexane | The filter was immersed in dichloromethane, acetone, and hexane (volume ratio 2:4:4) for 1 h. The solvent mixture was changed after 30 min and stirred gently regularly. | ambient sample | 81% | [62] |
| A mixture of ultra-pure water, dichloromethane, and acetone | Non-BC material is removed by a two-step method, and the sample with a diameter of 47 mm is placed on the funnel sand core. First, the funnel is injected slowly with 50~200 mL distilled water, retained for 30 min, and then discharged. Then the 60 mL 1:1 mixture is injected into the funnel retained for 30 min and then released. | ambient sample | - | [98] |
| diesel vehicle emission | ||||
| Dichloromethane | - | marine engine emission | - | [97] |
| Ultra-pure water | The filter was dried at 180 °C for 1 h, and then extracted via vacuum funnel extraction. | marine engine emission | - | [107] |
| Toluene |
| Technology Classification | Instrument Names | Principle | Coverage Wavelength Range (nm) | Source of Deviation | Reference |
|---|---|---|---|---|---|
| In situ | PASS (Photoacoustic Soot Spectrometer) | Photoacoustic technology | 405, 532, 781 | BC aging | [113] |
| PAX (Photoacoustic Extinctiometer) | 405, 532, 870 | [116] | |||
| MSS (Micro Soot Sensor) | 808 | [114] | |||
| PTI (Photo thermal interferometry) | 450, 880 | [126] | |||
| TAP (Tricolor Absorption Photometer) | 365, 467, 528, 652 | [127] | |||
| DPAS (Differential Photoacoustic Spectrometer) | 473, 532, 671 | [117] | |||
| Filter-based | MAAP (Multi-Angle Absorption Photometer) | Quantitative mass according to the attenuation of transmitted light passing through the filter. | 637 | PM morphology | [115] |
| AE (Aethalometer) | 370, 470, 520, 590, 660, 880, 950 | Loading effect of PM, multiple scattering effects of filter, scattering effect of PM | [118] | ||
| PSAP (Particle Soot Absorption Photometer) | 467, 530, 660 | [119] | |||
| COSMOS (Continuous Soot Monitoring System) | 565 | Charring of low volatile organic compounds | [120] | ||
| CLAP (Continuous Light Absorption Photometer) | 467, 528, 652 | BC aging | [121] |
| Correction Scheme | Corrected Bias Effect | Instrument | Reference |
|---|---|---|---|
| Weingartner (2003) | filter loading effects, filter multiple scattering effects | AE30 | [137] |
| Arnott (2005) | filter loading effects, filter multiple scattering effects, loaded aerosol scattering effects | AE31 | [133] |
| Schmid (2006) | filter loading effects, filter multiple scattering effects, loaded aerosol scattering effects | AE30 | [135] |
| Virkkula (2007) | filter loading effects | AE16, AE30 | [136] |
| Coen (2010) | filter loading effects, filter multiple scattering effects, loaded aerosol scattering effects | AE10, AE16, AE31 | [134] |
| Virkkula (2010) | filter loading effects, loaded aerosol scattering effects | TAP | [119] |
| Ogren (2010) | filter loading effects, loaded aerosol scattering effects | TAP | [138] |
| Drinovec (2015) | filter loading effects | AE33 | [131] |
| Kim (2018) | filter multiple scattering effects, loaded aerosol scattering effects | AE31, CLAP | [130] |
| Technology Classification | Instruments Names | Principle | Detection Ability | Source of Deviation | Reference |
|---|---|---|---|---|---|
| Laser induced incandescence | SP2 (Single Particle Soot Photometer) | Laser induced incandescence method | Range of particle size: 65~600 nm | 1.PM particle size detection range; 2. calibration material | [30] |
| SP2-XR (Single Particle Soot Photometer–Extended Range) | Range of particle size: 50~800 nm | [160] | |||
| LII 300 (laser-Induced Incandescence Instrument System) | Range of laser flux: 0.6~3.2 mI/mm2 | Particles of different sizes would reach different peak temperatures at different times, bringing uncertainty to the effective peak temperature. | [158] | ||
| Single-particle aerosol mass spectrum | SP-AMS (Soot Particle Aerosol Mass Spectrometer) | Laser induced incandescence, mass spectrometry | Range of particle size: PM1.0 | Collection efficiency | [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Cheng, Y.; Liang, L.; Liu, J. The Measurement of Atmospheric Black Carbon: A Review. Toxics 2023, 11, 975. https://doi.org/10.3390/toxics11120975
Zhang Z, Cheng Y, Liang L, Liu J. The Measurement of Atmospheric Black Carbon: A Review. Toxics. 2023; 11(12):975. https://doi.org/10.3390/toxics11120975
Chicago/Turabian StyleZhang, Zhiqing, Yuan Cheng, Linlin Liang, and Jiumeng Liu. 2023. "The Measurement of Atmospheric Black Carbon: A Review" Toxics 11, no. 12: 975. https://doi.org/10.3390/toxics11120975
APA StyleZhang, Z., Cheng, Y., Liang, L., & Liu, J. (2023). The Measurement of Atmospheric Black Carbon: A Review. Toxics, 11(12), 975. https://doi.org/10.3390/toxics11120975