Antioxidant Compounds for the Inhibition of Enzymatic Browning by Polyphenol Oxidases in the Fruiting Body Extract of the Edible Mushroom Hericium erinaceus


Abstract
1. Introduction
2. Results and Discussion
2.1. Observation of Dark Brown Pigment in the Fruiting-Body Lysate
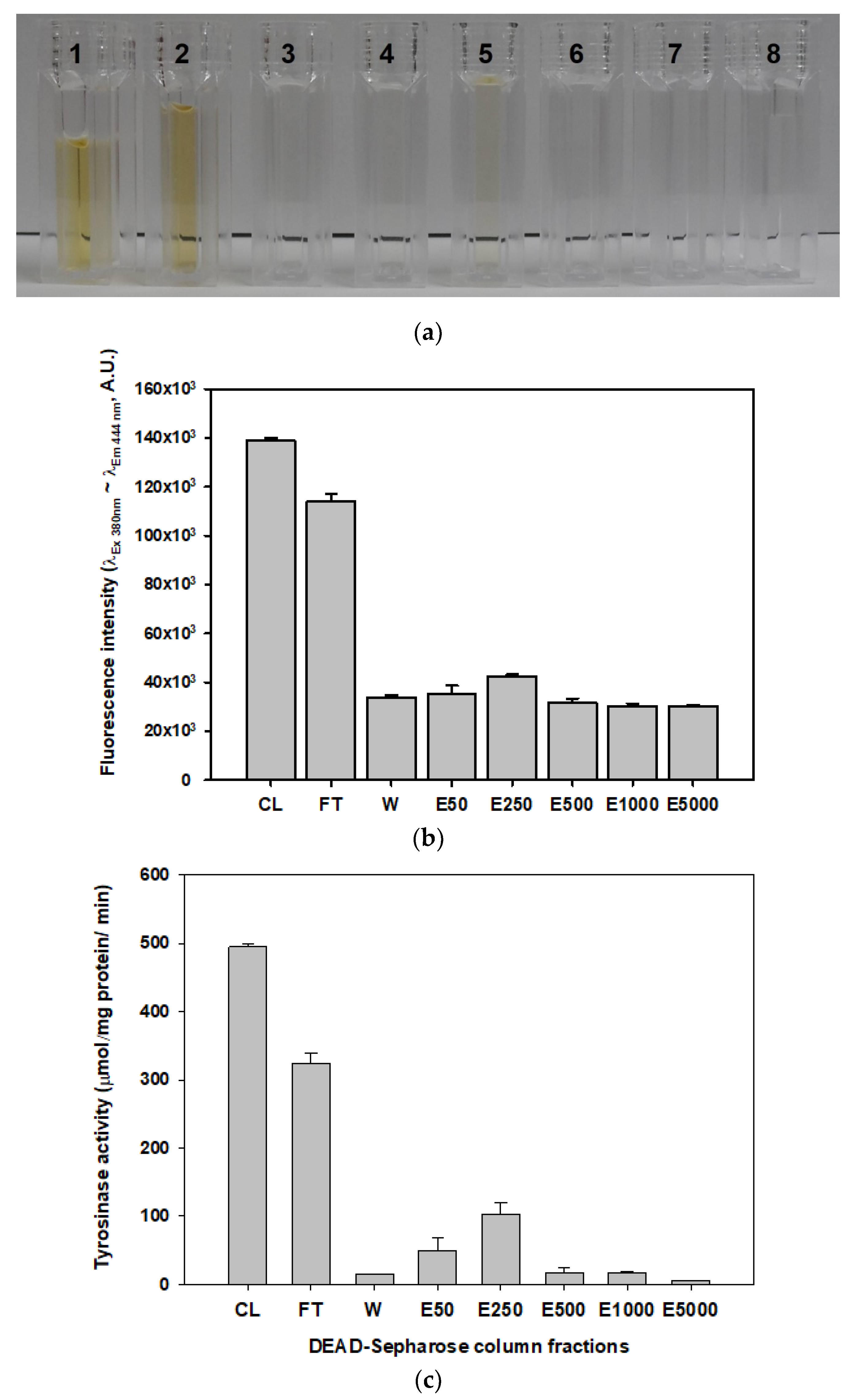
2.2. Measurement of Dark Brown Pigment
2.3. Measurement of Tyrosinase Activity and its Deactivation in the Mushroom Extract
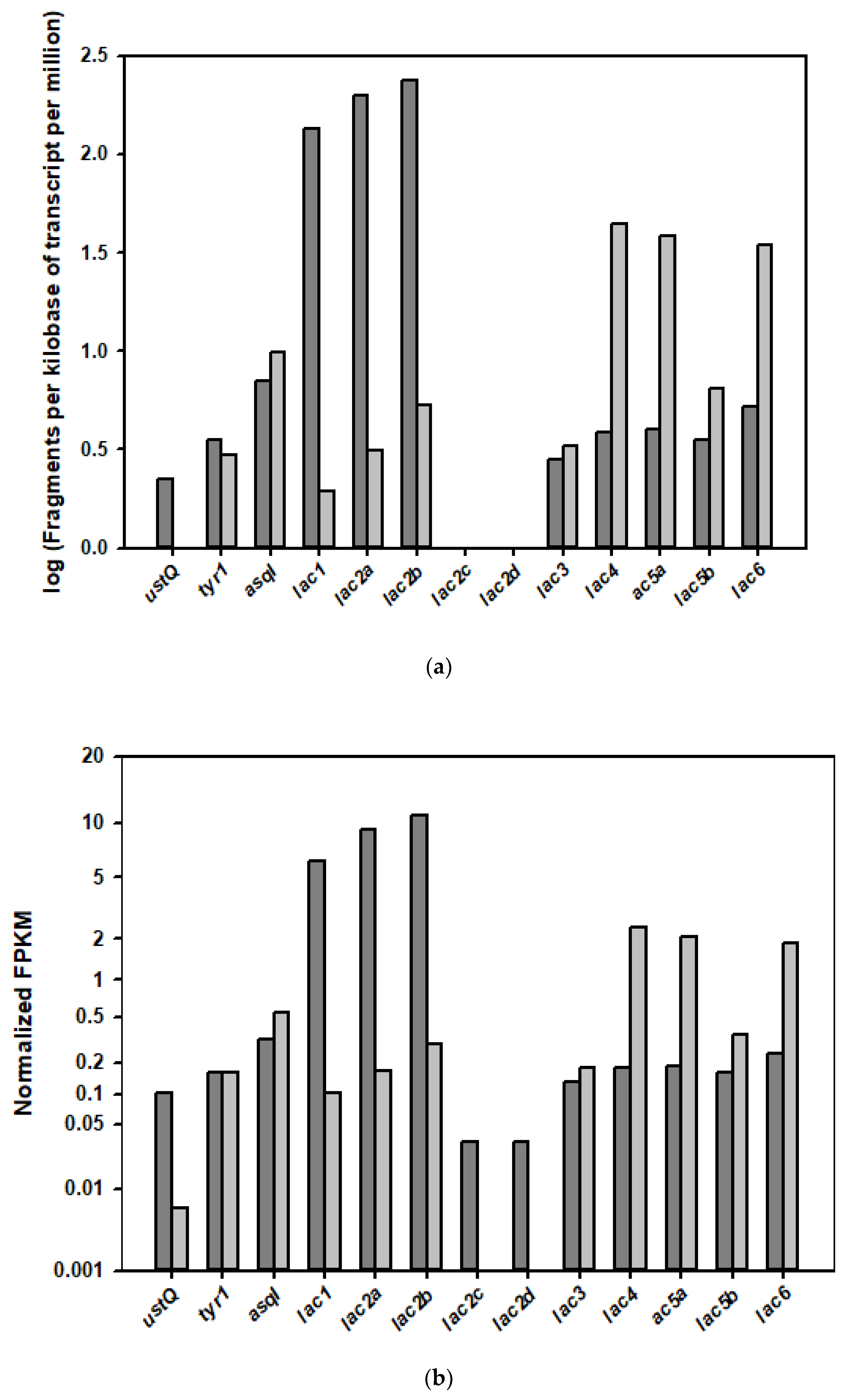
2.4. Analysis of the Transcription Levels of PPO-Coding Genes in the Different Growth Phases of the Mushroom
2.5. Analysis of the Main Metabolites in the Mushroom Fruiting Body and Mycelium
3. Material and Methods
3.1. Mushroom Strains and Culture Conditions
3.2. Preparation of the Mushroom Fruiting-Body Lysates
3.3. Fractionation of the Mushroom Fruiting-Body Lysate Using Ion-Exchange Chromatography
3.4. Browning Pigment Analysis
3.5. RNA Sequencing of the Mushroom Fruiting Body
3.6. Tyrosinase Activity Assay
3.7. Deactivation or Inactivation of the Polyphenol Oxidases’ Activities in the Mushroom Lysate
3.8. Metabolite Extracts and Analysis in the Mushroom Fruiting Body and Mycelium
4. Conclusions
Funding
Conflicts of Interest
References
- Erjavec, J.; Kos, J.; Ravnikar, M.; Dreo, T.; Sabotič, J. Proteins of higher fungi—From forest to application. Trends Biotechnol. 2012, 30, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Mushroom Polysaccharides: Chemistry and Antiobesity, Antidiabetes, Anticancer, and Antibiotic Properties in Cells, Rodents, and Humans. Foods 2016, 5, 80. [Google Scholar] [CrossRef]
- Chang, S.T.; Wasser, S.P. Current and Future Research Trends in Agricultural and Biomedical Applications of Medicinal Mushrooms and Mushroom Products (Review). Int. J. Med. Mushrooms 2018, 20, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, X.; Fang, J.; Chang, Y.; Ning, N.; Guo, H.; Huang, L.; Huang, X.; Zhao, Z. Structures, biological activities, and industrial applications of the polysaccharides from Hericium erinaceus (Lion’s Mane) mushroom: A review. Int. J. Boil. Macromol. 2017, 97, 228–237. [Google Scholar] [CrossRef]
- Friedman, M. Chemistry, Nutrition, and Health-Promoting Properties of Hericium erinaceus (Lion’s Mane) Mushroom Fruiting Bodies and Mycelia and Their Bioactive Compounds. J. Agric. Food Chem. 2015, 63, 7108–7123. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Bao, L.; Yang, Y.; Ma, K.; Liu, H. Three new cyathane diterpenes with neurotrophic activity from the liquid cultures of Hericium erinaceus. J. Antibiot. 2018, 71, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Hetland, G.; Tangen, J.-M.; Mahmood, F.; Mirlashari, M.R.; Nissen-Meyer, L.S.H.; Nentwich, I.; Therkelsen, S.P.; Tjønnfjord, G.E.; Johnson, E. Antitumor, Anti-inflammatory and Antiallergic Effects of Agaricus blazei Mushroom Extract and the Related Medicinal Basidiomycetes Mushrooms, Hericium erinaceus and Grifola frondosa: A Review of Preclinical and Clinical Studies. Nutrients 2020, 12, 1339. [Google Scholar] [CrossRef]
- Wong, J.H.; Ng, T.B.; Chan, H.H.L.; Liu, Q.; Man, G.C.W.; Zhang, C.Z.; Guan, S.; Ng, C.C.W.; Fang, E.F.; Wang, H.; et al. Mushroom extracts and compounds with suppressive action on breast cancer: Evidence from studies using cultured cancer cells, tumor-bearing animals, and clinical trials. Appl. Microbiol. Biotechnol. 2020, 104, 4675–4703. [Google Scholar] [CrossRef]
- Kim, S. A novel core 1 O-linked glycan-specific binding lectin from the fruiting body of Hericium erinaceus. Int. J. Boil. Macromol. 2018, 107, 1528–1537. [Google Scholar] [CrossRef]
- Sullivan, M.L. Beyond brown: Polyphenol oxidases as enzymes of plant specialized metabolism. Front. Plant Sci. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.M.; Kwon, E.-B.; Lee, B.; Kim, C.Y. Recent Trends in Controlling the Enzymatic Browning of Fruit and Vegetable Products. Molecules 2020, 25, 2754. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M. Polyphenol oxidases in plants and fungi: Going places? A review. Phytochemistry 2006, 67, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Soler-Rivas, C.; Arpin, N.; Olivier, J.M.; Wichers, H.J. Activation of tyrosinase in Agaricus bisporus strains following infection by Pseudomonas talaasii or treatment with a tolaasin-containing preparation. Mycol. Res. 1997, 101, 375–382. [Google Scholar] [CrossRef]
- Van Duuren, B.L. The Fluorescence Spectra of Aromatic Hydrocarbons and Heterocyclic Aromatic Compounds. Anal. Chem. 1960, 32, 1436–1442. [Google Scholar] [CrossRef]
- Halaouli, S.; Asther, M.; Sigoillot, J.-C.; Hamdi, M.; Lomascolo, A. Fungal tyrosinases: New prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 2006, 100, 219–232. [Google Scholar] [CrossRef]
- Soler-Rivas, C.; Olivier, J.; Wichers, H.J.; Jolivet, S.; Arpin, N. Biochemical and physiological aspects of brown blotch disease of Agaricus bisporus. FEMS Microbiol. Rev. 1999, 23, 591–614. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, S.; Chun, J.; Lee, H.; Lee, J. Influence of heat treatment on the antioxidant activities and polyphenolic compounds of Shiitake (Lentinus edodes) mushroom. Food Chem. 2006, 99, 381–387. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: Structure, inhibition mechanism and perspective for the future. Cell. Mol. Life Sci. 2005, 62, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, M.R.; Tundis, R.; Menichini, F. Natural and Synthetic Tyrosinase Inhibitors as Antibrowning Agents: An Update. Compr. Rev. Food Sci. Food Saf. 2012, 11, 378–398. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Khan, M.T.H.; Muñoz-Muñoz, J.L.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A comprehensive review on tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef]
- Cerny, A.C.; Davidek, T. Formation of Aroma Compounds from Ribose and Cysteine during the Maillard Reaction. J. Agric. Food Chem. 2003, 51, 2714–2721. [Google Scholar] [CrossRef] [PubMed]
- Skoog, D.A.; Holler, F.J.; Nieman, T. Molecular luminescence spectrometry. In Principle of Instrumental Analysis, 5th ed.; Mrssina, F., Sherman, M., Bortel, J., Eds.; Harcourt Brace & Company: Orlando, CA, USA, 1998; pp. 355–379. [Google Scholar]
- Zafar, K.S.; Siegel, D.; Ross, D. A Potential Role for Cyclized Quinones Derived from Dopamine, DOPA, and 3,4-Dihydroxyphenylacetic Acid in Proteasomal Inhibition. Mol. Pharmacol. 2006, 70, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Harrison, W.H.; Whisler, W.W.; Ko, S. Detection, and study by fluorescence spectrometry of stereospecificity in mushroom tyrosinase-catalyzed oxidations. Proposal of a copper-containing reaction rate control site. J. Boil. Chem. 1967, 242, 1660–1667. [Google Scholar]
- Friedman, M. Food Browning, and Its Prevention: An Overview. J. Agric. Food Chem. 1996, 44, 631–653. [Google Scholar] [CrossRef]
- Kuijpers, T.F.M.; van Herk, T.; Vincken, J.-P.; Janssen, R.H.; Narh, D.L.; van Berkel, W.J.H.; Gruppen, H. Potato and Mushroom Polyphenol Oxidase Activities Are Differently Modulated by Natural Plant Extracts. J. Agric. Food Chem. 2013, 62, 214–221. [Google Scholar] [CrossRef]
- Kaur, K.; Sharma, A.; Capalash, N.; Sharma, P. Multicopper oxidases: Biocatalysts in microbial pathogenesis and stress management. Microbiol. Res. 2019, 222, 1–13. [Google Scholar] [CrossRef]
- Smith, D.F.Q.; Casadevall, A. The Role of Melanin in Fungal Pathogenesis for Animal Hosts. Curr. Top. Microbiol. Immunol. 2019, 422, 1–30. [Google Scholar] [CrossRef]
- Cordero, R.J.; Casadevall, A. Functions of fungal melanin beyond virulence. Fungal Boil. Rev. 2017, 31, 99–112. [Google Scholar] [CrossRef]
- Pezzella, C.; Guarino, L.; Piscitelli, A. How to enjoy laccases. Cell. Mol. Life Sci. 2015, 72, 923–940. [Google Scholar] [CrossRef]
- Tobimatsu, Y.; Schuetz, M. Lignin polymerization: How do plants manage the chemistry so well? Curr. Opin. Biotechnol. 2019, 56, 75–81. [Google Scholar] [CrossRef]
- Pourcel, L.; Routaboul, J.-M.; Cheynier, V.; Lepiniec, L.; Debeaujon, I. Flavonoid oxidation in plants: From biochemical properties to physiological functions. Trends Plant Sci. 2007, 12, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Coman, C.; Moț, A.C.; Gal, E.; Parvu, M.; Silaghi-Dumitrescu, R. Laccase is upregulated via stress pathways in the phytopathogenic fungus Sclerotinia sclerotiorum. Fungal Boil. 2013, 117, 528–539. [Google Scholar] [CrossRef]
- Upadhyay, P.; Shrivastava, R.; Agrawal, P.K. Bioprospecting, and biotechnological applications of fungal laccase. 3 Biotech 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Rescigno, A.; Sollai, F.; Sanjust, E.; Rinaldi, A.C.; Curreli, N.; Rinaldi, A. Diafiltration in the presence of ascorbate in the purification of mushroom tyrosinase. Phytochemistry 1997, 46, 21–22. [Google Scholar] [CrossRef]
- Pero, R.W.; Lund, H.; Leanderson, T. Antioxidant metabolism induced by quinic acid. increased urinary excretion of tryptophan and nicotinamide. Phytother. Res. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- Stampfli, A.R.; Blankenfeldt, W.; Seebeck, F.P. Structural basis of ergothioneine biosynthesis. Curr. Opin. Struct. Biol. 2020, 65, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, D.P. Interaction of ergothioneine with metal ions and metalloenzymes. J. Med. Chem. 1971, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Cheah, I.K.; Tang, R.M.Y. Ergothioneine—A diet-derived antioxidant with therapeutic potential. FEBS Lett. 2018, 592, 3357–3366. [Google Scholar] [CrossRef]
- Kalač, P. A review of chemical composition and nutritional value of wild-growing and cultivated mushrooms. J. Sci. Food Agric. 2012, 93, 209–218. [Google Scholar] [CrossRef]
- Heleno, S.A.; Barros, L.; Martins, A.; Queiroz, M.J.R.P.; Morales, P.; Fernández-Ruiz, V.; Ferreira, I.C.F.R.; Morales, P. Chemical composition, antioxidant activity and bioaccessibility studies in phenolic extracts of two Hericium wild edible species. LWT 2015, 63, 475–481. [Google Scholar] [CrossRef]
- Taofiq, O.; González-Paramás, A.M.; Martins, A.; Barreiro, M.F.; Ferreira, I.C.F.R. Mushrooms extracts and compounds in cosmetics, cosmeceuticals and nutricosmetics—A review. Ind. Crop. Prod. 2016, 90, 38–48. [Google Scholar] [CrossRef]
- Muszyńska, B.; Grzywacz-Kisielewska, A.; Kała, K.; Gdula-Argasińska, J. Anti-inflammatory properties of edible mushrooms: A review. Food Chem. 2018, 243, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Taofiq, O.; Heleno, S.A.; Calhelha, R.C.; Alves, M.J.; Barros, L.; Barreiro, M.F.; González-Paramás, A.; Ferreira, I.C. Development of Mushroom-Based Cosmeceutical Formulations with Anti-Inflammatory, Anti-Tyrosinase, Antioxidant, and Antibacterial Properties. Molecules 2016, 21, 1372. [Google Scholar] [CrossRef] [PubMed]
- Kim, S. Hericium erinaceus isolectins recognize mucin-type O-glycans as tumor-associated carbohydrate antigens on the surface of K562 human leukemia cells. Int. J. Boil. Macromol. 2018, 120, 1093–1102. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Relative Peak Area 1 | m/z2 | MT/RT 3 | |
|---|---|---|---|---|
| Fruiting Body | Mycelium | |||
| Antioxidant | ||||
| Tyrosinase inhibitor | ||||
| Ascorbic acid | 640 | - | 175.025 | 8.34 |
| Caffeic acid | 88 | - | 179.035 | 8.88 |
| Chlorogenic acid | 92 | - | 355.107 | 5.51 |
| Cucumin | 45 | - | 269.134 | 10.32 |
| Pyridoxal | 6 | - | 166.066 | 8.52 |
| Quinic acid | 6400 | - | 191.058 | 8.07 |
| Terephtalic acid | 110 | 7.4 | 165.020 | 16.38 |
| Vanillic acid | - | 4.0 | 167.035 | 8.71 |
| Antioxidant, free radical scavenger | ||||
| Adenosine | 510 | 560 | 268.105 | 9.60 |
| Ergosterol | 1300 | 1300 | 379.339 | 15.63 |
| Ergothioneine | 2900 | 100 | 230.096 | 17.29 |
| Glutathione (GSH) | 12 | 240 | 308.092 | 12.98 |
| 5-Hydroxytryptophan | 20 | 3.7 | 221.094 | 11.20 |
| Hypotaurine | 33 | - | 110.027 | 18.06 |
| Tartaric acid | 180 | - | 149.011 | 22.05 |
| Polyamine | ||||
| 1,3-Diaminopropane | 19 | - | 75.091 | 4.31 |
| Putrescine | 16 | 1.3 | 89.107 | 4.58 |
| Spermidine | 190 | 2.4 | 146.165 | 4.40 |
| Compound | Relative Peak Area | m/z2 | MT/RT 3 | |
|---|---|---|---|---|
| Fruiting Body 1 | Mycelium 1 | |||
| Enzymatic browning substrates | ||||
| Amino acid and its derivative | ||||
| Indole-3-carboxyaldehyde | - | 170 | 146.062 | 6.91 |
| Kynurenine | 100 | 5.5 | 209.093 | 9.32 |
| Phenylalanine | 9400 | 1500 | 166.086 | 10.44 |
| Quinolinic acid | 160 | - | 166.016 | 15.47 |
| Tryptamine | 6.9 | - | 161.107 | 8.15 |
| Tryptophan | 520 | 110 | 205.097 | 10.69 |
| Tyrosine | 4900 | 280 | 182.081 | 11.09 |
| Phenolic compounds | ||||
| o-Aminophenol | - | 2.7 | 110.060 | 7.4 |
| o-Hydroxybenzoic acid | - | 2.1 | 137.025 | 10.10 |
| 2-Phenylethylamine | 19 | - | 122.097 | 7.52 |
| 3-Phenylpropionic acid | - | 5.6 | 149.062 | 8.60 |
| p-, m-, o-Toluic acid | - | 33 | 135.046 | 8.91 |
| Purine and pyrimidine nucleobases | ||||
| Purine base | ||||
| 1-Methyladenosine | 4.8 | 3.0 | 282.119 | 9.39 |
| 2′-, 5′-Deoxyadenosine | - | 2.1 | 252.111 | 9.09 |
| 3′-AMP | - | 2.9 | 346.059 | 9.42 |
| 5′-Deoxy-5′-methylthioadenosine | - | 2.2 | 298.097 | 9.49 |
| AMP | 290 | 11 | 346.058 | 9.02 |
| ADP | 260 | 33 | 426.024 | 10.52 |
| ATP | 270 | 33 | 505.991 | 11.36 |
| Adenine | - | 9.5 | 136.062 | 7.20 |
| GMP | 33 | - | 362.051 | 8.97 |
| GDP | - | 3.3 | 442.020 | 10.40 |
| GTP | 41 | - | 521.986 | 11.24 |
| Guanosine | 100 | 380 | 284.100 | 12.27 |
| Hypoxanthine | 23 | 14 | 137.046 | 10.45 |
| IMP | - | 1.8 | 347.038 | 9.28 |
| Inosine | - | 25 | 269.090 | 18.37 |
| Xanthine | - | 13 | 153.042 | 18.35 |
| Pyrimidine base | ||||
| Cytidine | 32 | 33 | 244.093 | 9.12 |
| UMP | 35 | 3.6 | 323.032 | 9.51 |
| UDP | - | 6.7 | 402.998 | 11.22 |
| UTP | - | 7.5 | 482.962 | 12.10 |
| Uracil | - | 17 | 113.036 | 20.08 |
| Uridine | 29 | 210 | 245.078 | 20.08 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S. Antioxidant Compounds for the Inhibition of Enzymatic Browning by Polyphenol Oxidases in the Fruiting Body Extract of the Edible Mushroom Hericium erinaceus. Foods 2020, 9, 951. https://doi.org/10.3390/foods9070951
Kim S. Antioxidant Compounds for the Inhibition of Enzymatic Browning by Polyphenol Oxidases in the Fruiting Body Extract of the Edible Mushroom Hericium erinaceus. Foods. 2020; 9(7):951. https://doi.org/10.3390/foods9070951
Chicago/Turabian StyleKim, Seonghun. 2020. "Antioxidant Compounds for the Inhibition of Enzymatic Browning by Polyphenol Oxidases in the Fruiting Body Extract of the Edible Mushroom Hericium erinaceus" Foods 9, no. 7: 951. https://doi.org/10.3390/foods9070951
APA StyleKim, S. (2020). Antioxidant Compounds for the Inhibition of Enzymatic Browning by Polyphenol Oxidases in the Fruiting Body Extract of the Edible Mushroom Hericium erinaceus. Foods, 9(7), 951. https://doi.org/10.3390/foods9070951