Sulfonic Acid-Functionalized SBA-15 as Strong Cation-Exchange Sorbent for Solid-Phase Extraction of Atropine and Scopolamine in Gluten-Free Grains and Flours

 ,
, 
 ,
,  and
and 
Abstract
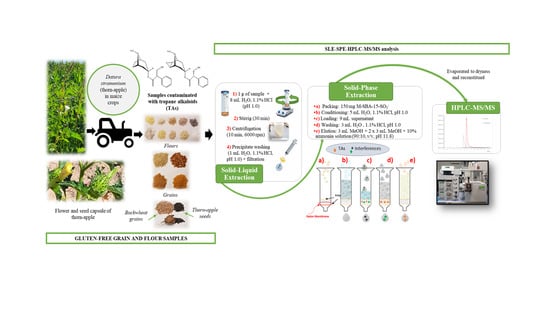
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Samples
2.3. Synthesis of Mesostructured Silicas
2.4. Characterization and Evaluation of Mesostructured Silicas
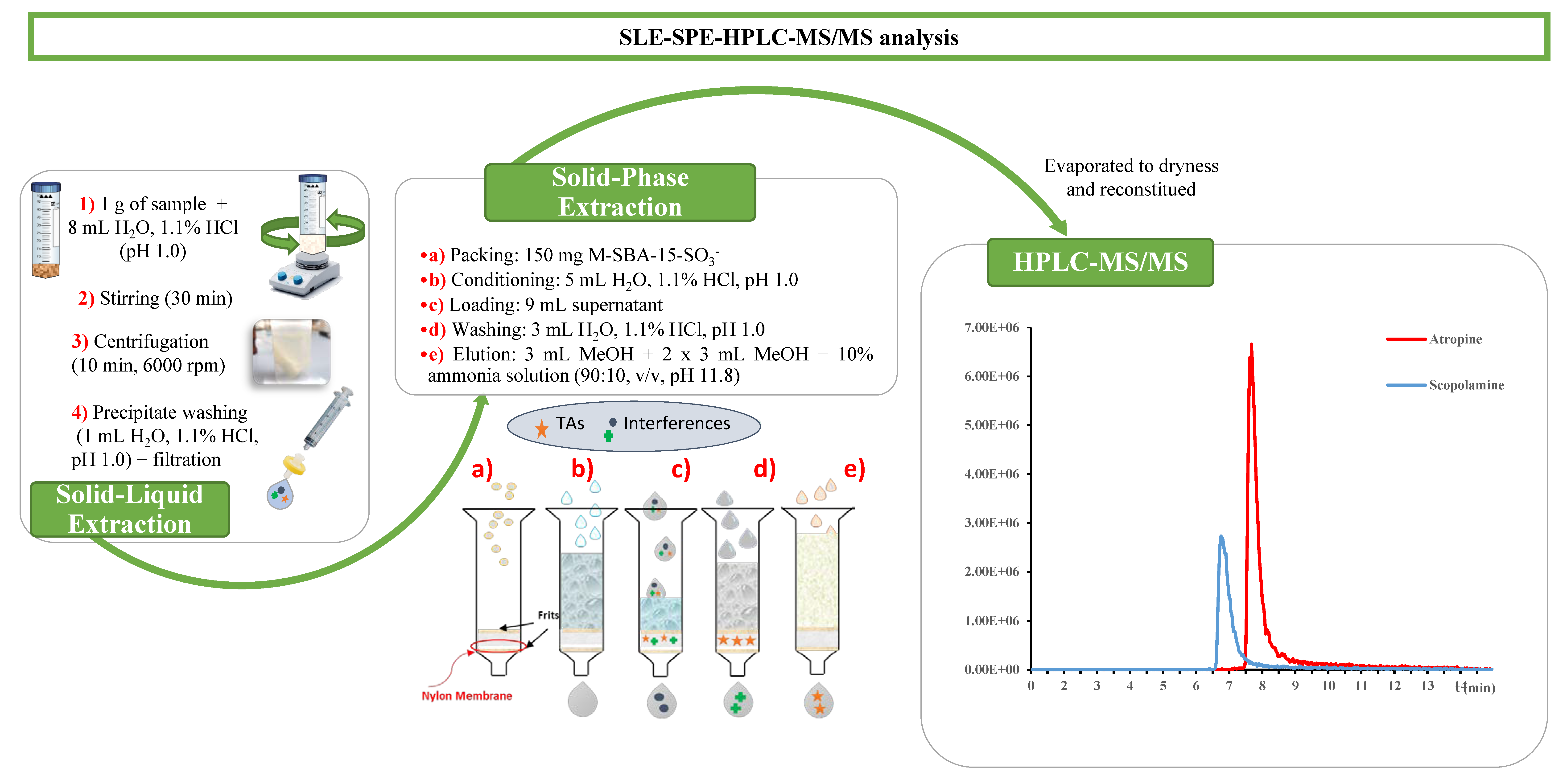
2.5. Optimized SLE-SPE Sample Extraction Procedure
2.6. Chromatographic Analysis and Instrumental Parameters
2.7. Method Validation
3. Results and Discussion
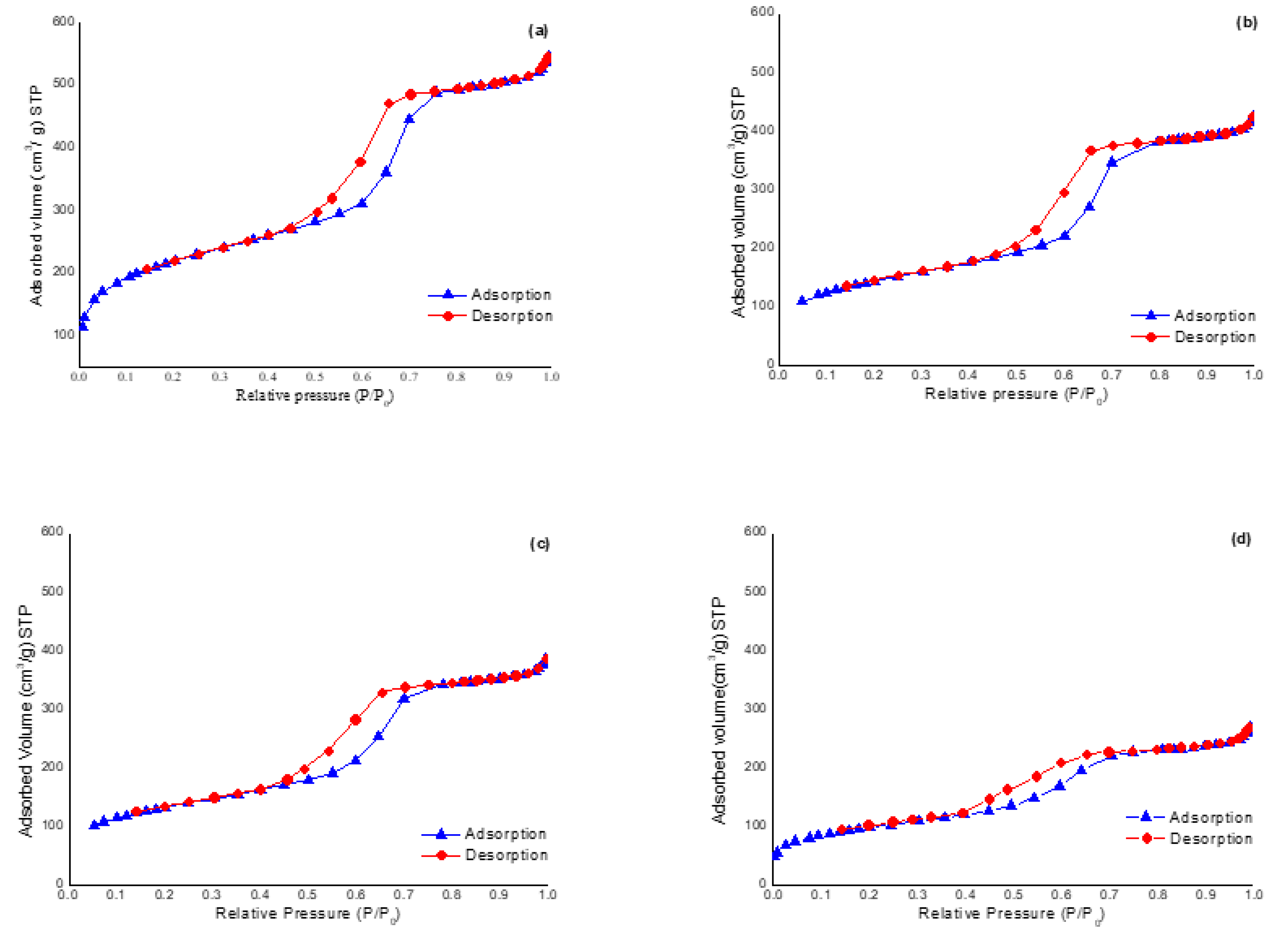
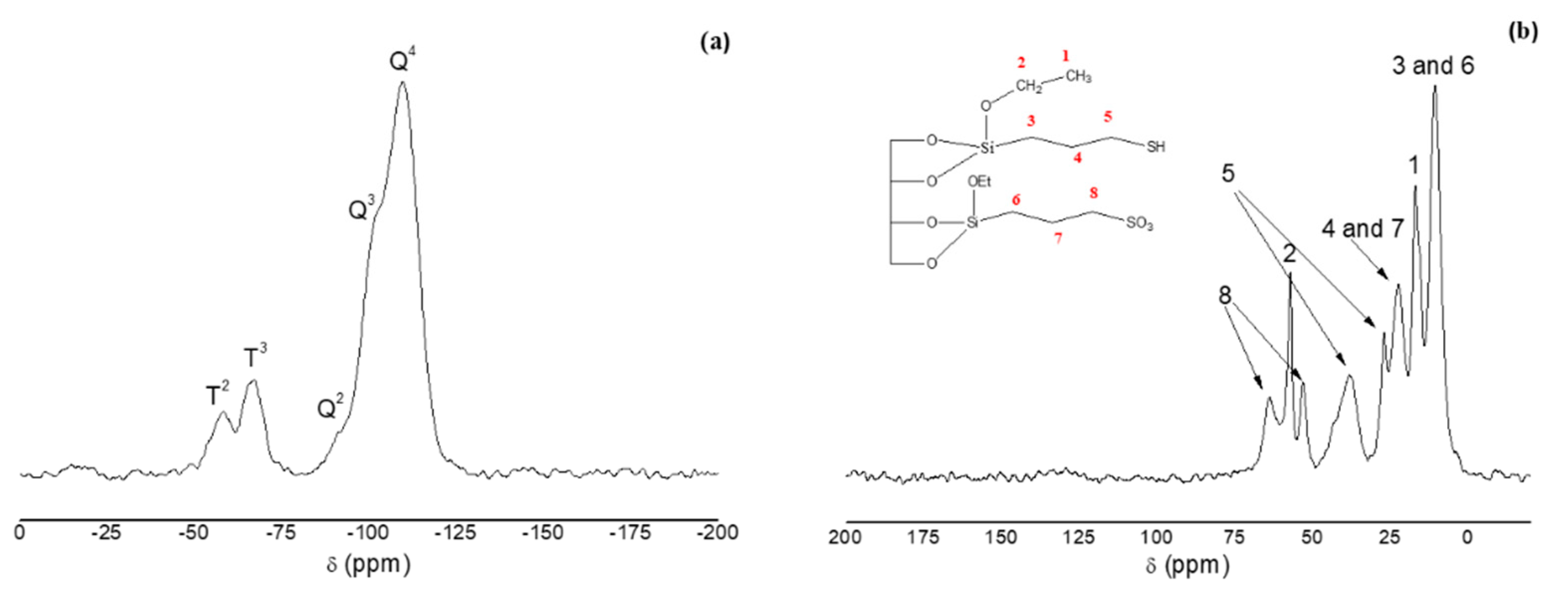
3.1. Characterization of Mesostructured Silicas and Evaluation as SCX-SPE Sorbent
3.2. Optimization of the Sample Treatment
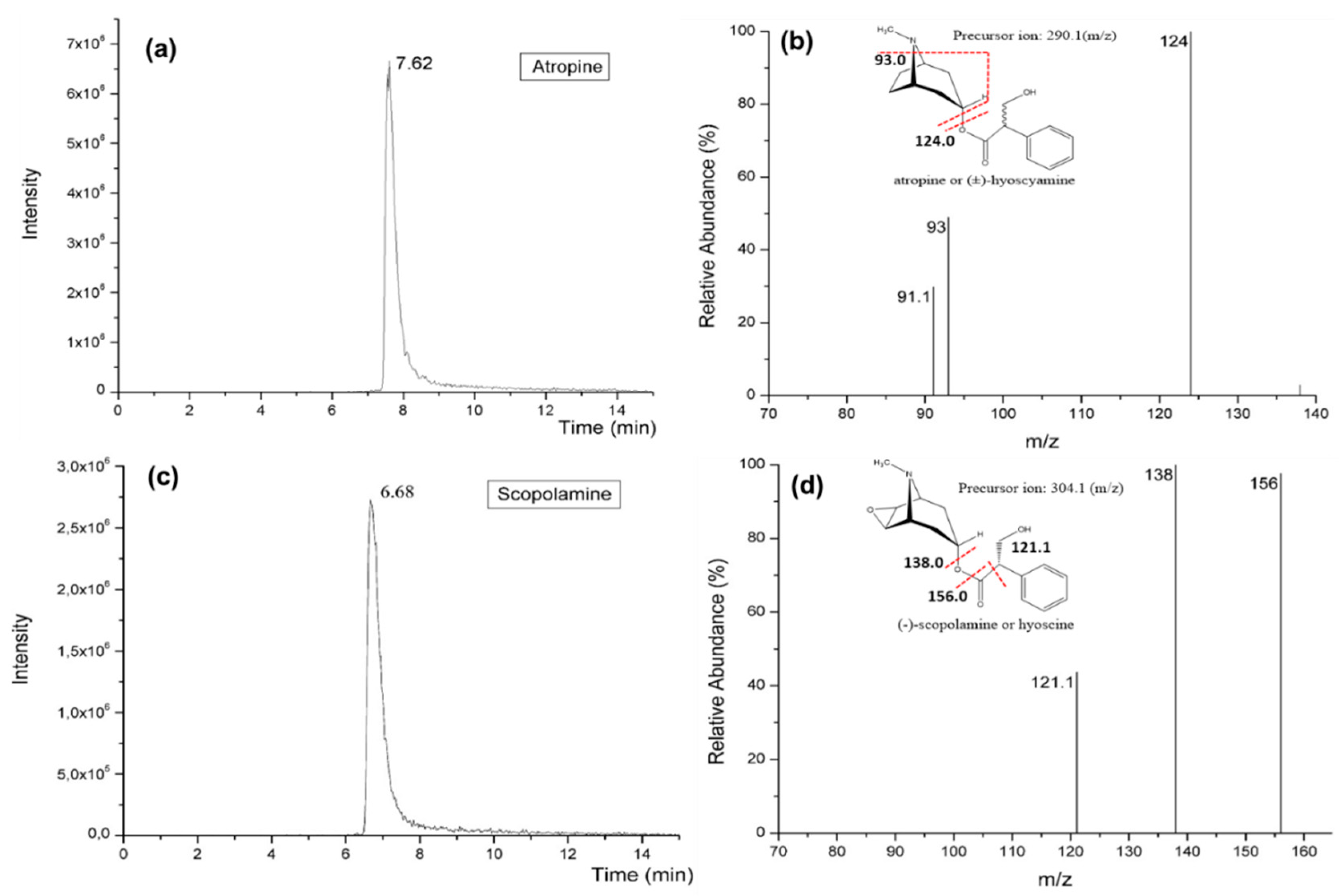
3.3. Optimization of HPLC-MS/MS Conditions and Instrumental Parameters
3.4. Method Performance Parameters
3.4.1. Evaluation of Matrix Effect, Calibrations and Limits
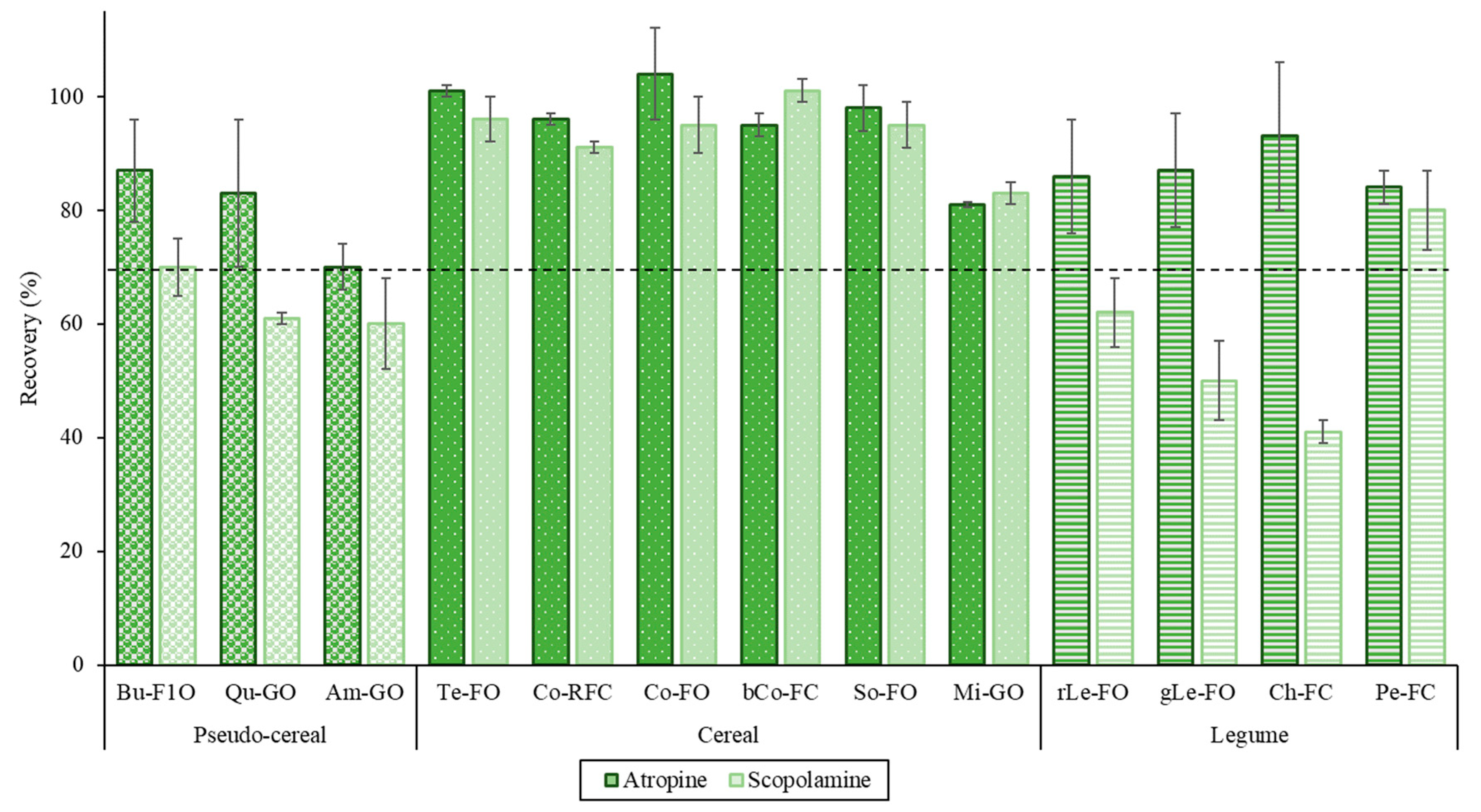
3.4.2. Selectivity, Accuracy and Precision
3.4.3. Robustness
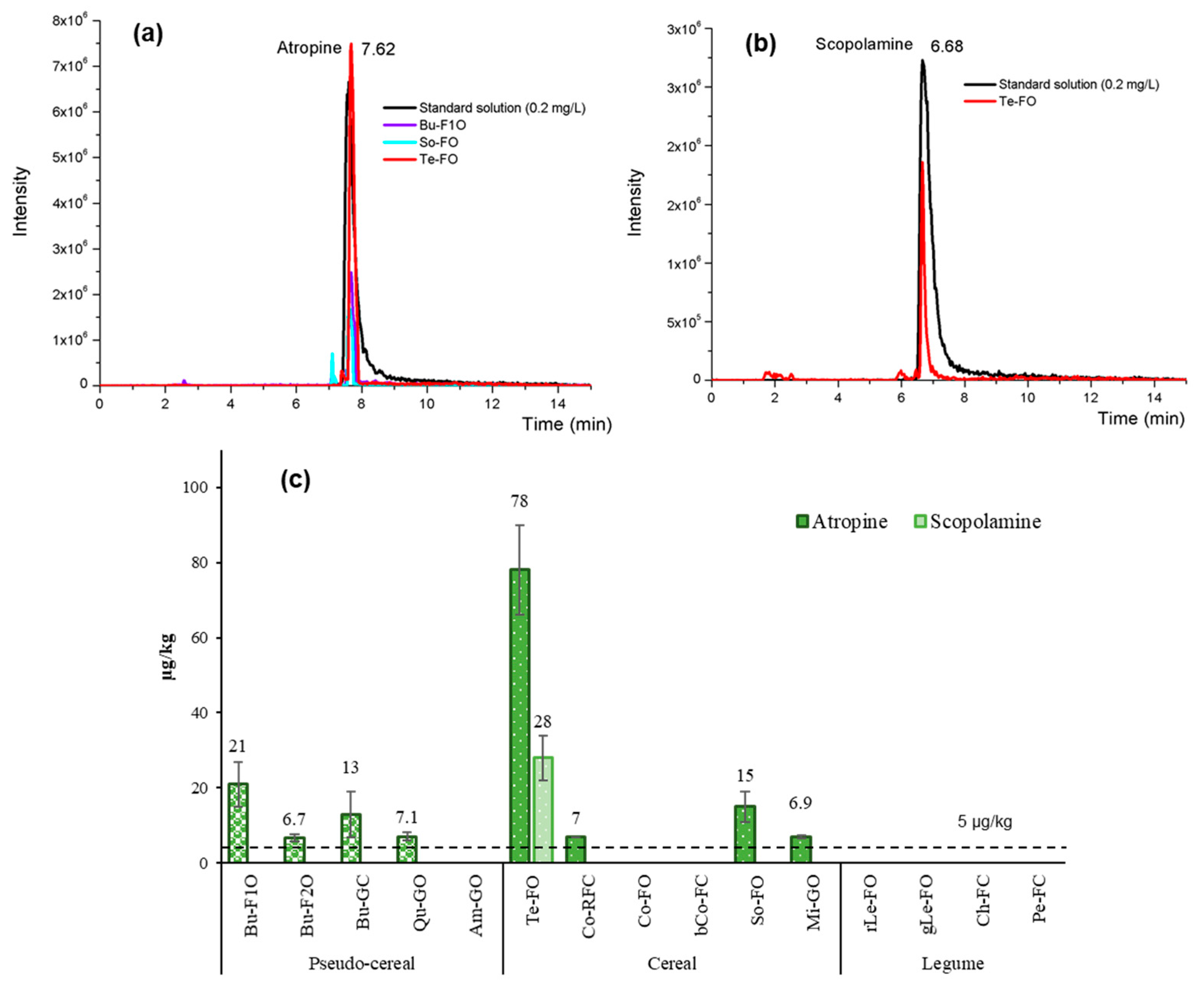
3.5. Application of the Validated Method to Commercial Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Giménez-Bastida, J.A.; Zielinski, H. Buckwheat as a Functional Food and Its Effects on Health. J. Agric. Food Chem. 2015, 63, 7896–7913. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P.; Arya, S.S. Nutritional, functional, phytochemical and structural characterization of gluten-free flours. J. Food Meas. Charact. 2017, 11, 1284–1294. [Google Scholar] [CrossRef]
- Barba, F.J.; Giuberti, G.; Rodriguez, J.M.L.; Barba, F.J.; Lorenzo, J.M. Gluten-free flours from cereals, pseudocereals and legumes: Phenolic fingerprints and in vitro antioxidant properties. Food Chem. 2019, 271, 157–164. [Google Scholar] [CrossRef]
- Burešová, I.; Tokár, M.; Mareček, J.; Hřivna, L.; Faměra, O.; Šottníková, V. The comparison of the effect of added amaranth, buckwheat, chickpea, corn, millet and quinoa flour on rice dough rheological characteristics, textural and sensory quality of bread. J. Cereal Sci. 2017, 75, 158–164. [Google Scholar] [CrossRef]
- Taylor, J.R.N.; Schober, T.J.; Bean, S.R. Novel food and non-food uses for sorghum and millets. J. Cereal Sci. 2006, 44, 252–271. [Google Scholar] [CrossRef]
- López-Martínez, A.; Azuara-Pugliese, V.; Sánchez-Macias, A.; Sosa-Mendoza, G.; Dibildox-Alvarado, E.; Grajales-Lagunes, A. High protein and low-fat chips (snack) made out of a legume mixture. CyTA J. Food 2019, 17, 661–668. [Google Scholar] [CrossRef]
- Pasqualone, A.; Costantini, M.; Coldea, T.E.; Summo, C. Use of Legumes in Extrusion Cooking: A Review. Foods 2020, 9, 958. [Google Scholar] [CrossRef]
- Gebremariam, M.M.; Zarnkow, M.; Becker, T. Teff (Eragrostis tef) as a raw material for malting, brewing and manufacturing of gluten-free foods and beverages: A review. J. Food Sci. Technol. 2012, 51, 2881–2895. [Google Scholar] [CrossRef]
- European Food Safety Authority. Scientific Opinion on Tropane alkaloids in food and feed. EFSA J. 2013, 11, 1–113. [Google Scholar] [CrossRef]
- Mulder, P.P.; De Nijs, M.; Castellari, M.; Hortós, M.; Macdonald, S.; Crews, C.; Hajslova, J.; Stranska, M. Occurrence of tropane alkaloids in food. EFSA Support. Publ. 2016, 13. [Google Scholar] [CrossRef]
- Mulder, P.P.J.; Pereboom-de Fauw, D.P.K.H.; Hoogenboom, R.L.A.P.; de Stoppelaar, J.; de Nijs, M. Tropane and ergot alkaloids in grain-based products for infants and young children in the Netherlands in 2011–2014. Food Addit. Contam. Part B Surveill. 2015, 8, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Bond, W.; Grundy, A.C. Non-chemical weed management in organic farming systems. Weed Res. 2001, 41, 383–405. [Google Scholar] [CrossRef]
- RASFF Portal—Food and Feed Safety Alerts. Available online: https://webgate.ec.europa.eu/rasff-window/portal/? (accessed on 28 October 2020).
- European Union. Commision Recommendation (EU) 2015/976 of 19 June 2015 on the monitoring of the presence of tropane alkaloids in food. Off. J. Eur. Union 2015, L157, 97. [Google Scholar]
- Caligiani, A.; Palla, G.; Bonzanini, F.; Bianchi, A.; Bruni, R. A validated GC–MS method for the detection of tropane alkaloids in buckwheat (Fagopyron esculentum L.) fruits, flours and commercial foods. Food Chem. 2011, 127, 204–209. [Google Scholar] [CrossRef]
- Marín-Sáez, J.; Romero-González, R.; Frenich, A.G. Degradation of tropane alkaloids in baked bread samples contaminated with Solanaceae seeds. Food Res. Int. 2019, 122, 585–592. [Google Scholar] [CrossRef]
- Chen, H.; Marín-Sáez, J.; Romero-González, R.; Frenich, A.G. Simultaneous determination of atropine and scopolamine in buckwheat and related products using modified QuEChERS and liquid chromatography tandem mass spectrometry. Food Chem. 2017, 218, 173–180. [Google Scholar] [CrossRef]
- Marín-Sáez, J.; Romero-González, R.; Frenich, A.G. Multi-analysis determination of tropane alkaloids in cereals and solanaceaes seeds by liquid chromatography coupled to single stage Exactive-Orbitrap. J. Chromatogr. A 2017, 1518, 46–58. [Google Scholar] [CrossRef]
- Casado, N.; Pérez-Quintanilla, D.; Morante-Zarcero, S.; Sierra, I. Current development and applications of ordered mesoporous silicas and other sol–gel silica-based materials in food sample preparation for xenobiotics analysis. TrAC Trends Anal. Chem. 2017, 88, 167–184. [Google Scholar] [CrossRef]
- Sierra, I.; Morante-Zarcero, S. New Advances in Food Sample Preparation with Nanomaterials for Organic Contaminants Analysis by Liquid Chromatography. In Nanomaterials in Chromatography, 1st ed.; Hussain, C.M., Ed.; Elsevier: Amsterdam, Netherlands, 2018; pp. 118–154. [Google Scholar] [CrossRef]
- Chen, Q.; Zhu, L.; Chen, J.; Jiang, T.; Ye, H.; Ji, H.; Tsang, S.; Zhao, Z.; Yi, T.; Chen, H.-B. Recent progress in nanomaterial-based assay for the detection of phytotoxins in foods. Food Chem. 2019, 277, 162–178. [Google Scholar] [CrossRef]
- Maciel, E.V.S.; De Toffoli, A.L.; Neto, E.S.; Nazario, C.E.D.; Lanças, F.M. New materials in sample preparation: Recent advances and future trends. TrAC Trends Anal. Chem. 2019, 119, 115633. [Google Scholar] [CrossRef]
- Casado, N.; Pérez-Quintanilla, D.; Morante-Zarcero, S.; Sierra, I. Evaluation of bi-functionalized mesoporous silicas as reversed phase/cation-exchange mixed-mode sorbents for multi-residue solid phase extraction of veterinary drug residues in meat samples. Talanta 2017, 165, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Casado, N.; Pérez-Quintanilla, D.; Morante-Zarcero, S.; Sierra, I. Bi-functionalized mesostructured silicas as reversed-phase/strong anion-exchange sorbents. Application to extraction of polyphenols prior to their quantitation by UHPLC with ion-trap mass spectrometry detection. Microchim. Acta 2019, 186, 164. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Hydroxymethylfurfural determination in cereal and insect bars by high-performance liquid chromatography-mass spectrometry employing a functionalized mesostructured silica as sorbent in solid-phase extraction. J. Chromatogr. A 2020, 1622, 461124. [Google Scholar] [CrossRef] [PubMed]
- Casado, N.; Gañán, J.; Morante-Zarcero, S.; Sierra, I. New advanced materials and sorbent-based microextraction techniques as strategies in sample preparation to improve the determination of natural toxins in food samples. Molecules 2020, 25, 702. [Google Scholar] [CrossRef]
- Cirlini, M.; DeMuth, T.M.; Biancardi, A.; Rychlik, M.; Dall’Asta, C.; Bruni, R. Are tropane alkaloids present in organic foods? Detection of scopolamine and atropine in organic buckwheat (Fagopyron esculentum L.) products by UHPLC–MS/MS. Food Chem. 2018, 239, 141–147. [Google Scholar] [CrossRef]
- Jandric, Z.; Rathor, M.; Švarc-Gajić, J.; Maestroni, B.; Sasanya, J.; Djurica, R.; Cannavan, A. Development of a liquid chromatography-tandem mass spectrometric method for the simultaneous determination of tropane alkaloids and glycoalkaloids in crops. Food Addit. Contam. Part A 2011, 28, 1205–1219. [Google Scholar] [CrossRef]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic Triblock and Star Diblock Copolymer and Oligomeric Surfactant Syntheses of Highly Ordered, Hydrothermally Stable, Mesoporous Silica Structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Luo, G.; Dai, Y. Functionalization of SBA-15 mesoporous silica with thiol or sulfonic acid groups under the crystallization conditions. Microporous Mesoporous Mater. 2005, 84, 275–282. [Google Scholar] [CrossRef]
- Margolese, D.; Melero, J.A.; Christiansen, S.C.; Chmelka, B.F.; Stucky, G.D. Direct Syntheses of Ordered SBA-15 Mesoporous Silica Containing Sulfonic Acid Groups. Chem. Mater. 2000, 12, 2448–2459. [Google Scholar] [CrossRef]
- European Commision SANTE/11813/2017. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues in Food and Feed. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/AqcGuidance_SANTE_2019_12682.pdf (accessed on 21 July 2020).
- AOAC International. Appendix K: Guidelines for Dietary Supplements and Botanicals. AOAC Off. Methods Anal. 2013, 2, 1–32. [Google Scholar]
- Dräger, B. Analysis of tropane and related alkaloids. J. Chromatogr. A 2002, 978, 1–35. [Google Scholar] [CrossRef]
- Wei, Y.-M.; Zhang, G.-Q.; Li, Z.-X. Study on nutritive and physico-chemical properties of buckwheat flour. Food/Nahr. 1995, 39, 48–54. [Google Scholar] [CrossRef]
- Long, Z.; Wang, C.; Guo, Z.; Zhang, X.; Nordahl, L.; Zeng, J.; Zeng, J.; Liang, X. A non-aqueous solid phase extraction method for alkaloid enrichment and its application in the determination of hyoscyamine and scopolamine. Analyst 2012, 137, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- European Union. Commission Regulation (EU) 2016/239 of 19 February 2016 amending Regulation (EC) No 1881/2006 as regards maximum levels of tropane alkaloids in certain cereal-based foods for infants and young children. Off. J. Eur. Union 2016, L45, 3. [Google Scholar]
- Naudé, T.W. Datura spp. and other related plants. In Veterinary Toxicology; Gupta, R.C., 1st ed.; Elsevier: New York, NY, USA, 2007; pp. 892–906. [Google Scholar] [CrossRef]
- Gerber, R.; Naudé, T.; De Kock, S. Confirmed Datura poisoning in a horse most probably due to D. ferox in contaminated tef hay: Clinical communication. J. South Afr. Vet. Assoc. 2006, 77, 86–89. [Google Scholar] [CrossRef][Green Version]
- Arcella, D.; Altieri, A.; Horváth, Z. Human acute exposure assessment to tropane alkaloids. EFSA J. 2018, 16. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | SBET a (m2/g) | Pore Volume b (cm3/g) | Pore Size c (Å) | L0 d (mmol S/g) | SO3− Groups e (mmol/g) | Recovery (% ± SD) f | |
|---|---|---|---|---|---|---|---|
| Atropine | Scopolamine | ||||||
| SBA-15 | 780 | 0.8 | 56 | - | - | - | - |
| L-SBA-15-SO3− | 587 | 0.7 | 55 | 0.5 | 0.3 | 38 ± 30 | 51 ± 13 |
| M-SBA-15-SO3− | 460 | 0.6 | 49 | 0.9 | 0.5 | 90 ± 8 | 91 ± 9 |
| H-SBA-15-SO3−- | 350 | 0.4 | 36 | 1.7 | 0.6 | 95 ± 0 | 104 ± 12 |
| MFE-PAK® SCX | 398 | 0.6 | 61 | 0.8 | 0.3 | 91 ± 2 | 91 ± 2 |
| Solvent | M-SBA-15-SO3− SCX-SPE Sorbent | MFE-PAK® SCX-SPE Sorbent | ||||||
|---|---|---|---|---|---|---|---|---|
| Atropine | Scopolamine | Atropine | Scopolamine | |||||
| Ss | Bu-F1O | So-FO | Ss | Bu-F1O | So-FO | Ss | Ss | |
| ACN/H2O, 0.5% HCOOH (3:5, v/v) | 74 ± 2 | 46± 22 | 10 ± 1 | 81 ± 4 | 5 ± 1 | 8 ± 1 | - | - |
| ACN/H2O, 1% HCOOH (3:5, v/v) | 78 ± 1 | 7 ± 3 | 7 ± 2 | 80 ± 5 | 4 ± 2 | 7 ± 3 | - | - |
| MeOH/H2O, 0.5% HAc (2:1, v/v) | 86 ± 3 | 16 ± 3 | 15 ± 2 | 105 ± 10 | 12 ± 1 | 16 ± 0 | 65 ± 3 | 65 ± 2 |
| H2O, 1.1% HCl (pH 1.0) | 90 ± 7 | 43 ± 10 | 66 ± 3 | 103 ± 1 | 25 ± 7 | 48± 3 | 36 ± 4 | 15 ± 2 |
| Sample | M-SBA-15-SO3− SCX-SPE Sorbent | MFE-PAK® SCX SPE Sorbent | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 50 mg | 100 mg | 150 mg | 200 mg | 150 mg | 200 mg | ||||||||
| At | Sc | At | Sc | At | Sc | At | Sc | At | Sc | At | Sc | ||
| Bu-F1O | 43 ± 10 | 25 ± 7 | 52 ± 21 | 33± 17 | 87 ± 9 | 70 ± 5 | - | - | 48 ± 6 | 10 ± 3 | 60 ± 16 | 14 ± 5 | |
| So-FO | 66 ± 3 | 48 ± 3 | 82 ± 16 | 85 ± 8 | 98 ± 4 | 95 ± 4 | - | - | 59 ± 8 | 21 ± 1 | 71 ± 11 | 34 ± 2 | |
| Ch-FC | - | - | 60 ± 1 | 27 ± 3 | 93 ± 13 | 41 ± 2 | 92 ± 9 | 72 ± 4 | 39 ± 2 | 17 ± 4 | 48 ± 2 | 20 ± 1 | |
| Atropine | Scopolamine | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Linearity (mg/L) | Matrix Matched Calibration (R2) | Cm * (%) | MDL * (µg/kg) | MQL * (µg/kg) | ME * (%) | Matrix Matched Calibration (R2) | Cm (%) | MDL (µg/kg) | MQL (µg/kg) | ME (%) |
| Co-RFC | 0.002–1 | 8.6 × 108x −8.5 × 106 (0.998) | 98 | 0.1 | 0.4 | 17 a | 3.3 × 108x + 4.3 × 104 (0.999) | 94 | 0.2 | 0.7 | 1 d |
| So-FO | 0.01–0.2 | 7.1 × 108x −3.9 × 106 (0.996) | 98 | 0.5 | 1.5 | −3 a | 3.0 × 108x +6.0 × 105 (0.999) | 99 | 0.7 | 2.4 | −7 d |
| Te-FO | 0.01–0.2 | 7.0 × 108x −1.24 × 107 (0.994) | 92 | 0.04 | 0.1 | −5 a | 2.9 × 108x +3.2 × 104 (0.994) | 98 | 1 | 3.3 | −11 d |
| Co-FO | 0.002–1 | 8.3 × 108x −5.8 × 106 (0.997) | 97 | 0.1 | 0.4 | 14 a | 3.1 × 108 −4.92 × 106 (0.996) | 96 | 0.2 | 0.7 | −5 d |
| Pe-FC | 0.01–0.4 | 5.6 × 108x −1.1 × 107 (0.993) | 95 | 0.3 | 1.1 | −35 b | 2.1 × 108x −2.7 × 106 (0.993) | 98 | 1.3 | 4.4 | −51 e |
| Am-GO | 0.01–1 | 3.62 × 108x +1.18 × 106 (1.000) | 96 | 0.2 | 0.5 | −51 c | 1.0 × 108x +1.4 × 106 (0.996) | 96 | 0.5 | 1.7 | −69 f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Gómez, L.; Gañán, J.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Sulfonic Acid-Functionalized SBA-15 as Strong Cation-Exchange Sorbent for Solid-Phase Extraction of Atropine and Scopolamine in Gluten-Free Grains and Flours. Foods 2020, 9, 1854. https://doi.org/10.3390/foods9121854
González-Gómez L, Gañán J, Morante-Zarcero S, Pérez-Quintanilla D, Sierra I. Sulfonic Acid-Functionalized SBA-15 as Strong Cation-Exchange Sorbent for Solid-Phase Extraction of Atropine and Scopolamine in Gluten-Free Grains and Flours. Foods. 2020; 9(12):1854. https://doi.org/10.3390/foods9121854
Chicago/Turabian StyleGonzález-Gómez, Lorena, Judith Gañán, Sonia Morante-Zarcero, Damián Pérez-Quintanilla, and Isabel Sierra. 2020. "Sulfonic Acid-Functionalized SBA-15 as Strong Cation-Exchange Sorbent for Solid-Phase Extraction of Atropine and Scopolamine in Gluten-Free Grains and Flours" Foods 9, no. 12: 1854. https://doi.org/10.3390/foods9121854
APA StyleGonzález-Gómez, L., Gañán, J., Morante-Zarcero, S., Pérez-Quintanilla, D., & Sierra, I. (2020). Sulfonic Acid-Functionalized SBA-15 as Strong Cation-Exchange Sorbent for Solid-Phase Extraction of Atropine and Scopolamine in Gluten-Free Grains and Flours. Foods, 9(12), 1854. https://doi.org/10.3390/foods9121854