Supporting the Sensory Panel to Grade Virgin Olive Oils: An In-House-Validated Screening Tool by Volatile Fingerprinting and Chemometrics

 ,
,  , , , and
, , , and 
Abstract
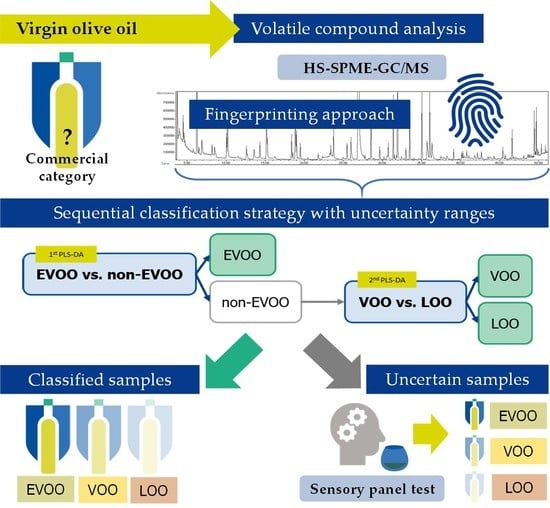
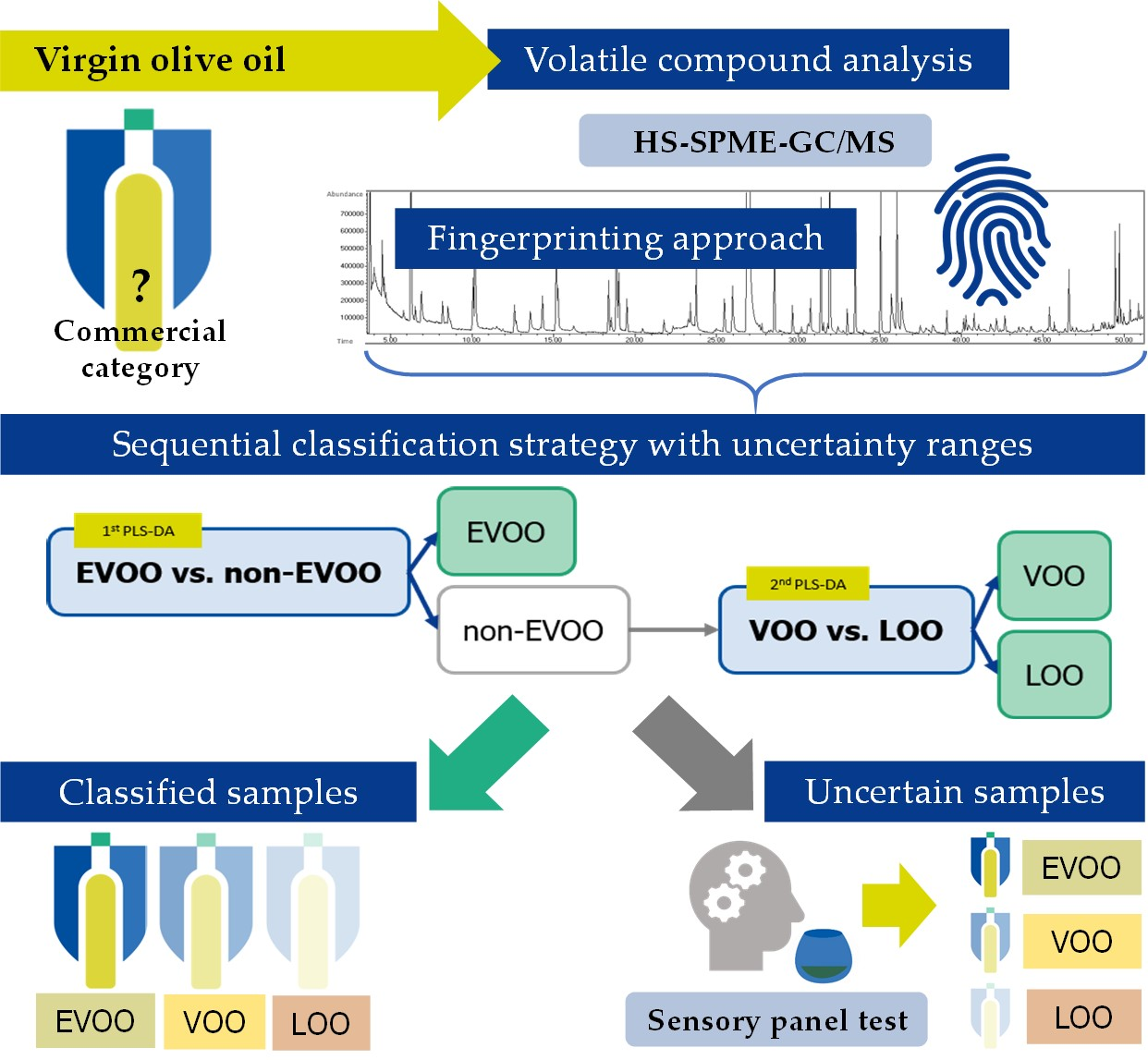
1. Introduction
2. Materials and Methods
2.1. Olive Oil Samples
2.2. Chemicals and Reagents
2.3. Head Space-Solid Phase Microextraction (HS-SPME)
2.4. Gas Chromatography–Mass Spectrometry (GC–MS)
2.4.1. Instruments and Procedures
2.4.2. Analytical System Suitability
2.5. In-House Validation of the Analytical Outcome
2.6. Chemometrics
3. Results and Discussion
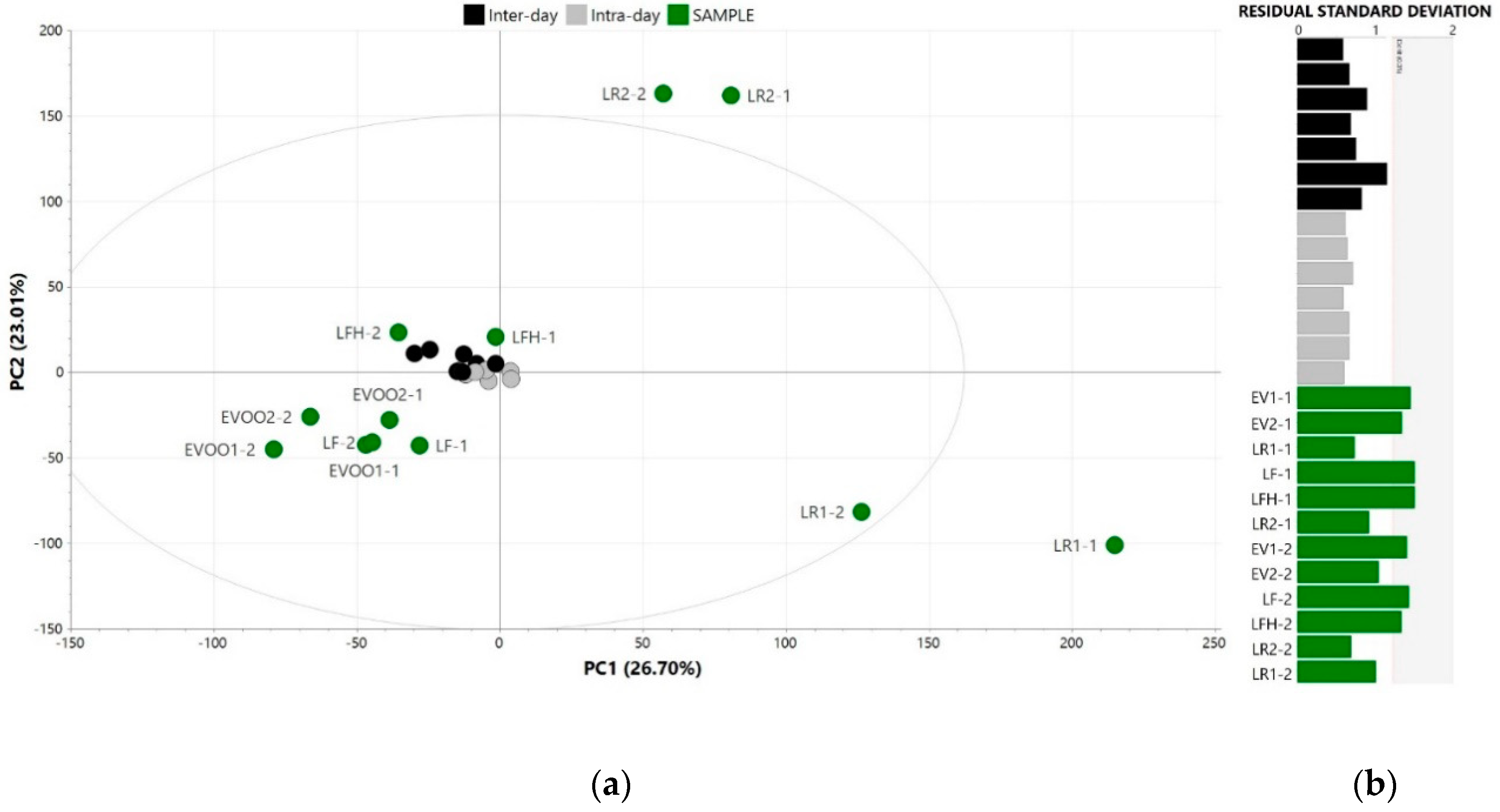
3.1. In-House Validation of the Analytical Outcome
3.2. Raw Data Alignment, Pre-Processing and Exploration
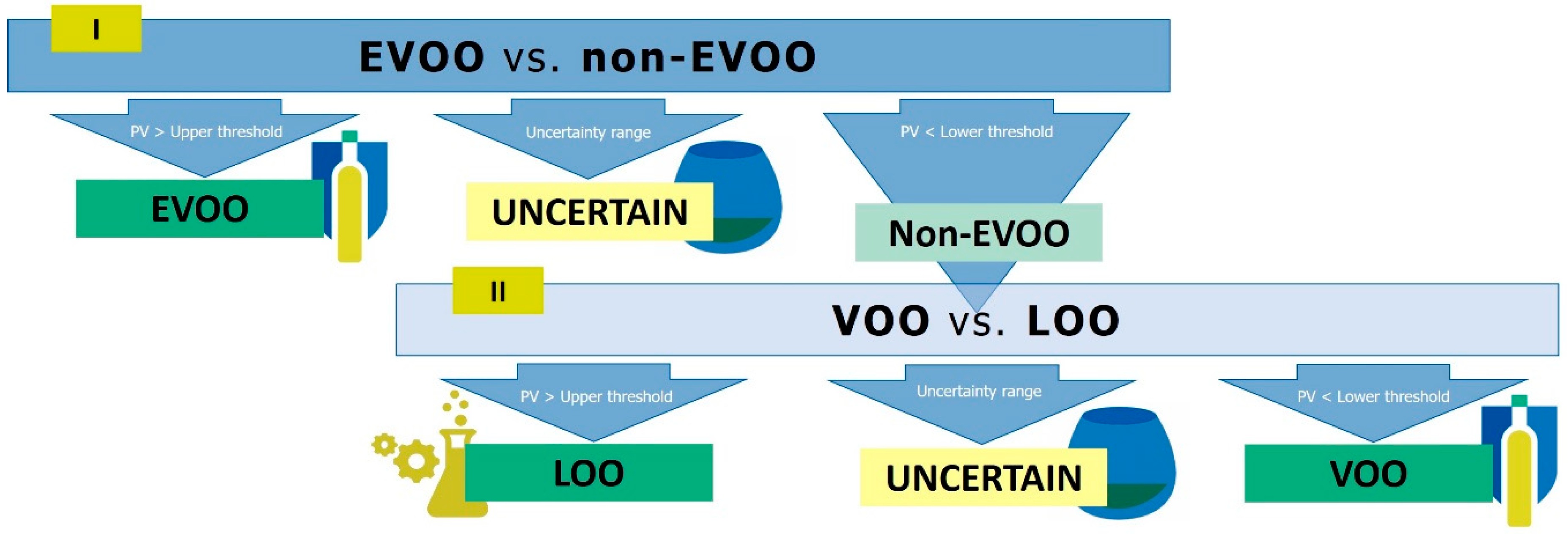
3.3. Development of the Hierarchical Classification Strategy
3.4. External Validation of the Instrumental Screening Tool
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Olive Council (IOC). Trade Standard Applying to Olive Oils and Olive Pomace Oils; International Olive Council (IOC): Madrid, Spain, 2019. [Google Scholar]
- Official Journal of the European Communities. Commission Regulation (EEC) No 2568/91; Official Journal of the European Union: Brussels, Belgium, 1991. [Google Scholar]
- International Olive Council (IOC). Sensory Analysis of Olive Oil-Method for the Organoleptic Assessment of Virgin Olive Oil; International Olive Council (IOC): Madrid, Spain, 2018. [Google Scholar]
- Circi, S.; Capitani, D.; Randazzo, A.; Ingallina, C.; Mannina, L.; Sobolev, A.P. Panel Test and Chemical Analyses of Commercial Olive Oils: A Comparative Study. Chem. Biol. Technol. Agric. 2017, 4, 18. [Google Scholar] [CrossRef]
- Barbieri, S.; Bubola, K.B.; Bendini, A.; Bučar-Miklavčič, M.; Lacoste, F.; Tibet, U.; Winkelmann, O.; García-González, D.L.; Gallina Toschi, T. Alignment and Proficiency of Virgin Olive Oil Sensory Panels: The Oleum Approach. Foods 2020, 9, 355. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Ruiz, R.; Morales, M.T.; Aparicio, R. Does Authenticity of Virgin Olive Oil Sensory Quality Require Input from Chemistry? Eur. J. Lipid Sci. Technol. 2019, 121, 1–10. [Google Scholar] [CrossRef]
- Conte, L.; Bendini, A.; Valli, E.; Lucci, P.; Moret, S.; Maquet, A.; Lacoste, F.; Brereton, P.; García-González, D.L.; Moreda, W.; et al. Olive Oil Quality and Authenticity: A Review of Current EU Legislation, Standards, Relevant Methods of Analyses, Their Drawbacks and Recommendations for the Future. Trends Food Sci. Technol. 2019, in press. [Google Scholar] [CrossRef]
- Angerosa, F.; Servili, M.; Selvaggini, R.; Taticchi, A.; Esposto, S.; Montedoro, G. Volatile Compounds in Virgin Olive Oil: Occurrence and Their Relationship with the Quality. J. Chromatogr. A 2004, 1054, 17–31. [Google Scholar] [CrossRef]
- Kalua, C.M.; Allen, M.S.; Bedgood, D.R., Jr.; Bishop, A.G.; Prenzler, P.D.; Robards, K. Olive Oil Volatile Compounds, Flavour Development and Quality: A Critical Review. Food Chem. 2007, 100, 273–286. [Google Scholar] [CrossRef]
- Aparicio, R.; Morales, M.T.; García-González, D.L. Towards New Analyses of Aroma and Volatiles to Understand Sensory Perception of Olive Oil. Eur. J. Lipid Sci. Technol. 2012, 114, 1114–1125. [Google Scholar] [CrossRef]
- Sales, C.; Cervera, M.I.; Gil, R.; Portolés, T.; Pitarch, E.; Beltran, J. Quality Classification of Spanish Olive Oils by Untargeted Gas Chromatography Coupled to Hybrid Quadrupole-Time of Flight Mass Spectrometry with Atmospheric Pressure Chemical Ionization and Metabolomics-Based Statistical Approach. Food Chem. 2017, 216, 365–373. [Google Scholar] [CrossRef]
- Sales, C.; Portolés, T.; Johnsen, L.G.; Danielsen, M.; Beltran, J. Olive Oil Quality Classification and Measurement of Its Organoleptic Attributes by Untargeted GC–MS and Multivariate Statistical-Based Approach. Food Chem. 2019, 271, 488–496. [Google Scholar] [CrossRef]
- Cecchi, L.; Migliorini, M.; Giambanelli, E.; Rossetti, A.; Cane, A.; Melani, F.; Mulinacci, N. Headspace Solid-Phase Microextraction–Gas Chromatography–Mass Spectrometry Quantification of the Volatile Profile of More than 1200 Virgin Olive Oils for Supporting the Panel Test in Their Classification: Comparison of Different Chemometric Approaches. J. Agric. Food Chem. 2019, 67, 9112–9120. [Google Scholar] [CrossRef]
- Quintanilla-Casas, B.; Bustamante, J.; Guardiola, F.; García-González, D.L.; Barbieri, S.; Bendini, A.; Gallina Toschi, T.; Vichi, S.; Tres, A. Virgin Olive Oil Volatile Fingerprint and Chemometrics: Towards an Instrumental Screening Tool to Grade the Sensory Quality. LWT-Food Sci. Technol. 2020, 121, 108936. [Google Scholar] [CrossRef]
- Contreras, M.M.; Arroyo-Manzanares, N.; Arce, C.; Arce, L. HS-GC-IMS and Chemometric Data Treatment for Food Authenticity Assessment: Olive Oil Mapping and Classification through Two Different Devices as an Example. Food Control 2019, 98, 82–93. [Google Scholar] [CrossRef]
- Contreras, M.; Jurado-Campos, N.; Arce, L. A Robustness Study of Calibration Models for Olive Oil Classification: Targeted and Non-Targeted Fingerprint Approaches Based on GC-IMS. Food Chem. 2019, 288, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, N.; Schwolow, S.; Rohn, S.; Pérez-Cacho, P.R.; Galán-Soldevilla, H.; Arce, L.; Weller, P. Quality Assessment of Olive Oils Based on Temperature-Ramped HS-GC-IMS and Sensory Evaluation: Comparison of Different Processing Approaches by LDA, KNN, and SVM. Food Chem. 2019, 278, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Valli, E.; Panni, F.; Casadei, E.; Barbieri, S.; Cevoli, C.; Bendini, A.; García-González, D.L.; Gallina Toschi, T. An HS-GC-IMS Method for the Quality Classification of Virgin Olive Oils as Screening Support for the Panel Test. Foods 2020, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Vega-Márquez, B.; Nepomuceno-Chamorro, I.; Jurado-Campos, N.; Rubio-Escudero, C. Deep Learning Techniques to Improve the Performance of Olive Oil Classification. Front. Chem. 2020, 7. [Google Scholar] [CrossRef]
- Taiti, C.; Marone, E. EVOO or Not EVOO? A New Precise and Simple Analytical Tool to Discriminate Extra Virgin Olive Oils. Adv. Hortic. Sci. 2017, 31, 329–337. [Google Scholar] [CrossRef]
- López-Feria, S.; Cárdenas, S.; García-Mesa, J.A.; Valcárcel, M. Usefulness of the Direct Coupling Headspace–Mass Spectrometry for Sensory Quality Characterization of Virgin Olive Oil Samples. Anal. Chim. Acta 2007, 583, 411–417. [Google Scholar] [CrossRef]
- Borràs, E.; Ferré, J.; Boqué, R.; Aceña, L.; Calvo, A.; Busto, O. Prediction of Olive Oil Sensory Descriptors Using Instrumental Data Fusion and Partial Least Squares (PLS) Regression. Talanta 2016, 155, 116–123. [Google Scholar] [CrossRef]
- Borràs, E.; Ferré, J.; Boqué, R.; Mestres, M.; Aceña, L.; Calvo, A.; Busto, O. Olive Oil Sensory Defects Classification with Data Fusion of Instrumental Techniques and Multivariate Analysis (PLS-DA). Food Chem. 2016, 203, 314–322. [Google Scholar] [CrossRef]
- Piñero, M.Y.; Amo-González, M.; Ballesteros, R.D.; Pérez, L.R.; de la Mora, G.F.; Arce, L. Chemical Fingerprinting of Olive Oils by Electrospray Ionization-Differential Mobility Analysis-Mass Spectrometry: A New Alternative to Food Authenticity Testing. J. Am. Soc. Mass Spectrom. 2020, 31, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Melucci, D.; Bendini, A.; Tesini, F.; Barbieri, S.; Zappi, A.; Vichi, S.; Conte, L.; Gallina Toschi, T. Rapid direct analysis to discriminate geographic origin of extra virgin olive oils by flash gas chromatography electronic nose and chemometrics. Food Chem. 2016, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, S.; Cevoli, C.; Bendini, A.; Quintanilla-Casas, B.; García-González, D.L.; Gallina Toschi, T. Flash Gas Chromatography in Tandem with Chemometrics: A Rapid Screening Tool for Quality Grades of Virgin Olive Oils. Foods 2020, 9, 862. [Google Scholar] [CrossRef] [PubMed]
- Veloso, A.C.A.; Silva, L.M.; Rodrigues, N.; Rebello, L.P.G.; Dias, L.G.; Pereira, J.A.; Peres, A.M. Perception of Olive Oils Sensory Defects Using a Potentiometric Taste Device. Talanta 2018, 176, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; Gabriel, F.; Carpio, A.; Arce, L. Use of Whole Electrophoretic Profile and Chemometric Tools for the Differentiation of Three Olive Oil Qualities. Talanta 2019, 197, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Vichi, S.; Isabel, A.; Pizzale, L.; Conte, L.S.; Buxaderas, S.; Lopez-Tamames, E. Analysis of Virgin Olive Oil Volatile Compounds by Headspace Solid-Phase Microextraction Coupled to Gas Chromatography with Mass Spectrometric and Flame Ionization Detection. J. Chromatogr. A 2003, 983, 19–33. [Google Scholar] [CrossRef]
- Vichi, S.; Guadayol, J.M.; Caixach, J.; López-Tamames, E.; Buxaderas, S. Comparative Study of Different Extraction Techniques for the Analysis of Virgin Olive Oil Aroma. Food Chem. 2007, 105, 1171–1178. [Google Scholar] [CrossRef]
- Romero, I.; García-González, D.L.; Aparicio-Ruiz, R.; Morales, M.T. Validation of SPME-GCMS Method for the Analysis of Virgin Olive Oil Volatiles Responsible for Sensory Defects. Talanta 2015, 134, 394–401. [Google Scholar] [CrossRef]
- Berrueta, L.A.; Alonso-Salces, R.M. Supervised Pattern Recognition in Food Analysis. J. Chromatogr. A 2007, 1158, 196–214. [Google Scholar] [CrossRef]
- Thompson, M.; Ellison, S.L.R.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC technical report). Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- US FDA Bioanalytical Method Validation, Guidance for Industry. 2018. Available online: http://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-For_industry.pdf (accessed on 3 June 2019).
- Riedl, J.; Esslinger, S.; Fauhl-Hassek, C. Review of Validation and Reporting of Non-Targeted Fingerprinting Approaches for Food Authentication. Anal. Chim. Acta 2015, 885, 17–32. [Google Scholar] [CrossRef]
- Sangster, T.; Major, H.; Plumb, R.; Wilson, A.J.; Wilson, I.D. A Pragmatic and Readily Implemented Quality Control Strategy for HPLC-MS and GC-MS-Based Metabonomic Analysis. Analyst 2006, 10, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Vallejo, M.; García, A.; Barbas, C. Method Validation Strategies Involved in Non-Targeted Metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [Google Scholar] [CrossRef]
- Fawcett, T. An Introduction to ROC Analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Tomasi, G.; Savorani, F.; Engelsen, S.B. icoshift: An Effective Tool for the Alignment of Chromatographic Data. J. Chromatogr. A 2011, 1218, 7832–7840. [Google Scholar] [CrossRef]
- Allwood, J.W.; Erban, A.; de Koning, S.; Dunn, W.B.; Luedemann, A.; Lommen, A.; Kay, L.; Löscher, R.; Kopka, J.; Goodacre, R. Inter-Laboratory Reproducibility of Fast Gas Chromatography-Electron Impact-Time of Flight Mass Spectrometry (GC-EI-TOF/MS) Based Plant Metabolomics. Metabolomics 2009, 5, 479–496. [Google Scholar] [CrossRef]
- Dudzik, D.; Barbas-Bernardos, C.; García, A.; Barbas, C. Quality Assurance Procedures for Mass Spectrometry Untargeted Metabolomics. A Review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. [Google Scholar] [CrossRef]
- Horwitz, W. Evaluation of Analytical Methods Used for Regulation of Foods and Drugs. Anal. Chem. 1982, 54, 67–76. [Google Scholar] [CrossRef]
- López, M.I.; Colomer, N.; Ruisánchez, I.; Callao, M.P. Validation of Multivariate Screening Methodology. Case Study: Detection of Food Fraud. Anal. Chim. Acta 2014, 827, 28–33. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| 1st PLS-DA (EVOO vs. Non-EVOO) 7 LVs; Q2 >0.4; RMSEcv < 0.37; ANOVA p-Value < 0.05 | 2nd PLS-DA (VOO vs. LOO) 5–7 LVs; Q2 >0.4; RMSEcv < 0.33; ANOVA p-Value < 0.05 | |||||
|---|---|---|---|---|---|---|
| Set | Lower Threshold 1 | Upper Threshold 2 | Uncertain Samples 3 (%) | Lower Threshold 1 | Upper Threshold 2 | Uncertain Samples 3 (%) |
| Full model | 0.354 | 0.752 | 15 (49/301) | 0.434 | - | |
| Subsets | ||||||
| Set 1 | 0.397 | 0.503 | 4.2 (10/241) | 0.473 | - | |
| Set 2 | 0.427 | 0.836 | 17.4 (42/241) | 0.418 | 0.514 | 2.8 (4/143) |
| Set 3 | 0.383 | 0.771 | 15.4 (37/241) | 0.468 | - | |
| Set 4 | 0.310 | 0.497 | 6.6 (16/241) | 0.442 | - | |
| Set 5 | 0.370 | 0.748 | 14.5 (35/241) | 0.491 | - | |
| Set 6 | 0.426 | 0.812 | 16.6 (40/241) | 0.483 | - | |
| Set 7 | 0.411 | 0.760 | 22.2 (27/241) | 0.425 | 0.502 | 2.1 (3/143) |
| Mean | 0.389 | 0.704 | 13.8 | 0.457 | 0.482 | 2.5 |
| SD | 0.041 | 0.142 | 6.31 | 0.029 | 0.024 | 0.5 |
| Instrumental Screening Tool | Screening Tool + Reference Method 1 | |||||||
|---|---|---|---|---|---|---|---|---|
| Uncertain Samples 2 (% of Total Samples) | Samples Assigned to a Category 3 (% of Total Samples) | Correctly Classified 4 (% of Assigned Samples) | Reliable Assignment (%) | |||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| EVOO | 31.0 | 7.9 | 69.0 | 7.9 | 82.3 | 15.7 | 87.5 | 11.3 |
| VOO | 26.0 | 14.4 | 74.0 | 14.4 | 74.6 | 17.2 | 80.5 | 15.5 |
| LOO | 6.1 | 6.4 | 93.9 | 6.4 | 80.5 | 9.7 | 83.7 | 9.9 |
| Total | 23.3 | 7.4 | 76.7 | 7.4 | 78.9 5 | 3.83 5 | 84.0 | 4.4 |
| MPD | Validation Set 1 | Uncertain Samples (%) | Samples Assigned to a Category | Non-EVOO Samples Assigned to a Category | ||
|---|---|---|---|---|---|---|
| Total (%) | Correct (%) as Non-EVOO 2 | Correct (%) as VOO 3 | Correct (%) as LOO 3 | |||
| RANCID | 1 (n = 14) | 0 (0/14) | 100 (14/14) | 71.4 (10/14) | 40.0 (4/10) | 50.0 (2/4) |
| 2 (n = 13) | 46.2 (6/13) | 53.8 (7/13) | 85.7 (6/7) | 100 (2/2) | 80.0 (4/5) | |
| 3 (n = 12) | 16.7 (2/12) | 83.3 (10/12) | 90.0 (9/10) | 80.0 (4/5) | 80.0 (4/5) | |
| 4 (n = 14) | 14.3 (2/14) | 85.7 (12/14) | 75.0 (9/12) | 57.1 (4/7) | 60.0 (3/5) | |
| 5 (n = 14) | 35.7 (5/14) | 64.3 (9/14) | 88.9 (8/9) | 60.0 (3/5) | 75.0 (3/4) | |
| 6 (n = 12) | 33.3 (4/12) | 66.7 (8/12) | 100 (8/8) | 75.0 (3/4) | 100 (4/4) | |
| 7 (n = 10) | 50.0 (5/10) | 50.0 (5/10) | 100 (5/5) | 100 (2/2) | 100 (3/3) | |
| Weighted mean | 26.9 | 73.0 | 84.6 | 62.9 | 76.7 | |
| FUSTY-MUDDY | 1 (n = 15) | 0 (0/15) | 100 (15/15) | 93.3 (14/15) | 77.8 (7/9) | 83.3 (5/6) |
| 2 (n = 19) | 21.1 (4/19) | 78.9 (15/19) | 100 (15/15) | 100 (12/12) | 66.7 (2/3) | |
| 3 (n = 17) | 35.3 (6/17) | 64.7 (11/17) | 100 (11/11) | 83.3 (5/6) | 100 (5/5) | |
| 4 (n = 20) | 20.0 (4/20) | 80.0 (16/20) | 87.5 (14/16) | 62.5 (5/8) | 87.5 (7/8) | |
| 5 (n = 15) | 20.0 (3/15) | 80.0 (12/15) | 91.7 (11/12) | 66.7 (4/6) | 83.3 (5/6) | |
| 6 (n = 19) | 10.5 (2/19) | 89.5 (17/19) | 94.1 (16/17) | 92.3 (12/13) | 75.0 (3/4) | |
| 7 (n = 20) | 5.0 (1/20) | 95.0 (19/20) | 90.0 (18/20) | 85.7 (12/14) | 100 (5/5) | |
| Weighted Mean | 16.0 | 84.0 | 93.4 | 83.8 | 86.5 | |
| MUSTY-HUMID-EARTHY | 1 (n = 7) | 0 (0/7) | 100 (7/7) | 85.7 (6/7) | 33.3 (1/3) | 75.00 (3/4) |
| 2 (n = 4) | 0 (0/4) | 100 (4/4) | 100 (4/4) | 100 (1/1) | 66.7 (2/3) | |
| 3 (n = 7) | 28.6 (2/7) | 71.4 (5/7) | 100 (5/5) | 0 (0/2) | 100 (3/3) | |
| 4 (n = 2) | 0 (0/2) | 10 (2/2) | 100 (2/2) | 100 (1/1) | 100 (1/1) | |
| 5 (n = 7) | 14.3 (1/7) | 85.7 (6/7) | 100 (6/6) | 50 (1/2) | 75.0 (3/4) | |
| 6 (n = 5) | 20.0 (1/5) | 80.0 (4/5) | 100 (4/4) | (0/0) | 75.0 (3/4) | |
| 7 (n = 6) | 33.3 (2/6) | 66.7 (4/6) | 100 (4/4) | (0/0) | 100 (4/4) | |
| Weighted Mean | 15.8 | 84.2 | 97 | 44.4 | 82.6 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintanilla-Casas, B.; Marin, M.; Guardiola, F.; García-González, D.L.; Barbieri, S.; Bendini, A.; Gallina Toschi, T.; Vichi, S.; Tres, A. Supporting the Sensory Panel to Grade Virgin Olive Oils: An In-House-Validated Screening Tool by Volatile Fingerprinting and Chemometrics. Foods 2020, 9, 1509. https://doi.org/10.3390/foods9101509
Quintanilla-Casas B, Marin M, Guardiola F, García-González DL, Barbieri S, Bendini A, Gallina Toschi T, Vichi S, Tres A. Supporting the Sensory Panel to Grade Virgin Olive Oils: An In-House-Validated Screening Tool by Volatile Fingerprinting and Chemometrics. Foods. 2020; 9(10):1509. https://doi.org/10.3390/foods9101509
Chicago/Turabian StyleQuintanilla-Casas, Beatriz, Marco Marin, Francesc Guardiola, Diego Luis García-González, Sara Barbieri, Alessandra Bendini, Tullia Gallina Toschi, Stefania Vichi, and Alba Tres. 2020. "Supporting the Sensory Panel to Grade Virgin Olive Oils: An In-House-Validated Screening Tool by Volatile Fingerprinting and Chemometrics" Foods 9, no. 10: 1509. https://doi.org/10.3390/foods9101509
APA StyleQuintanilla-Casas, B., Marin, M., Guardiola, F., García-González, D. L., Barbieri, S., Bendini, A., Gallina Toschi, T., Vichi, S., & Tres, A. (2020). Supporting the Sensory Panel to Grade Virgin Olive Oils: An In-House-Validated Screening Tool by Volatile Fingerprinting and Chemometrics. Foods, 9(10), 1509. https://doi.org/10.3390/foods9101509