
Analysis of Human Gut Microbiota Enzymes for Biotechnological and Food Industrial Applications

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. In Silico Whole Genome Sequences (WGS) Enzyme Searching Approach
2.2. In Vitro Culturing and Enzyme Activities
3. Results
3.1. In Silico WGS Enzyme Activity in Microbiota
3.1.1. Amylases
3.1.2. Xylanases
3.1.3. Cellulases
3.1.4. Nucleases
3.1.5. Esterase/Lipase
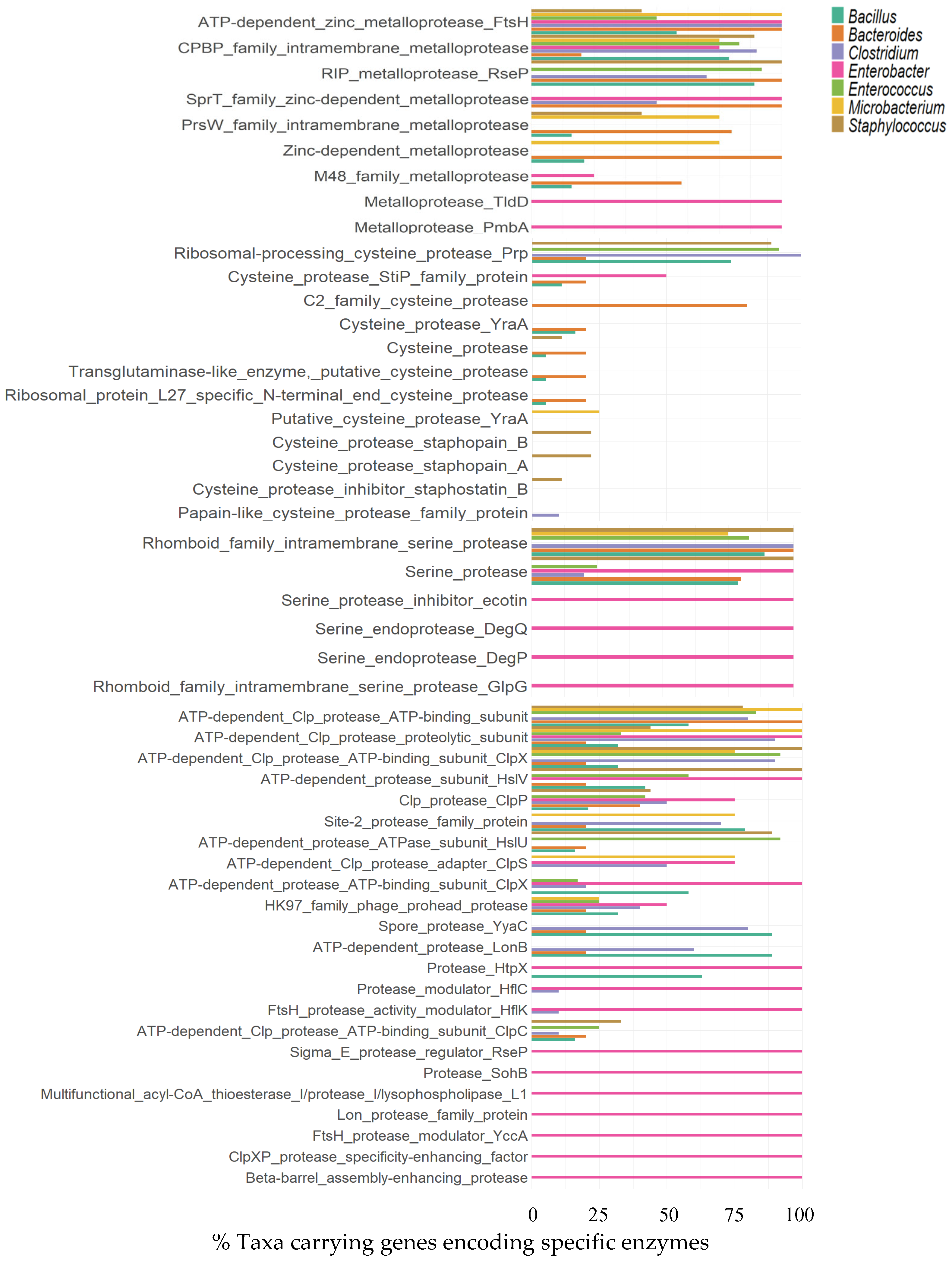
3.1.6. Proteases
Metalloproteases
Cysteine Proteases
Serine Proteases
Other Proteases
3.1.7. Laccases
3.2. In Vitro Enzyme Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ursell, L.K.; Haiser, H.J.; Van Treuren, W.; Garg, N.; Reddivari, L.; Vanamala, J.; Dorrestein, P.C.; Turnbaugh, P.J.; Knight, R. The Intestinal Metabolome—An Intersection Between Microbiota and Host. Gastroenterology 2014, 146, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Han, X.; Kim, K.H.; Jeon, C.O. Discovery and mining of enzymes from the human gut microbiome. Trends Biotechnol. 2022, 40, 240–254. [Google Scholar] [CrossRef]
- Choix, F.J.; Palacios, O.A.; Nevarez-Moorillón, G.V. Traditional and new proposals for environmental microbial indicators—A review. Environ Monit Assess 2023 195, 1521. [CrossRef]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.; Cryan, J.F.; Hyland, N.P. Gut Reactions: Breaking Down Xenobiotic-Microbiome Interactions. Pharmacol Rev 2019, 71, 198–224. [CrossRef] [PubMed]
- Wardman, J.F.; Bains, R.K.; Rahfeld, P.; Withers, S.G. Carbohydrate-active enzymes (CAZymes) in the gut microbiome. Nat. Rev. Microbiol. 2022, 20, 542–556. [Google Scholar] [CrossRef]
- Mafra, D.; Barros, A.F.; Fouque, D. Dietary Protein Metabolism by Gut Microbiota and its Consequences for Chronic Kidney Disease Patients. Future Microbiol. 2013, 8, 1317–1323. [Google Scholar] [CrossRef]
- Brown, E.M.; Clardy, J.; Xavier, R.J. Gut microbiome lipid metabolism and its impact on host physiology. Cell Host Microbe 2023, 31, 173–186. [Google Scholar] [CrossRef]
- Kadyan, S.; Park, G.; Wang, B.; Singh, P.; Arjmandi, B.; Nagpal, R. Resistant starches from dietary pulses modulate the gut metabolome in association with microbiome in a humanized murine model of ageing. Sci. Rep. 2023, 13, 10566. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, X.; Liu, H.; Brown, M.A.; Qiao, S. Dietary Protein and Gut Microbiota Composition and Function. Curr. Protein Pept. Sci. 2019, 20, 145–154. [Google Scholar] [CrossRef]
- Chapman, J.; Ismail, A.E.; Dinu, C.Z. Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks. Catalysts 2018, 8, 238. [Google Scholar] [CrossRef]
- Kim, H.S.; Noh, M.H.; White, E.M.; Kandefer, M.V.; Wright, A.F.; Datta, D.; Lim, H.G.; Smiggs, E.; Locklin, J.J.; Rahman, M.A.; et al. Biocomposite thermoplastic polyurethanes containing evolved bacterial spores as living fillers to facilitate polymer disintegration. Nat. Commun. 2024, 15, 3338. [Google Scholar] [CrossRef]
- Nam, N.N.; Do, H.D.K.; Loan Trinh, K.T.; Lee, N.Y. Metagenomics: An Effective Approach for Exploring Microbial Diversity and Functions. Foods 2023, 12, 2140. [Google Scholar] [CrossRef] [PubMed]
- Orsi, E.; Schada von Borzyskowski, L.; Noack, S.; Nikel, P.I.; Lindner, S.N. Automated in vivo enzyme engineering accelerates biocatalyst optimization. Nat. Commun. 2024, 15, 3447. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Moreno, A.; Cerk, K.; Rodrigo, L.; Suarez, A.; Aguilera, M.; Ruiz-Rodriguez, A. Bisphenol A exposure affects specific gut taxa and drives microbiota dynamics in childhood obesity. mSystems 2024, 9, :e0095723. [Google Scholar] [CrossRef]
- Luque, G.; Ortiz, P.; Torres-Sánchez, A.; Ruiz-Rodríguez, A.; López-Moreno, A.; Aguilera, M. Impact of Ex Vivo Bisphenol A Exposure on Gut Microbiota Dysbiosis and Its Association with Childhood Obesity. J. Xenobiot. 2025, 15, 14. [Google Scholar] [CrossRef]
- López-Moreno, A.; Ruiz-Moreno, Á.; Pardo-Cacho, J.; Cerk, K.; Torres-Sánchez, A.; Ortiz, P.; Úbeda, M.; Aguilera, M. Culturing and Molecular Approaches for Identifying Microbiota Taxa Impacting Children’s Obesogenic Phenotypes Related to Xenobiotic Dietary Exposure. Nutrients 2022, 14, 241. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 11 March 2025).
- Latorre, J.D.; Hernandez-Velasco, X.; Wolfenden, R.E.; Vicente, J.L.; Wolfenden, A.D.; Menconi, A.; Bielke, L.R.; Hargis, B.M.; Tellez, G. Evaluation and Selection of Bacillus Species Based on Enzyme Production, Antimicrobial Activity, and Biofilm Synthesis as Direct-Fed Microbial Candidates for Poultry. Front. Vet. Sci. 2016, 3, 95. [Google Scholar] [CrossRef]
- Menasria, T.; Aguilera, M.; Hocine, H.; Benammar, L.; Ayachi, A.; Si Bachir, A.; Dekak, A.; Monteoliva-Sánchez, M. Diversity and bioprospecting of extremely halophilic archaea isolated from Algerian arid and semi-arid wetland ecosystems for halophilic-active hydrolytic enzymes. Microbiol. Res. 2018, 207, 289–298. [Google Scholar] [CrossRef]
- Montalvo-Rodríguez, R.; Vreeland, R.H.; Oren, A.; Kessel, M.; Betancourt, C.; López-Garriga, J. Halogeometricum borinquense gen. nov., sp. nov., a novel halophilic archaeon from Puerto Rico. Int. J. Syst. Evol. Microbiol. 1998, 48 Pt 4, 1305–1312. [Google Scholar] [CrossRef]
- Kasana, R.C.; Salwan, R.; Dhar, H.; Dutt, S.; Gulati, A. A Rapid and Easy Method for the Detection of Microbial Cellulases on Agar Plates Using Gram’s Iodine. Curr. Microbiol. 2008, 57, 503–507. [Google Scholar] [CrossRef]
- Allais, J.-J.; Kammoun, S.; Blanc, P.; Girard, C.; Baratti, J.C. Isolation and Characterization of Bacterial Strains with Inulinase Activity. Appl. Environ. Microbiol. 1986, 52, 1086–1090. [Google Scholar] [CrossRef]
- Sierra, G. A simple method for the detection of lipolytic activity of micro-organisms and some observations on the influence of the contact between cells and fatty substrates. Antonie Leeuwenhoek 1957, 23, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, C.D.; Holtman, D.F.; Guse, D.G. Rapid method for determining the activity of microorgan-isms on nucleic acids. J. Bacteriol. 1957, 73, 590–591. [Google Scholar] [CrossRef]
- Frazier, W.C. A Method for the Detection of Changes in Gelatin Due to Bacteria: Two Plates. J. Infect. Dis. 1926, 39, 302–309. [Google Scholar] [CrossRef]
- Pailin, T.; Kang, D.H.; Schmidt, K.; Fung, D.Y.C. Detection of extracellular bound proteinase in EPS-producing lactic acid bacteria cultures on skim milk agar. Lett. Appl. Microbiol. 2001, 33, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Mandic, M.; Djokic, L.; Nikolaivits, E.; Prodanovic, R.; O’Connor, K.; Jeremic, S.; Topakas, E.; Nikodinovic-Runic, J. Identification and Characterization of New Laccase Biocatalysts from Pseudomonas Species Suitable for Degradation of Synthetic Textile Dyes. Catalysts 2019, 9, 629. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Malard, F.; Dore, J.; Gaugler, B.; Mohty, M. Introduction to host microbiome symbiosis in health and disease. Mucosal Immunol. 2021, 14, 547–554. [Google Scholar] [CrossRef]
- Ross, F.C.; Patangia, D.; Grimaud, G.; Lavelle, A.; Dempsey, E.M.; Ross, R.P.; Stanton, C. The interplay between diet and the gut microbiome: Implications for health and disease. Nat. Rev. Microbiol. 2024, 22, 671–686. [Google Scholar] [CrossRef]
- Zhang, F.; He, F.; Li, L.; Guo, L.; Zhang, B.; Yu, S.; Zhao, W. Bioavailability Based on the Gut Microbiota: A New Perspective. Microbiol. Mol. Biol. Rev. 2020, 84, e00072-19. [Google Scholar] [CrossRef] [PubMed]
- Graham, A.E.; Ledesma-Amaro, R. The microbial food revolution. Nat. Commun. 2023, 14, 2231. [Google Scholar] [CrossRef] [PubMed]
- Divakaran, D.; Chandran, A.; Pratap Chandran, R. Comparative study on production of a-Amylase from Bacillus licheniformis strains. Braz. J. Microbiol. 2011, 42, 1397–1404. [Google Scholar] [CrossRef]
- Ibrahim, S.; Amin, H.; Hassan, E.; Sulieman, A.M. Amylase Production on Solid State Fermentation by Bacillus spp. Food Public Health 2012, 2, 30–35. [Google Scholar] [CrossRef]
- Zhao, J.; Shao, T.; Chen, S.; Tao, X.; Li, J. Characterization and identification of cellulase-producing Enterococcus species isolated from Tibetan yak (Bos grunniens) rumen and their application in various forage silages. J. Appl. Microbiol. 2021, 131, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Seper, A.; Fengler, V.H.I.; Roier, S.; Wolinski, H.; Kohlwein, S.D.; Bishop, A.L.; Camilli, A.; Reidl, J.; Schild, S. Extracellular nucleases and extracellular DNA play important roles in Vibrio cholerae biofilm formation. Mol. Microbiol. 2011, 82, 1015–1037. [Google Scholar] [CrossRef]
- Liao, C.; Mao, F.; Qian, M.; Wang, X. Pathogen-Derived Nucleases: An Effective Weapon for Escaping Extracellular Traps. Front. Immunol. 2022, 13, 899890. [Google Scholar] [CrossRef]
- Li, Y.; Jia, J.; Man, S.; Ye, S.; Ma, L. Emerging programmable nuclease-based detection for food safety. Trends Biotechnol. 2024, 42, 151–155. [Google Scholar] [CrossRef]
- Price, M.N.; Deutschbauer, A.M.; Skerker, J.M.; Wetmore, K.M.; Ruths, T.; Mar, J.S.; Kuehl, J.V.; Shao, W.; Arkin, A.P. Indirect and suboptimal control of gene expression is widespread in bacteria. Mol. Syst. Biol. 2013, 9, 660. [Google Scholar] [CrossRef]
- Rollof, J.; Hedström, S.A.; Nilsson-Ehle, P. The Tween 80 reaction does not correlate to triglyceride lipase production of Staphylococcus aureus. APMIS 1988, 96, 732–734. [Google Scholar] [CrossRef]
- Sipiczki, G.; Micevic, S.S.; Kohari-Farkas, C.; Nagy, E.S.; Nguyen, Q.D.; Gere, A.; Bujna, E. Effects of Olive Oil and Tween 80 on Production of Lipase by Yarrowia Yeast Strains. Processes 2024, 12, 1206. [Google Scholar] [CrossRef]
- Najjar, A.; Hassan, E.A.; Zabermawi, N.; Saber, S.H.; Bajrai, L.H.; Almuhayawi, M.S.; Abujamel, T.S.; Almasaudi, S.B.; Azhar, L.E.; Moulay, M.; et al. Optimizing the catalytic activities of methanol and thermotolerant Kocuria flava lipases for biodiesel production from cooking oil wastes. Sci. Rep. 2021, 11, 13659. [Google Scholar] [CrossRef] [PubMed]
- Mekonnen, M.; Girma, S.; Atnafu, A. Hydrolysis of Gelatin from Animal Hoof Using Bacterial Gelatinase. Int. J. Microbiol. Biotechnol. 2022, 7, 135–142. [Google Scholar] [CrossRef]
- Song, P.; Zhang, X.; Wang, S.; Xu, W.; Wang, F.; Fu, R.; Wei, F. Microbial proteases and their applications. Front. Microbiol. 2023, 14, 1236368. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-T.; Chen, X.; Huo, D.; Arifuzzaman, M.; Qiao, S.; Jin, W.-B.; Shi, H.; Li, X.V.; Iliev, I.D.; Artis, D.; et al. Microbiota metabolism of intestinal amino acids impacts host nutrient homeostasis and physiology. Cell Host Microbe 2024, 32, 661–675.e10. [Google Scholar] [CrossRef] [PubMed]
- Balint, D.; Brito, I.L. Human–gut bacterial protein–protein interactions: Understudied but impactful to human health. Trends Microbiol. 2024, 32, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Caminero, A.; Guzman, M.; Libertucci, J.; Lomax, A.E. The emerging roles of bacterial proteases in intestinal diseases. Gut Microbes 2023, 15, 2181922. [Google Scholar] [CrossRef]
- da Cruz, T.A.; Donatelli Muro, B.B.; Machado Costa Lima, E.; dos Santos Moreira, V.; de Carvalho, J.C.C.; Pospissil Garbossa, C.A.; Batista Costa, L. Aspartic protease supplementation enhancing the performance, carcass characteristics, nutrient digestibility and economic viability, without changing blood parameters and salivary cortisol of pigs. Sci. Rep. 2024, 14, 11238. [Google Scholar] [CrossRef]
- Thuy-Boun, P.S.; Wang, A.Y.; Crissien-Martinez, A.; Xu, J.H.; Chatterjee, S.; Stupp, G.S.; Su, A.I.; Coyle, W.J.; Wolan, D.W. Quantitative Metaproteomics and Activity-based Protein Profiling of Patient Fecal Microbiome Identifies Host and Microbial Serine-type Endopeptidase Activity Associated With Ulcerative Colitis. Mol. Cell. Proteom. 2022, 21, 100197. [Google Scholar] [CrossRef]
- Jariwala, P.B.; Pellock, S.J.; Goldfarb, D.; Cloer, E.W.; Artola, M.; Simpson, J.B.; Bhatt, A.P.; Walton, W.G.; Roberts, L.R.; Major, M.B.; et al. Discovering the Microbial Enzymes Driving Drug Toxicity with Activity-Based Protein Profiling. ACS Chem Biol. 2020, 15, 217–225. [Google Scholar] [CrossRef]
- Danilova, I.; Sharipova, M. The Practical Potential of Bacilli and Their Enzymes for Industrial Production. Front. Microbiol. 2020, 11, 1782. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, A.S.; Bhatia, S.; Batra, N. Laccase: Sustainable production strategies, heterologous expression and potential biotechnological applications. Int. J. Biol. Macromol. 2024, 280, 135745. [Google Scholar] [CrossRef] [PubMed]
- Shafana Farveen, M.; Madhavan, T.; Narayanan, R. Association of Laccase from Bacillus cereus O2-B and Pseudomonas aeruginosa O1-P with the bio-degradation of polymers: An in vitro to in silico approach. Biodegradation 2023, 34, 383–403. [Google Scholar] [CrossRef] [PubMed]
- Gangola, S.; Sharma, A.; Bhatt, P.; Khati, P.; Chaudhary, P. Presence of esterase and laccase in Bacillus subtilis facilitates biodegradation and detoxification of cypermethrin. Sci. Rep. 2018, 8, 12755. [Google Scholar] [CrossRef]
- Kyomuhimbo, H.D.; Brink, H.G. Applications and immobilization strategies of the copper-centred laccase enzyme; a review. Heliyon 2023, 9, e13156. [Google Scholar] [CrossRef]
- Khan, S.I.; Sahinkaya, M.; Colak, D.N.; Zada, N.S.; Uzuner, U.; Belduz, A.O.; Çanakçi, S.; Khan, A.Z.; Khan, S.; Badshah, M.; et al. Production and characterization of novel thermostable CotA-laccase from Bacillus altitudinis SL7 and its application for lignin degradation. Enzym. Microb. Technol. 2024, 172, 110329. [Google Scholar] [CrossRef]
- EFSA Panel on Food Contact Materials, Enzymes and Processing Aids (CEP); Lambré, C.; Barat Baviera, J.M.; Bolognesi, C.; Cocconcelli, P.S.; Crebelli, R.; Gott, D.M.; Grob, K.; Lampi, E.; Mengelers, M.; et al. Safety evaluation of the food enzyme α-amylase from the genetically modified Bacillus licheniformis strain NZYM-BC. EFSA J. 2022, 20, e07370. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Name | EC Number |
|---|---|
| Alpha-amylase | 3.2.1.1 |
| Alpha-amylase family glycosyl hydrolase | 3.2.1.- |
| Alpha amylase C-terminal domain-containing protein | 3.2.1.- |
| Neopullulanase/maltogenic alpha-amylase | 3.2.1.135/3.2.1.133 |
| Maltogenic alpha-amylase | 3.2.1.133 |
| Glucoamylase family protein | 3.2.1.- |
| Alpha-amylase family protein | 3.2.1.- |
| Alpha-amylase precursor | 3.2.1.- |
| Putative glycanase or glycogenase with amylase domain | 3.2.1.- |
| Maltohexaose-producing amylase | 3.2.1.- |
| Family 15 glucoamylase | 3.2.1.- |
| Alpha amylase, catalytic domain protein | 3.2.1.- |
| Cellulase | 3.2.1.4 |
| Cellulase family glycosylhydrolase | 3.2.1.- |
| Beta-1,4-glucanase/cellulase | 3.2.1.4 |
| Nuclease | 3.1.- |
| Esterase | 3.1.1.1 |
| Lipase | 3.1.1.3 |
| Phospholipase C, phosphocholine-specific | 3.1.4.3 |
| Phospholipase | 3.1.1.- |
| Phospholipase D family protein | 3.1.4.4 |
| Lipase chaperone | 3.1.1.- |
| Patatin-like phospholipase family protein | 3.1.1.3 |
| Triacylglycerol lipase | 3.1.1.3 |
| GDSL-type esterase/lipase family protein | 3.1.1.3 |
| Lysophospholipase | 3.1.1.5 |
| Carboxylesterase/lipase family protein | 3.1.1.1- |
| Phospholipase C | 3.1.4.3 |
| Serine protease, patatin-like phospholipase family protein | 3.1.1.5 |
| Lysophospholipase-like family protein, putative | 3.1.2.22 |
| Minor cardiolipin synthetase (phospholipase D family) | 3.1.4.- |
| Conserved lipase family protein | 3.1.1.3 |
| Zinc-dependent phospholipase C family protein | 3.1.4.3 |
| Lipase family protein | 3.1.1.3 |
| Phospholipase D-like domain-containing protein | 3.1.4.- |
| Spore germination lipase | 3.1.1.- |
| Phosphatidylinositol-specific phospholipase C | 3.1.4.11 |
| Phospholipase/carboxylesterase | 3.1.1.- |
| Phospholipase A2 family protein | 3.1.1.4- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Sánchez, A.; Luque, G.; Ortiz, P.; Ruiz-Rodríguez, A.; López-Moreno, A.; Aguilera, M. Analysis of Human Gut Microbiota Enzymes for Biotechnological and Food Industrial Applications. Foods 2025, 14, 1794. https://doi.org/10.3390/foods14101794
Torres-Sánchez A, Luque G, Ortiz P, Ruiz-Rodríguez A, López-Moreno A, Aguilera M. Analysis of Human Gut Microbiota Enzymes for Biotechnological and Food Industrial Applications. Foods. 2025; 14(10):1794. https://doi.org/10.3390/foods14101794
Chicago/Turabian StyleTorres-Sánchez, Alfonso, Gracia Luque, Pilar Ortiz, Alicia Ruiz-Rodríguez, Ana López-Moreno, and Margarita Aguilera. 2025. "Analysis of Human Gut Microbiota Enzymes for Biotechnological and Food Industrial Applications" Foods 14, no. 10: 1794. https://doi.org/10.3390/foods14101794
APA StyleTorres-Sánchez, A., Luque, G., Ortiz, P., Ruiz-Rodríguez, A., López-Moreno, A., & Aguilera, M. (2025). Analysis of Human Gut Microbiota Enzymes for Biotechnological and Food Industrial Applications. Foods, 14(10), 1794. https://doi.org/10.3390/foods14101794