
Construction of an MLR-QSAR Model Based on Dietary Flavonoids and Screening of Natural α-Glucosidase Inhibitors

 ,
,  ,
,  ,
, 
Abstract
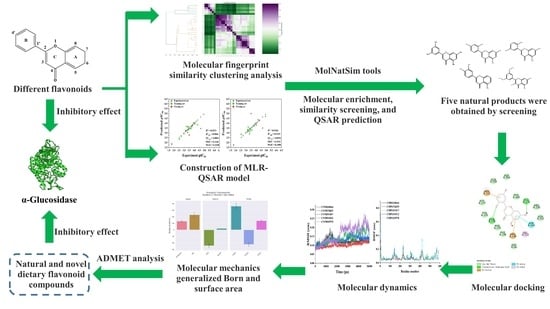
1. Introduction
2. Materials and Methods
2.1. Preparation of Datasets
2.2. Structural Modeling
2.3. Molecular Fingerprint Similarity Calculation
2.4. MLR-QSAR Modeling
2.5. Validation of the MLR-QSAR Model
2.6. Natural Product Screening
2.7. Molecular Docking and Molecular Dynamics Simulation
2.8. Combined Free Energy Calculation
3. Results and Discussion
3.1. Molecular Fingerprint Similarity Analysis
3.2. MLR-QSAR Analysis
3.3. Discovery of Natural α-Glucosidase Inhibitors and ADMET Analysis
3.4. Molecular Docking Analysis
3.5. Molecular Dynamics Simulation Analysis
3.6. Combining Free Energy Calculations by the MMGBSA Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fan, W. Epidemiology in diabetes mellitus and cardiovascular disease. Cardiovasc. Endocrinol. Metab. 2017, 6, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Rahim, F.; Zaman, K.; Selvaraj, M.; Khan, K.M. Synthesis, α-glycosidase inhibitory potential and molecular docking study of benzimidazole derivatives. Bioorg. Chem. 2019, 95, 103555. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.C.; Carson, M.L.; Miller, M.P.; Turowski, M.; Bell, M.; Wilder, D.M.; Reeves, M.S. High-Viscosity Hydroxypropylmethylcellulose Blunts Postprandial Glucose and Insulin Responses. Diabetes Care 2007, 30, 1039. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Anouar, E.H.; Ahmad, R.; Azman, N.; Marzouk, M.; Al-Salahi, R. Triazoloquinazolines as a new class of potent alpha-glucosidase inhibitors: In vitro evaluation and docking study. PLoS ONE 2019, 14, e0220379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, G.; Wu, D.; Yu, Y.; Hu, N.; Wang, H.; Li, X.; Wu, Y. Emerging strategies for the activity assay and inhibitor screening of alpha-glucosidase. Food Funct. 2020, 11, 66–82. [Google Scholar] [CrossRef]
- Chen, Y.G.; Li, P.; Li, P.; Yan, R.; Zhang, X.Q.; Wang, Y.; Zhang, X.T.; Ye, W.C.; Zhang, Q.W. alpha-Glucosidase inhibitory effect and simultaneous quantification of three major flavonoid glycosides in Microctis folium. Molecules 2013, 18, 4221–4232. [Google Scholar] [CrossRef]
- Poovitha, S.; Parani, M. In vitro and in vivo α-amylase and α-glucosidase inhibiting activities of the protein extracts from two varieties of bitter gourd (Momordica charantia L.). BMC Complement. Altern. Med. 2016, 16, 185. [Google Scholar] [CrossRef]
- Adib, M.; Peytam, F.; Rahmanian-Jazi, M.; Mahernia, S.; Bijanzadeh, H.R.; Jahani, M.; Mohammadi-Khanaposhtani, M.; Imanparast, S.; Faramarzi, M.A.; Mahdavi, M. New 6-amino-pyrido [2,3-d]pyrimidine-2,4-diones as novel agents to treat type 2 diabetes: A simple and efficient synthesis, α-glucosidase inhibition, molecular modeling and kinetic study. Eur. J. Med. Chem. 2018, 155, 353–363. [Google Scholar] [CrossRef]
- Saltos, M.B.V.; Puente, B.F.N.; Faraone, I.; Milella, L.; Tommasi, N.D.; Braca, A. Inhibitors of α-amylase and α-glucosidase from Andromachia igniaria Humb. & Bonpl. Phytochem. Lett. 2015, 14, 45–50. [Google Scholar]
- Tian, J.L.; Si, X.; Wang, Y.H.; Gong, E.S.; Xie, X.; Zhang, Y.; Li, B.; Shu, C. Bioactive flavonoids from Rubus corchorifolius inhibit alpha-glucosidase and alpha-amylase to improve postprandial hyperglycemia. Food Chem. 2021, 341 Pt 1, 128149. [Google Scholar] [CrossRef]
- Wafaa, B.; Sad, B.; Mohamed, B. Antidiabetic medicinal plants as a source of alpha glucosidase inhibitors. Curr. Diabetes Rev. 2010, 6, 247–254. [Google Scholar]
- Sun, H.; Li, Y.; Zhang, X.; Lei, Y.; Ding, W.; Zhao, X.; Wang, H.; Song, X.; Yao, Q.; Zhang, Y.; et al. Synthesis, alpha-glucosidase inhibitory and molecular docking studies of prenylated and geranylated flavones, isoflavones and chalcones. Bioorg. Med. Chem. Lett. 2015, 25, 4567–4571. [Google Scholar] [CrossRef]
- Bondonno, N.P.; Bondonno, C.P.; Rich, L.; Ward, N.C.; Hodgson, J.M.; Croft, K.D. Flavonoid-rich apple improves endothelial function in individuals at risk for cardiovascular disease. J. Nutr. Intermed. Metab. 2017, 8, 79–80. [Google Scholar] [CrossRef]
- Martens, S.; Mithöfer, A. Flavones and flavone synthases. Phytochemistry. Phytochemistry 2005, 66, 2399–2407. [Google Scholar] [CrossRef]
- Liu, M.; Yin, H.; Liu, G.; Dong, J.; Qian, Z.; Miao, J. Xanthohumol, a prenylated chalcone from beer hops, acts as an alpha-glucosidase inhibitor in vitro. J. Agric. Food Chem. 2014, 62, 5548–5554. [Google Scholar] [CrossRef]
- Imran, S.; Taha, M.; Ismail, N.H.; Kashif, S.M.; Rahim, F.; Jamil, W.; Hariono, M.; Yusuf, M.; Wahab, H. Synthesis of novel flavone hydrazones: In-vitro evaluation of α-glucosidase inhibition, QSAR analysis and docking studies. Eur. J. Med. Chem. 2015, 105, 156–170. [Google Scholar] [CrossRef]
- Sigalapalli, D.K.; Yele, V.; Jupudi, S.; Shaikh, A.S.; Kadagathur, M.; Tangellamudi, N.D.; Nagendra Babu, B. Insights into the pharmacophore-based 3D-QSAR modeling, molecular dynamics simulation studies of certain dihydroxy pyrrolidine/piperidine and aza-flavanone derivatives as α-glucosidase inhibitors. J. Mol. Struct. 2021, 1223, 129243. [Google Scholar] [CrossRef]
- Al-ghulikah, H.A.; Mughal, E.U.; Elkaeed, E.B.; Naeem, N.; Nazir, Y.; Alzahrani, A.Y.A.; Sadiq, A.; Shah, S.W.A. Discovery of chalcone derivatives as potential α-glucosidase and cholinesterase inhibitors: Effect of hyperglycemia in paving a path to dementia. J. Mol. Struct. 2022, 1275, 134658. [Google Scholar] [CrossRef]
- Hansch, C.; Maloney, P.P.; Fujita, T.; Muir, R.M. Correlation of Biological Activity of Phenoxyacetic Acids with Hammett Substituent Constants and Partition Coefficients. Nature 1962, 194, 178–180. [Google Scholar] [CrossRef]
- Xu, J.; Huang, S.; Luo, H.; Li, G.; Bao, J.; Cai, S.; Wang, Y. QSAR Studies on andrographolide derivatives as alpha-glucosidase inhibitors. Int. J. Mol. Sci. 2010, 11, 880–895. [Google Scholar] [CrossRef]
- Jia, Y.; Ma, Y.; Cheng, G.; Zhang, Y.; Cai, S. Comparative Study of Dietary Flavonoids with Different Structures as α-Glucosidase Inhibitors and Insulin Sensitizers. J. Agric. Food Chem. 2019, 67, 10521–10533. [Google Scholar] [CrossRef]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2021, 22, bbaa161. [Google Scholar] [CrossRef] [PubMed]
- GitHub. SourceForge RDKit: Open-Source Cheminformatics Software. Available online: http://www.rdkit.org (accessed on 19 August 2022).
- Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J. Comput. Chem. 1999, 20, 730–748. [Google Scholar] [CrossRef]
- Zhang, D.; Ouyang, S.; Cai, M.; Zhang, H.; Ding, S.; Liu, D.; Cai, P.; Le, Y.; Hu, Q.N. FADB-China: A molecular-level food adulteration database in China based on molecular fingerprints and similarity algorithms prediction expansion. Food Chem. 2020, 327, 127010. [Google Scholar] [CrossRef] [PubMed]
- Swami, A.; Jain, R. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2013, 12, 2825–2830. [Google Scholar]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Sculley, D. Web-scale k-means clustering. In Proceedings of the International Conference on World Wide Web, Raleigh, NC, USA, 26–30 April 2010. [Google Scholar]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Ehlert, S.; Mewes, J.M. rSCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 8208–8215. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 526128. [Google Scholar] [CrossRef]
- Tian, L.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar]
- Lu, T. Sobtop, Version Dev 3. Available online: http://sobereva.com/soft/Sobtop (accessed on 19 August 2022).
- Rizzuti, B. Molecular simulations of proteins: From simplified physical interactions to complex biological phenomena. Biochim. Biophys. Acta Proteins Proteom. 2022, 1870, 140757. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Simmerling, C.; Hauser, K.E.; Maier, J.A.; Kasavajhala, K. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. JCTC 2015, 11, 3696–3713. [Google Scholar]
- Pan, F.; Zhao, L.; Cai, S.; Tang, X.; Mehmood, A.; Alnadari, F.; Tuersuntuoheti, T.; Zhou, N.; Ai, X. Prediction and evaluation of the 3D structure of Macadamia integrifolia antimicrobial protein 2 (MiAMP2) and its interaction with palmitoleic acid or oleic acid: An integrated computational approach. Food Chem. 2022, 367, 130677. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Tsuji, P.A.; Stephenson, K.K.; Wade, K.L.; Liu, H.; Fahey, J.W. Structure-activity analysis of flavonoids: Direct and indirect antioxidant, and antiinflammatory potencies and toxicities. Nutr. Cancer 2013, 65, 1014–1025. [Google Scholar] [CrossRef]
- Roy, K.; Ambure, P. The “double cross-validation” software tool for MLR QSAR model development. Chemom. Intell. Lab. Syst. 2016, 159, 108–126. [Google Scholar] [CrossRef]
- Hollas, B. An Analysis of the Autocorrelation Descriptor for Molecules. J. Math. Chem. 2003, 33, 91–101. [Google Scholar] [CrossRef]
- Adedirin, O.; Uzairu, A.; Shallangwa, G.A.; Abechi, S.E. Qsar and molecular docking based design of some n-benzylacetamide as Γ-aminobutyrate-aminotransferase inhibitors. J. Eng. Exact Sci. 2018, 4, 0065–0084. [Google Scholar] [CrossRef]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Molecular Descriptors for Chemoinformatics: Volume I: Alphabetical Listing/Volume II: Appendices, References; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Todeschini, R.; Consonni, V. Molecular descriptors for chemoinformatics. In Molecular Descriptors for Chemoinformatics; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Li, X.; Pan, F.; Yang, Z.; Gao, F.; Li, J.; Zhang, F.; Wang, T. Construction of QSAR model based on cysteine-containing dipeptides and screening of natural tyrosinase inhibitors. J. Food Biochem. 2022, 46, e14338. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Hanwarinroj, C.; Thongdee, P.; Sukchit, D.; Taveepanich, S.; Kamsri, P.; Punkvang, A.; Ketrat, S.; Saparpakorn, P.; Hannongbua, S.; Suttisintong, K.; et al. In silico design of novel quinazoline-based compounds as potential Mycobacterium tuberculosis PknB inhibitors through 2D and 3D-QSAR, molecular dynamics simulations combined with pharmacokinetic predictions. J. Mol. Graph. Model. 2022, 115, 108231. [Google Scholar] [CrossRef] [PubMed]
- Kant, U.R. Drug Delivery Systems, CNS Protection, and the Blood Brain Barrier. BioMed Res. Int. 2014, 2014, 869269. [Google Scholar]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Khaldan, A.; Bouamrane, S.; El-mernissi, R.; Maghat, H.; Ajana, M.A.; Sbai, A.; Bouachrine, M.; Lakhlifi, T. 3D-QSAR modeling, molecular docking and ADMET properties of benzothiazole derivatives as α-glucosidase inhibitors. Mater. Today Proc. 2021, 45, 7643–7652. [Google Scholar] [CrossRef]
- Wu, X.; Ding, H.; Hu, X.; Pan, J.; Liao, Y.; Gong, D.; Zhang, G. Exploring inhibitory mechanism of gallocatechin gallate on a-amylase and a-glucosidase relevant to postprandial hyperglycemia. J. Funct. Foods 2018, 48, 200–209. [Google Scholar] [CrossRef]
- Park, H.; Hwang, K.Y.; Oh, K.H.; Kim, Y.H.; Lee, J.Y.; Kim, K. Discovery of novel alpha-glucosidase inhibitors based on the virtual screening with the homology-modeled protein structure. Bioorg. Med. Chem. 2008, 16, 284–292. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, Y.; Ma, Y.; Cheng, G.; Cai, S. Molecules phenolic composition, antioxidant properties, and inhibition toward digestive enzymes with molecular docking analysis of different fractions from Prinsepia utilis royle fruits. Molecules 2019, 23, 3373. [Google Scholar] [CrossRef]
- Shojapour, M.; Farahmand, S. Point mutation consideration in CcO protein of the electron transfer chain by MD simulation. J. Mol. Graph. Model. 2022, 117, 108309. [Google Scholar] [CrossRef]
- Ni, Z.; Wang, X.; Zhang, T.; Jin, R.Z. Molecular dynamics simulations reveal the allosteric effect of F1174C resistance mutation to ceritinib in ALK-associated lung cancer. Comput. Biol. Chem. 2016, 65, 54–60. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D-MLR-QSAR | 3D-MLR-QSAR | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PubChem CID | Name | pIC50 * | pIC50pre # | Residue | PubChem CID | Name | pIC50 | pIC50pre | Residue |
| 5280443 | Apigenin | 3.224 | 3.684 | −0.46 | 5280445 | Luteolin | 4.082 | 4.054 | 0.028 |
| 5281605 | Baicalein | 3.586 | 3.42 | 0.166 | 5280863 | Kaempferol | 3.814 | 3.746 | 0.068 |
| 5281616 | Galangin | 3.381 | 3.284 | 0.097 | 5280443 | Apigenin | 3.224 | 3.593 | −0.37 |
| 5281654 | Isorhamnetin | 3.17 | 3.271 | −0.102 | 5280343 | Quercetin | 3.939 | 4.008 | −0.07 |
| 5280445 | Luteolin | 4.082 | 3.938 | 0.144 | 5281654 | Isorhamnetin | 3.17 | 3.264 | −0.095 |
| 5281672 | Myricetin | 4.934 | 4.92 | 0.014 | 5281672 | Myricetin | 4.934 | 4.729 | 0.206 |
| 5280863 | Kaempferol | 3.814 | 3.751 | 0.064 | 5280704 | Apigenin-7-O-glucoside | 4.642 | 3.502 | 1.14 |
| 5280343 | Quercetin | 3.939 | 4.05 | −0.111 | 5281605 | Baicalein | 3.586 | 3.29 | 0.296 |
| 5280704 | Apigenin-7-O-glucoside | 4.642 | 3.468 | 1.174 | 5281614 | Fisetin | 4.334 | 4.271 | 0.063 |
| 72281 | Hesperitin | 2.72 | 2.654 | 0.067 | 440735 | Eriodictyol | 2.986 | 3.035 | −0.049 |
| 5280441 | Vitexin | 3.448 | 3.353 | 0.095 | 5281616 | Galangin | 3.381 | 3.122 | 0.259 |
| 5281614 | Fisetin | 4.334 | 3.839 | 0.494 | 439533 | Taxifolin | 2.531 | 2.869 | −0.338 |
| 439533 | Taxifolin | 2.531 | 2.371 | 0.16 | 72281 | Hesperitin | 2.72 | 2.708 | 0.012 |
| 440735 | Eriodictyol | 2.986 | 2.911 | 0.075 | 5280378 | Formononetin | 3.126 | 3.101 | 0.025 |
| 4788 | Phloretin | 3.85 | 3.806 | 0.045 | 5280637 | Luteoloside | 3.365 | 3.463 | −0.098 |
| 9064 | (+)-Catechin | 1.789 | 1.888 | −0.098 | 5280961 | Genistein | 3.192 | 3.357 | −0.164 |
| 5280637 | Luteoloside | 3.365 | 3.582 | −0.217 | 5280441 | Vitexin | 3.448 | 3.302 | 0.147 |
| 5280378 | Formononetin | 3.126 | 3.189 | −0.063 | 3084995 | Isoschaftoside | 3.411 | 3.366 | 0.045 |
| 5280961 | Genistein | 3.192 | 3.219 | −0.027 | 172648475 | Kaempferol-7-O-β-glucoside | 3.689 | 3.654 | 0.035 |
| 172648475 | Kaempferol-7-O-β-glucoside | 3.689 | 3.47 | 0.219 | 9064 | (+)-Catechin | 1.789 | 1.729 | 0.06 |
| 5281673 | Myricetin | 3.51 | 2.776 | 0.734 | 5281673 | Myricetin | 3.51 | 4.997 | −1.487 |
| 441667 | Cyanidin-3-O-glucoside | 4.197 | 4.204 | −0.006 | 5280805 | Rutin | 3.761 | 3.771 | −0.01 |
| 65064 | Epigallocatechin Gallate | 4.011 | 4.009 | 0.002 | 5481663 | Isorhamnetin-3-O-rutinoside | 3.268 | 3.196 | 0.072 |
| 3084995 | Isoschaftoside | 3.411 | 3.646 | −0.234 | 56776173 | Vitexin-4′-O-glucoside | 3.354 | 2.898 | 0.456 |
| 5280805 | Rutin | 3.761 | 3.623 | 0.138 | 4788 | Phloretin | 3.85 | 3.865 | −0.015 |
| 56776173 | Vitexin-4′-O-glucoside | 3.354 | 3.049 | 0.306 | 65064 | Epigallocatechin Gallate | 4.011 | 3.826 | 0.185 |
| 5481663 | Isorhamnetin-3-O-rutinoside | 3.268 | 3.245 | 0.023 | 441667 | Cyanidin-3-O-glucoside | 4.197 | 4.365 | −0.168 |
| Parameters | 2D-MLR-QSAR | 3D-MLR-QSAR |
|---|---|---|
| Q2LOO | 0.8861 | 0.8991 |
| R2 fitting | 0.9273 | 0.9336 |
| R2 adjusted | 0.9046 | 0.9129 |
| RSS | 0.6906 | 0.5679 |
| PRESS | 1.0829 | 0.8633 |
| SDEC | 0.1772 | 0.1607 |
| SDEP | 0.2219 | 0.1981 |
| MSE | 0.1636 | 0.2545 |
| MAE | 0.3138 | 0.349 |
| MLR | pIC50 = 4.12506 + (−0.15599) × MIC1 + (−0.00011) × ATS4v + (0.05492) × AATS7m + (3.06615) × CIC3 + (−3.00189) × minssCH2 | pIC50 = 8.97844 + (0.49135) LOBMAX + (−0.03043) × RDF35i + (0.00505) × TDB10i + (−0.01383) × TDB9i + (0.01155) × TDB6m |
| Compounds | Molecular Structure | IC50pre # (μM) | Water Solubility | Bioavailability Score | GI Absorption | BBB Permeant | Pgp Substrate | log Kp (cm/s) | Toxicity | |
|---|---|---|---|---|---|---|---|---|---|---|
| Carcino_Mouse | Carcino_Rat | |||||||||
| 2-(3,5-Dihydroxyphenyl)-5,7-dihydroxy-4H-chromen-4-one |  | 8.98 | Soluble | 0.55 | High | No | No | −6.05 | Negative | Negative |
| Norartocarpetin |  | 31.95 | Soluble | 0.55 | High | No | No | −6.16 | Negative | Negative |
| 2-(2,5-Dihydroxyphenyl)-5,7-dihydroxy-4H-chromen-4-one |  | 78.57 | Soluble | 0.55 | High | No | No | −6.16 | Negative | Positive |
| 2-(3,4-Dihydroxyphenyl)-5-hydroxy-4H-chromen-4-one |  | 87.87 | Moderately soluble | 0.55 | High | No | No | −5.6 | Negative | Positive |
| Morelosin |  | 94.14 | Moderately soluble | 0.55 | High | No | No | −6.76 | Negative | Positive |
| Energy (kJ/mol) | 2-(3,5-Dihydroxyphenyl)-5,7-Dihydroxy-4H-Chromen-4-one | Norartocarpetin | 2-(2,5-Dihydroxyphenyl)-5,7-Dihydroxy-4H-Chromen-4-one | 2-(3,4-Dihydroxyphenyl)-5-Hydroxy-4H-Chromen-4-one | Morelosin |
|---|---|---|---|---|---|
| ΔEvdw | −23.24 ± 1.95 | −30.29 ± 2.98 | −26.31 ± 4.52 | −35.28 ± 2.77 | −29.34 ± 2.45 |
| ∆Eele | −38.62 ± 5.45 | −53.66 ± 5.59 | −46.07 ± 9.18 | −15.2 ± 10.68 | −7.1 ± 3.49 |
| ∆Gpol | 41.82 ± 3.12 | 51.32 ± 3.38 | 48.7 ± 8.00 | 32.38 ± 7.23 | 23.99 ± 3.64 |
| ∆Gnon-pol | −3.84 ± 0.27 | −5.15 ± 0.13 | −4.13 ± 0.34 | −4.43 ± 0.15 | −4.11 ± 0.23 |
| ∆Ggas | −61.86 ± 5.44 | 83.95 ± 4.93 | −72.38 ± 10.14 | −50.48 ± 9.98 | −36.43 ± 4.19 |
| ∆Gsol | 37.98 ± 3.01 | 46.17 ± 3.36 | 44.57 ± 7.72 | 27.95 ± 7.25 | 19.88 ± 3.52 |
| ∆Gbind | −23.87 ± 3.18 | −37.78 ± 3.28 | −27.81 ± 3.45 | −22.53 ± 3.40 | −16.56 ± 2.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.; Yang, Z.; Pan, F.; Jia, Y.; Cai, S.; Zhao, L.; Zhao, L.; Wang, O.; Wang, C. Construction of an MLR-QSAR Model Based on Dietary Flavonoids and Screening of Natural α-Glucosidase Inhibitors. Foods 2022, 11, 4046. https://doi.org/10.3390/foods11244046
Yang T, Yang Z, Pan F, Jia Y, Cai S, Zhao L, Zhao L, Wang O, Wang C. Construction of an MLR-QSAR Model Based on Dietary Flavonoids and Screening of Natural α-Glucosidase Inhibitors. Foods. 2022; 11(24):4046. https://doi.org/10.3390/foods11244046
Chicago/Turabian StyleYang, Ting, Zichen Yang, Fei Pan, Yijia Jia, Shengbao Cai, Liang Zhao, Lei Zhao, Ou Wang, and Chengtao Wang. 2022. "Construction of an MLR-QSAR Model Based on Dietary Flavonoids and Screening of Natural α-Glucosidase Inhibitors" Foods 11, no. 24: 4046. https://doi.org/10.3390/foods11244046
APA StyleYang, T., Yang, Z., Pan, F., Jia, Y., Cai, S., Zhao, L., Zhao, L., Wang, O., & Wang, C. (2022). Construction of an MLR-QSAR Model Based on Dietary Flavonoids and Screening of Natural α-Glucosidase Inhibitors. Foods, 11(24), 4046. https://doi.org/10.3390/foods11244046