
Qualitative and Quantitative Correlation of Microstructural Properties and In Vitro Glucose Adsorption and Diffusion Behaviors of Pea Insoluble Dietary Fiber Induced by Ultrafine Grinding

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of PIDF Samples with Different Particle Sizes
2.3. Microstructure Characterization of PIDF
2.3.1. Particle-Size Distribution
2.3.2. Scanning Electron Microscopy (SEM) Images
2.3.3. Specific Surface Area (SSA) and Pore-Size Distribution
2.3.4. Cellulose Crystallinity (CrI)
2.3.5. Surface Element Analysis
2.4. Determination of Glucose Adsorption Capacity
2.5. Measurement of Glucose Diffusion and Glucose Dialysis Retardation Index
2.6. Statistical Analysis
3. Results and Discussion
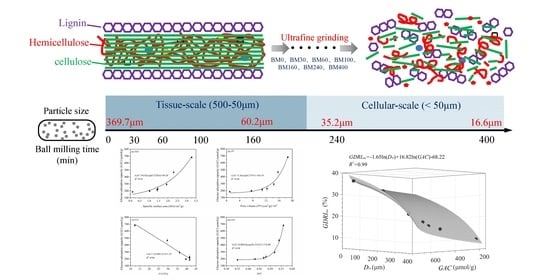
3.1. Microstructural Characterization of PIDF at Different Scales
3.1.1. Particle-Size Distribution
3.1.2. SEM Images
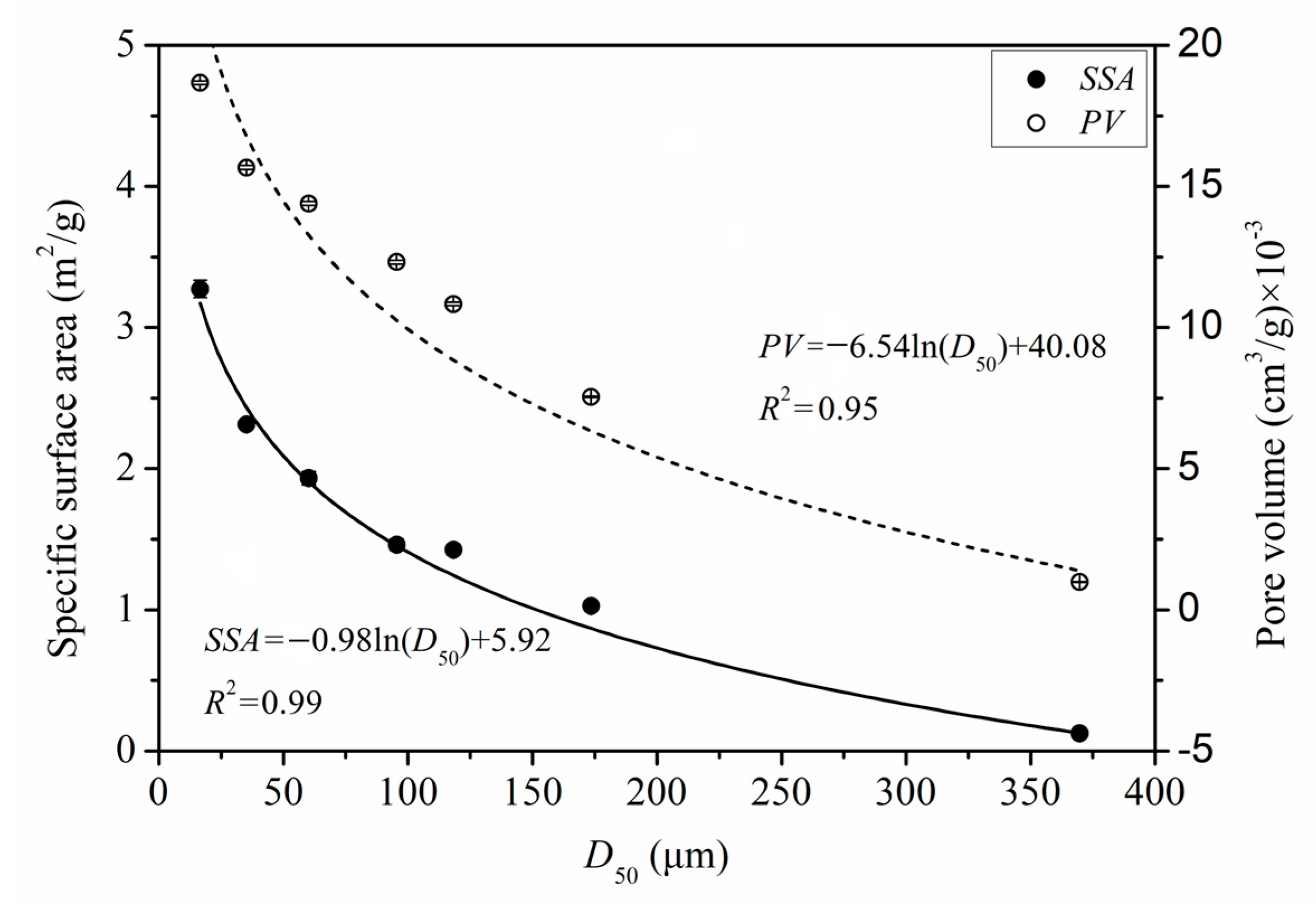
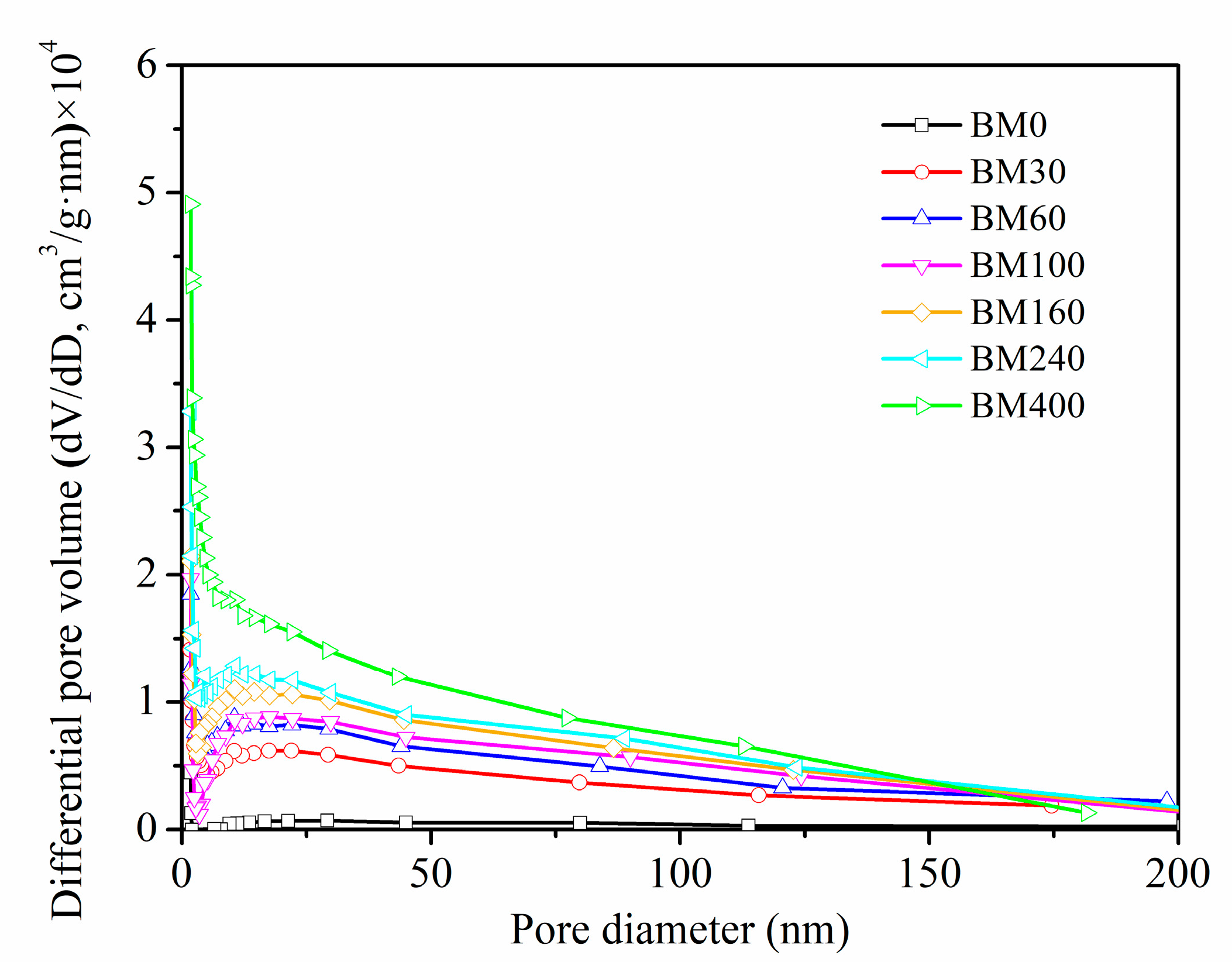
3.1.3. Specific Surface Area and Pores
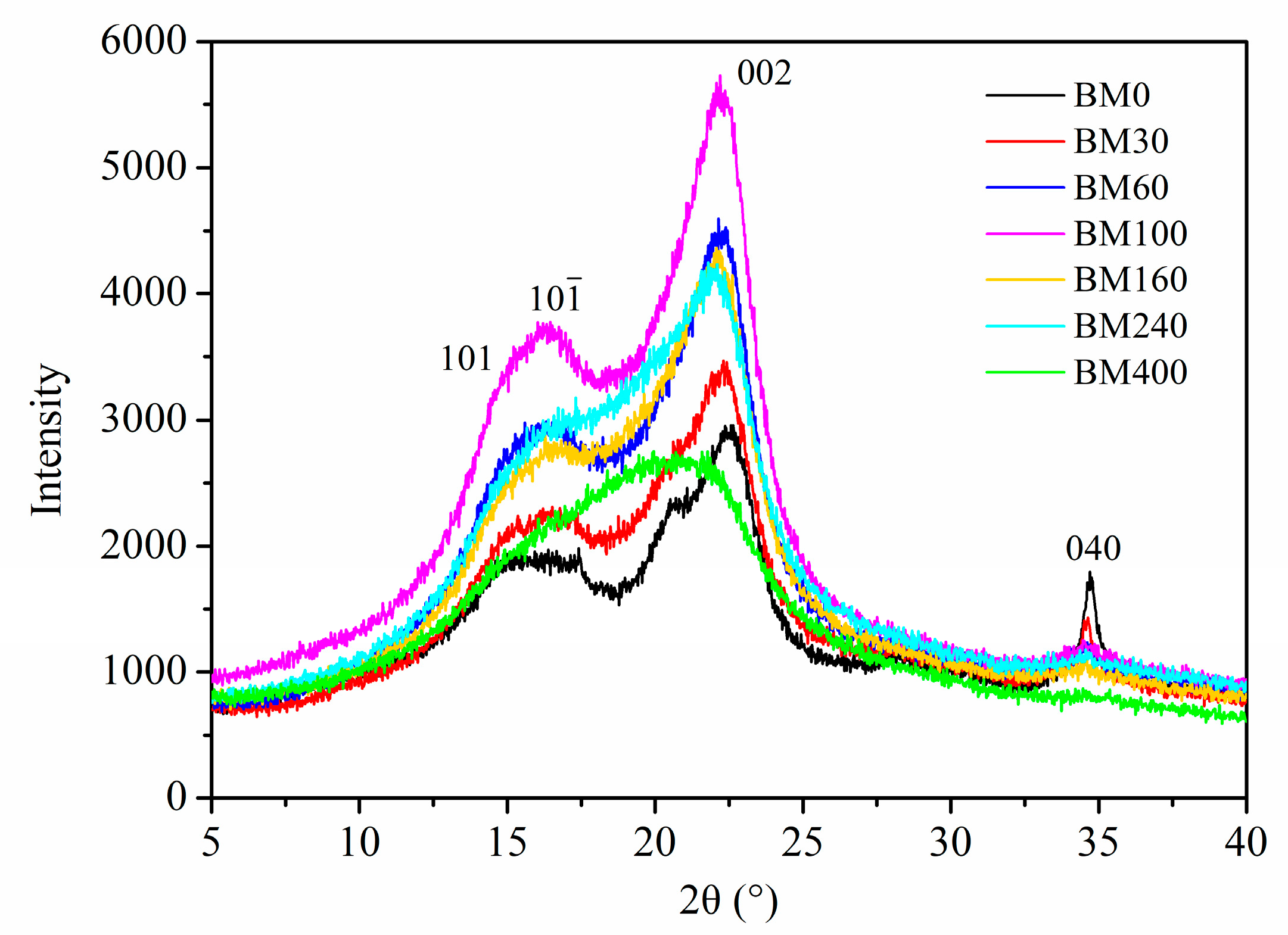
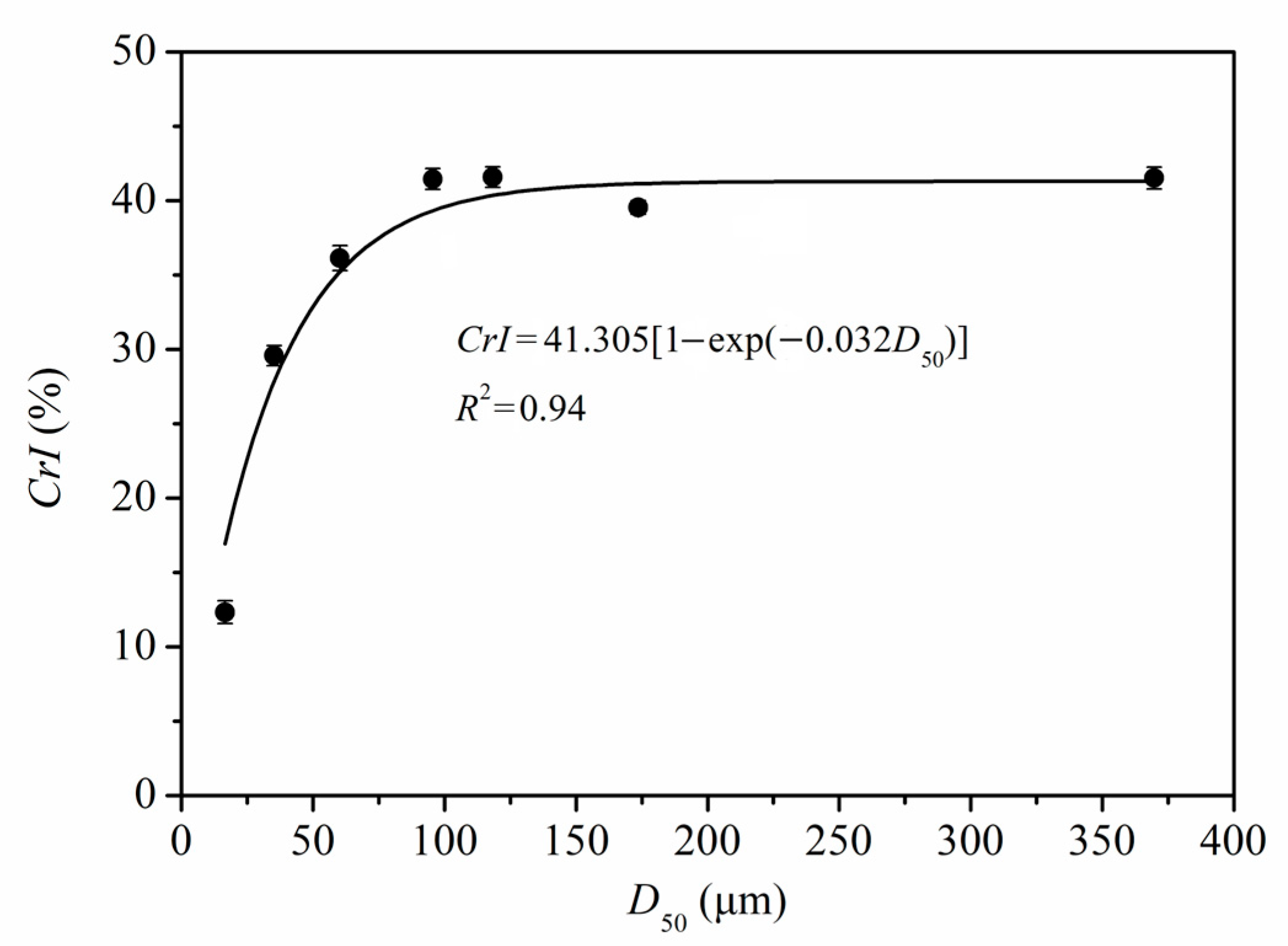
3.1.4. Cellulose Crystallinity
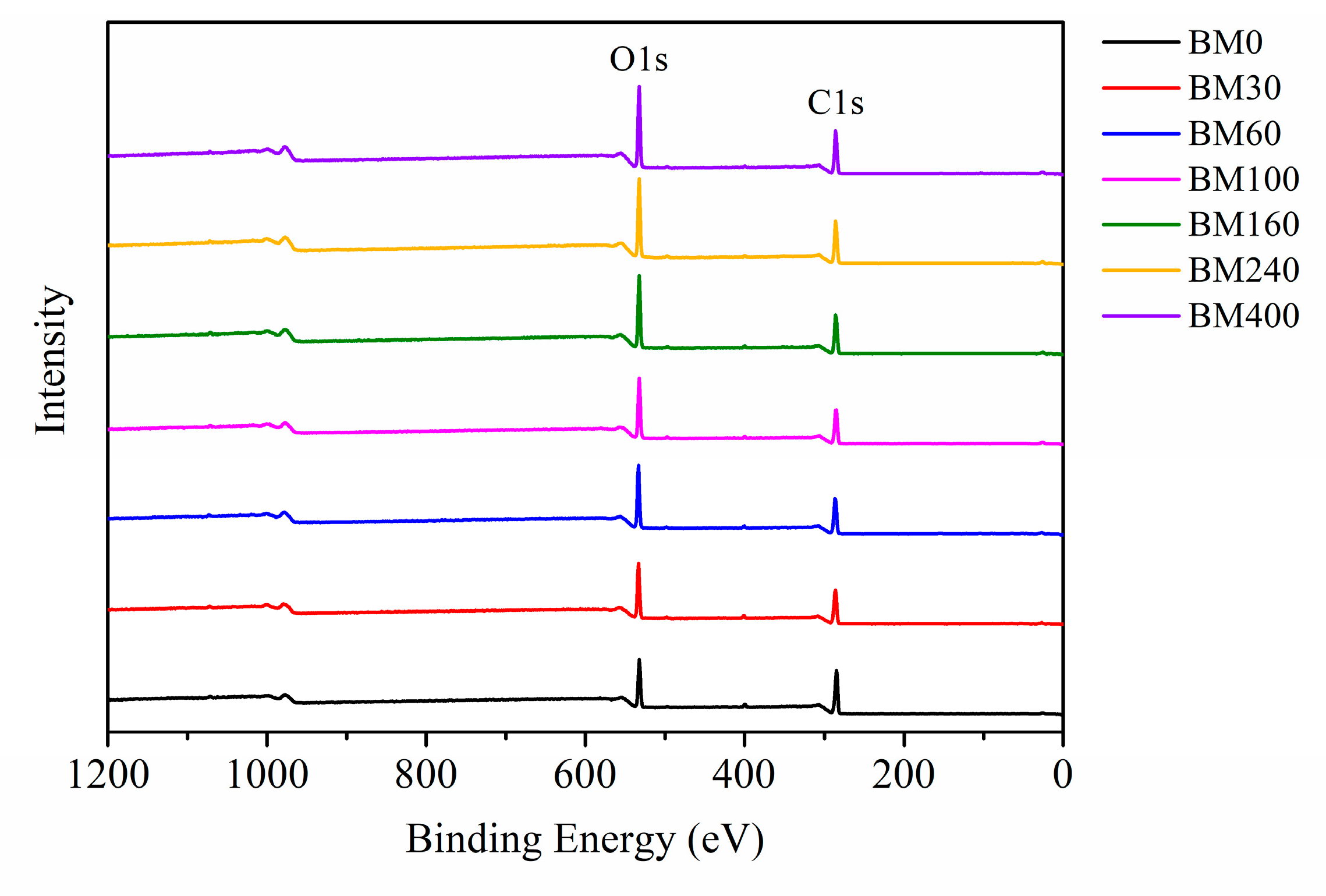
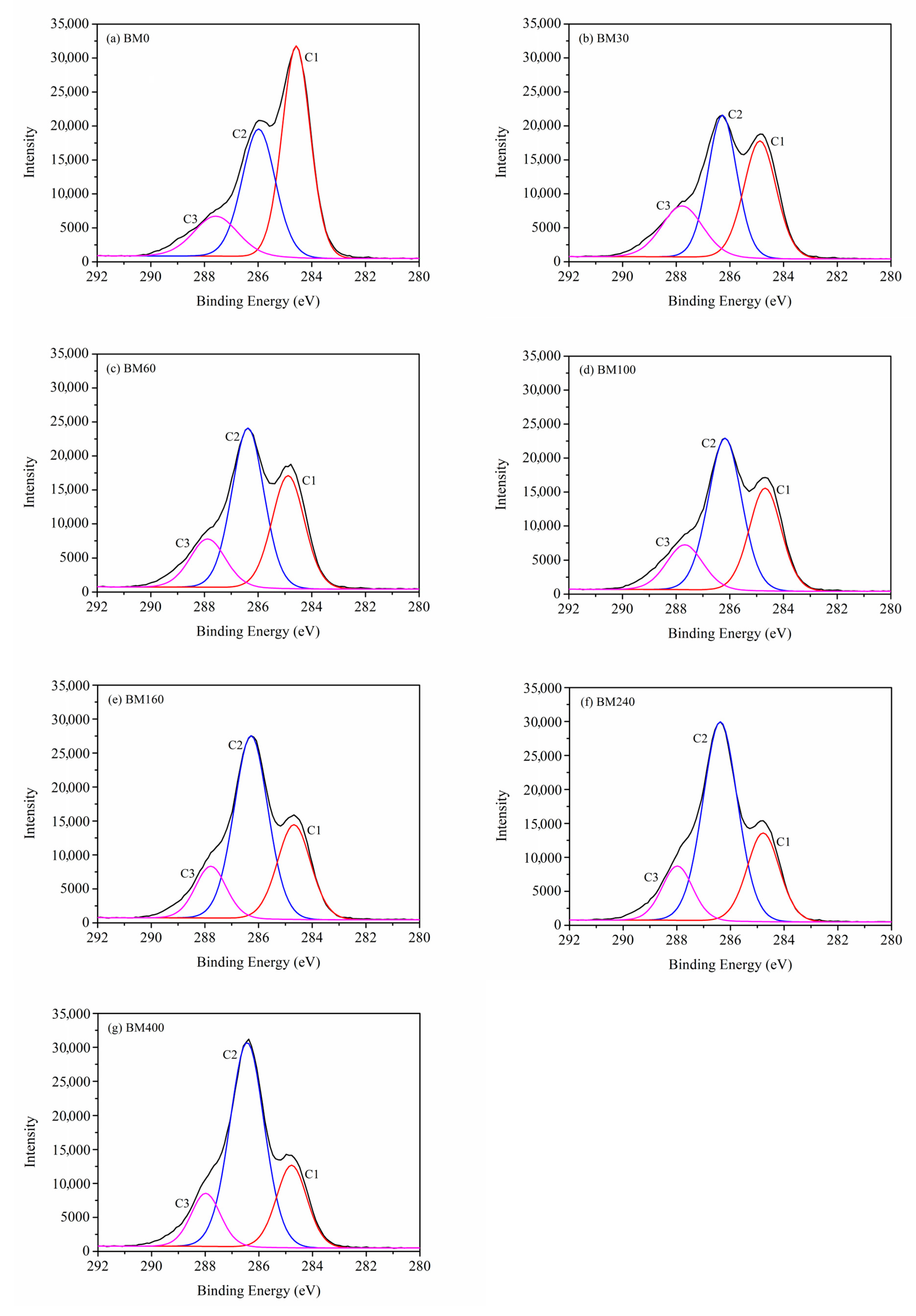
3.1.5. Surface Elemental Analysis
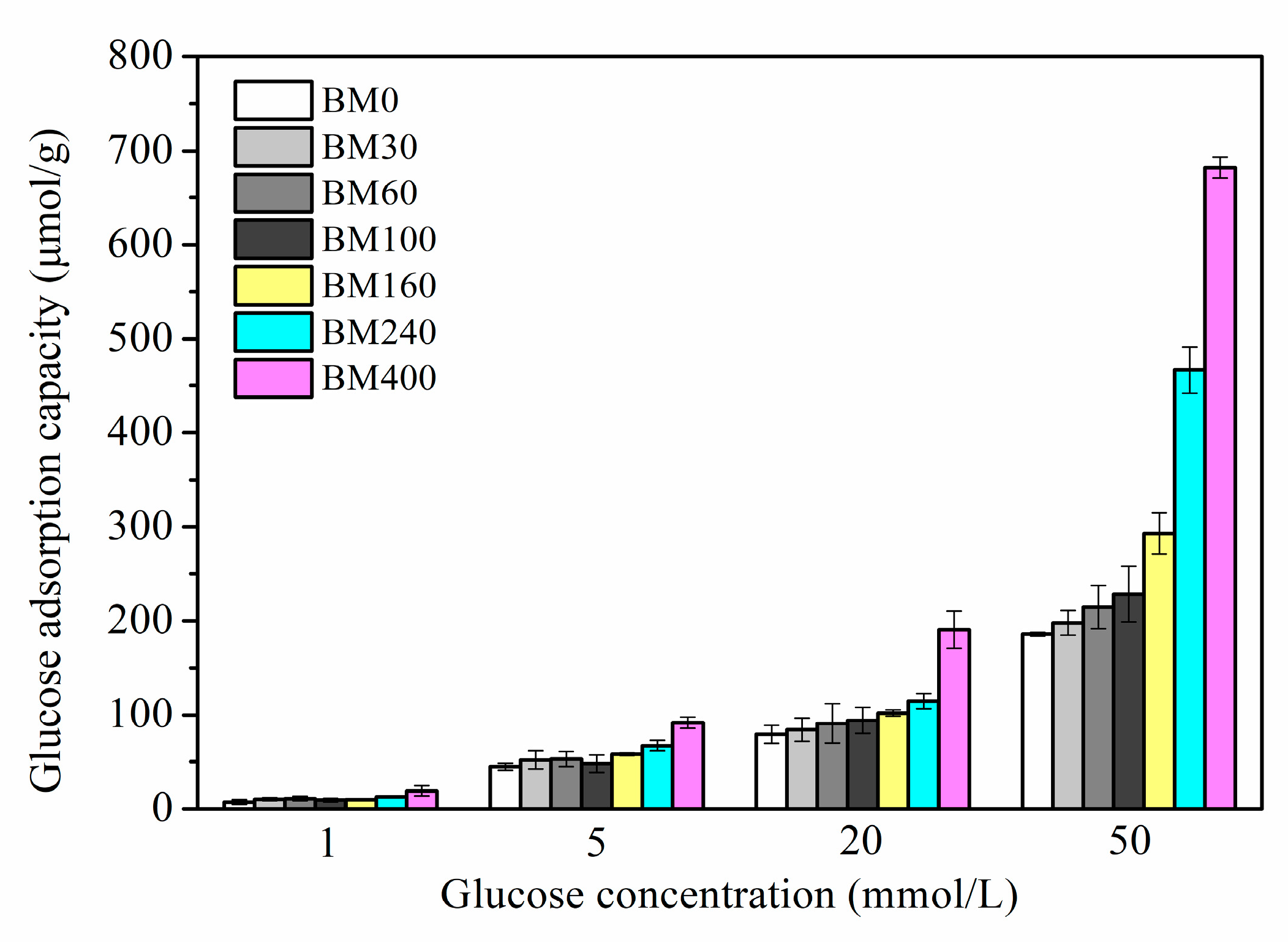
3.2. Glucose Adsorption Capacity
3.3. Glucose Diffusion and Glucose Dialysis Retardation Index
3.4. Correlation and Quantitative Relationship Analysis of Microstructure Parameters, GAC, and GDRI
3.4.1. Pearson Correlation Analysis of Microstructure Parameters, GAC, and GDRI
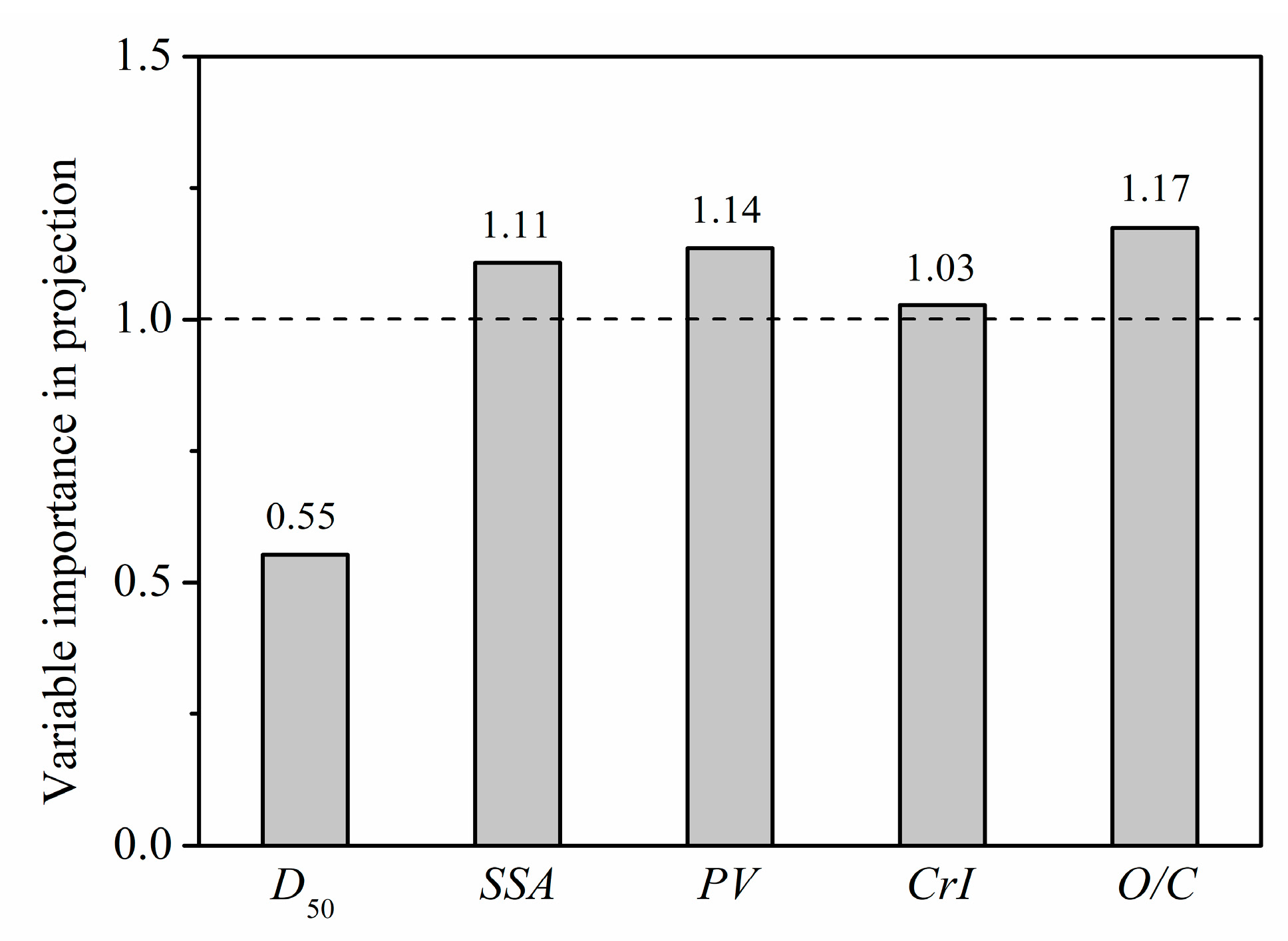
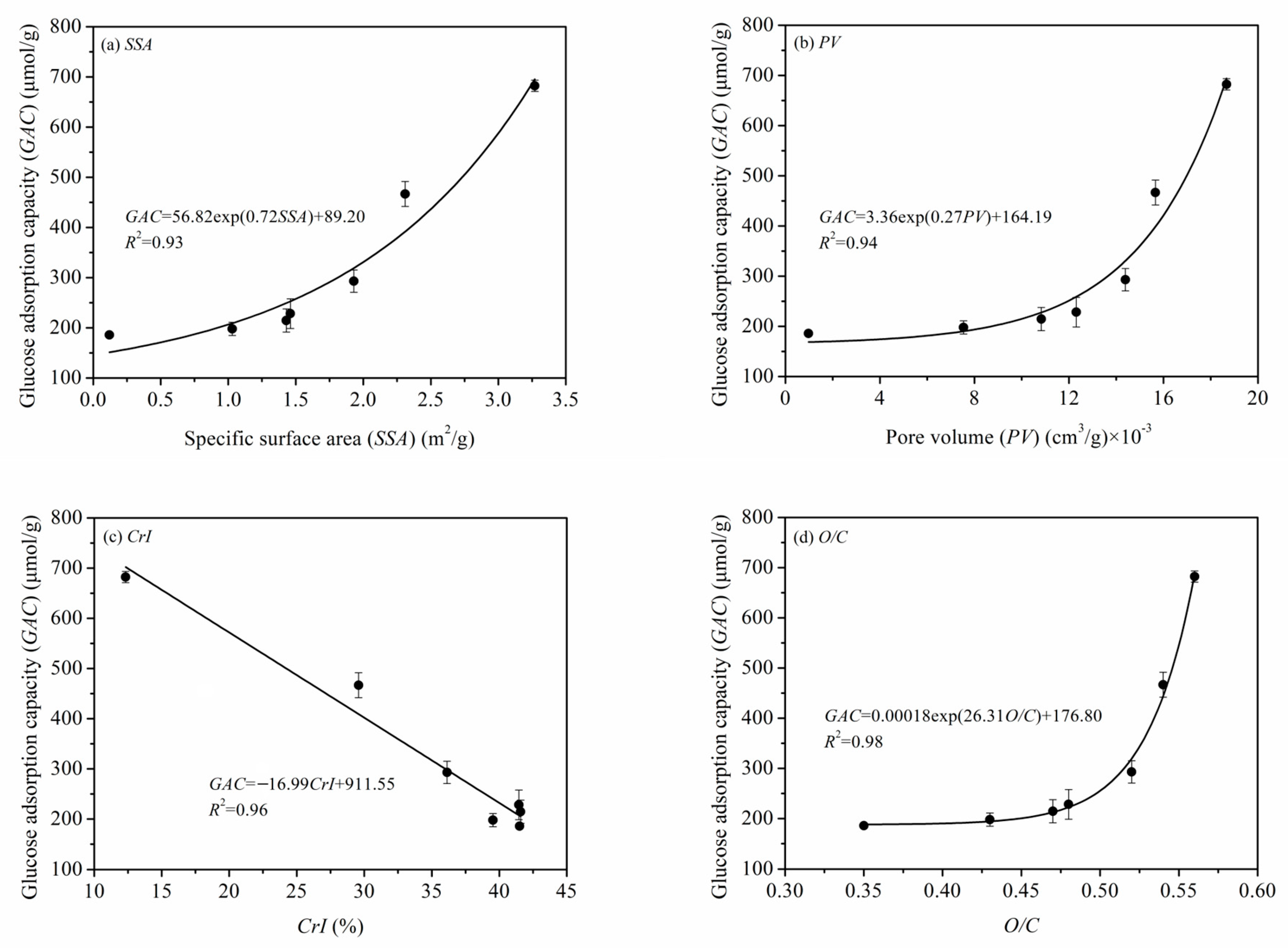
3.4.2. Quantitative Relationships between the Microstructure Parameters and GAC
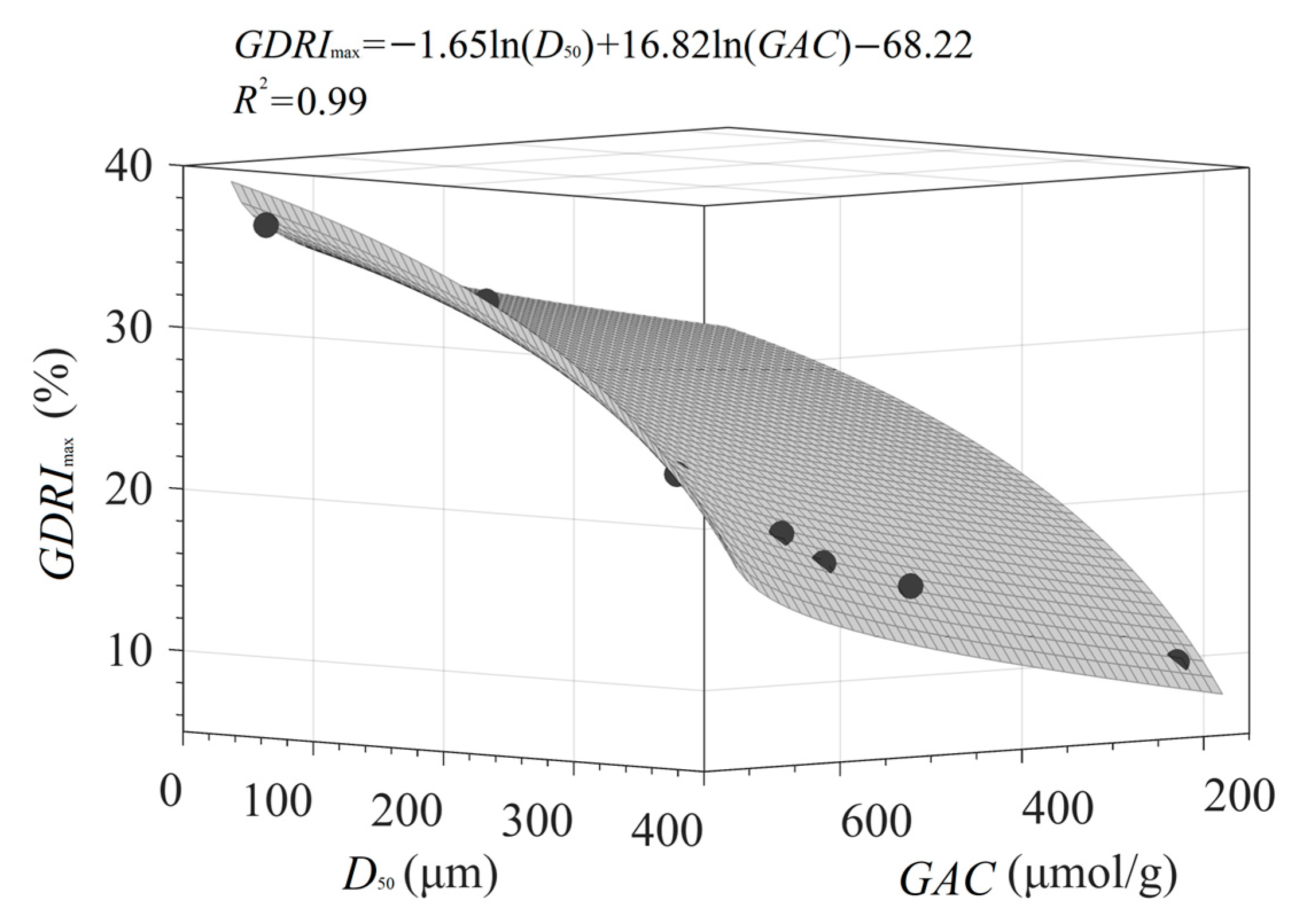
3.4.3. Quantitative Relationship of D50, GAC, and GDRImax
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martens, L.G.; Nilsen, M.M.; Provan, F. Pea hull fibre: Novel and sustainable fibre with important health and functional properties. EC Nutr. 2017, 10, 139–148. [Google Scholar]
- Hashemi, Z.; Fouhse, J.; Im, H.; Chan, C.; Willing, B. Dietary pea fiber supplementation improves glycemia and induces changes in the composition of gut microbiota, serum short chain fatty acid profile and expression of mucins in glucose intolerant rats. Nutrients 2017, 9, 1236. [Google Scholar] [CrossRef]
- Jia, M.; Yu, Q.; Chen, J.; He, Z.; Chen, Y.; Xie, J.; Nie, S.; Xie, M. Physical quality and in vitro starch digestibility of biscuits as affected by addition of soluble dietary fiber from defatted rice bran. Food Hydrocoll. 2020, 99, 105349. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Q.; Huang, J.; Fang, D.; Zhuang, W.; Luo, X.; Zou, X.; Zheng, B.; Cao, H. Hypoglycemic effect of dietary fibers from bamboo shoot shell: An in vitro and in vivo study. Food Chem. Toxicol. 2019, 127, 120–126. [Google Scholar] [CrossRef]
- Arun, K.B.; Thomas, S.; Reshmitha, T.R.; Akhil, G.C.; Nisha, P. Dietary fibre and phenolic-rich extracts from Musa paradisiaca inflorescence ameliorates type 2 diabetes and associated cardiovascular risks. J. Funct. Foods 2017, 31, 198–207. [Google Scholar] [CrossRef]
- Qi, J.; Li, Y.; Masamba, K.G.; Shoemaker, C.F.; Zhong, F.; Majeed, H.; Ma, J. The effect of chemical treatment on the In vitro hypoglycemic properties of rice bran insoluble dietary fiber. Food Hydrocoll. 2016, 52, 699–706. [Google Scholar] [CrossRef]
- Du, B.; Meenu, M.; Xu, B. Insights into improvement of physiochemical and biological properties of dietary fibers from different sources via micron technology. Food Rev. Int. 2020, 36, 367–383. [Google Scholar] [CrossRef]
- Ramachandraiah, K.; Chin, K.B. Evaluation of ball-milling time on the physicochemical and antioxidant properties of persimmon by-products powder. Innov. Food Sci. Emerg. 2016, 37, 115–124. [Google Scholar] [CrossRef]
- Barakat, A.; Monlau, F.; Solhy, A.; Carrere, H. Mechanical dissociation and fragmentation of lignocellulosic biomass: Effect of initial moisture, biochemical and structural proprieties on energy requirement. Appl. Energy 2015, 142, 240–246. [Google Scholar] [CrossRef]
- Barakat, A.; Mayer-Laigle, C.; Solhy, A.; Arancon, R.A.D.; de Vries, H.; Luque, R. Mechanical pretreatments of lignocellulosic biomass: Towards facile and environmentally sound technologies for biofuels production. RSC Adv. 2014, 4, 48109–48127. [Google Scholar] [CrossRef]
- Ji, G.; Gao, C.; Xiao, W.; Han, L. Mechanical fragmentation of corncob at different plant scales: Impact and mechanism on microstructure features and enzymatic hydrolysis. Bioresour. Technol. 2016, 205, 159–165. [Google Scholar] [CrossRef]
- Ji, G.; Han, L.; Gao, C.; Xiao, W.; Zhang, Y.; Cao, Y. Quantitative approaches for illustrating correlations among the mechanical fragmentation scales, crystallinity and enzymatic hydrolysis glucose yield of rice straw. Bioresour. Technol. 2017, 241, 262–268. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, L.; Lu, M.; Li, J.; Han, L. A novel film-pore-surface diffusion model to explain the enhanced enzyme adsorption of corn stover pretreated by ultrafine grinding. Biotechnol. Biofuels 2016, 9, 181. [Google Scholar] [CrossRef]
- Protonotariou, S.; Mandala, I.; Rosell, C.M. Jet milling effect on functionality, quality and in vitro digestibility of whole wheat flour and bread. Food Bioprocess Technol. 2015, 8, 1319–1329. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Liu, F.; Pan, S. Effect of grinding methods on structural, physicochemical, and functional properties of insoluble dietary fiber from orange peel. Int. J. Polym. Sci. 2016, 2016, 6269302. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Y. Physicochemical and functional properties of coconut (Cocos nucifera L.) cake dietary fibres: Effects of cellulase hydrolysis, acid treatment and particle size distribution. Food Chem. 2018, 257, 135–142. [Google Scholar] [CrossRef]
- Srichamroen, A.; Chavasit, V. In vitro retardation of glucose diffusion with gum extracted from malva nut seeds produced in Thailand. Food Chem. 2011, 127, 455–460. [Google Scholar] [CrossRef]
- Chen, H.; Li, J.; Yao, R.; Yan, S.; Wang, Q. Mechanism of lipid metabolism regulation by soluble dietary fibre from micronized and non-micronized powders of lotus root nodes as revealed by their adsorption and activity inhibition of pancreatic lipase. Food Chem. 2020, 305, 125435. [Google Scholar] [CrossRef]
- Zhu, F.; Du, B.; Li, R.; Li, J. Effect of micronization technology on physicochemical and antioxidant properties of dietary fiber from buckwheat hulls. Biocatal. Agric. Biotechnol. 2014, 3, 30–34. [Google Scholar] [CrossRef]
- Chen, J.; Gao, D.; Yang, L.; Gao, Y. Effect of microfluidization process on the functional properties of insoluble dietary fiber. Food Res. Int. 2013, 54, 1821–1827. [Google Scholar] [CrossRef]
- Siqueira, G.; Arantes, V.; Saddler, J.N.; Ferraz, A.; Milagres, A.M.F. Limitation of cellulose accessibility and unproductive binding of cellulases by pretreated sugarcane bagasse lignin. Biotechnol. Biofuels 2017, 10, 176. [Google Scholar] [CrossRef]
- Silva, G.G.D.; Couturier, M.; Berrin, J.; Buléon, A.; Rouau, X. Effects of grinding processes on enzymatic degradation of wheat straw. Bioresour. Technol. 2012, 103, 192–200. [Google Scholar] [CrossRef]
- Liu, T.Y.; Ma, Y.; Yu, S.F.; Shi, J.; Xue, S. The effect of ball milling treatment on structure and porosity of maize starch granule. Innov. Food Sci. Emerg. 2011, 12, 586–593. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Segal, L.; Creely, J.J.; Martin, A.E.; Conrad, C.M. An empirical method for estimating the degree of crystallinity of native cellulose using the X-Ray diffractometer. Text. Res. J. 1959, 29, 786–794. [Google Scholar] [CrossRef]
- Ji, G.; Xiao, W.; Gao, C.; Cao, Y.; Zhang, Y.; Han, L. Mechanical fragmentation of wheat and rice straw at different scales: Energy requirement in relation to microstructure properties and enzymatic hydrolysis. Energy Convers. Manag. 2018, 171, 38–47. [Google Scholar] [CrossRef]
- Ou, S.; Kwok, K.; Li, Y.; Fu, L. In vitro study of possible role of dietary fiber in lowering postprandial serum glucose. J. Agric. Food Chem. 2001, 49, 1026–1029. [Google Scholar] [CrossRef]
- Arantes, V.; Saddler, J.N. Cellulose accessibility limits the effectiveness of minimum cellulase loading on the efficient hydrolysis of pretreated lignocellulosic substrates. Biotechnol. Biofuels 2011, 4, 3–20. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, L.; Li, J.; Lu, M.; Han, L. Quantitative characterization of enzyme adsorption and hydrolytic performance for ultrafine grinding pretreated corn stover. Bioresour. Technol. 2017, 234, 23–32. [Google Scholar] [CrossRef]
- Li, H.; Ye, C.; Liu, K.; Gu, H.; Du, W.; Bao, J. Analysis of particle size reduction on overall surface area and enzymatic hydrolysis yield of corn stover. Bioprocess Biosyst. Eng. 2015, 38, 149–154. [Google Scholar] [CrossRef]
- Tumuluru, J.S.; Tabil, L.G.; Song, Y.; Iroba, K.L.; Meda, V. Grinding energy and physical properties of chopped and hammer-milled barley, wheat, oat, and canola straws. Biomass Bioenergy 2014, 60, 58–67. [Google Scholar] [CrossRef]
- Park, S.; Baker, J.O.; Himmel, M.E.; Parilla, P.A.; Johnson, D.K. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels Bioprod. 2010, 3, 10. [Google Scholar] [CrossRef]
- Peng, H.; Li, H.; Luo, H.; Xu, J. A novel combined pretreatment of ball milling and microwave irradiation for enhancing enzymatic hydrolysis of microcrystalline cellulose. Bioresour. Technol. 2013, 130, 81–87. [Google Scholar] [CrossRef]
- Yang, Y.; Ji, G.; Xiao, W.; Han, L. Changes to the physicochemical characteristics of wheat straw by mechanical ultrafine grinding. Cellulose 2014, 21, 3257–3268. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Zhu, J.Y.; Ralph, S.A. Enzymatic hydrolysis of loblolly pine: Effects of cellulose crystallinity and delignification. Holzforschung 2013, 67, 371–377. [Google Scholar] [CrossRef]
- Sain, M.; Panthapulakkal, S. Bioprocess preparation of wheat straw fibers and their characterization. Ind. Crop. Prod. 2006, 23, 1–8. [Google Scholar] [CrossRef]
- Chundawat, S.P.S.; Venkatesh, B.; Dale, B.E. Effect of particle size based separation of milled corn stover on AFEX pretreatment and enzymatic digestibility. Biotechnol. Bioeng. 2007, 96, 219–231. [Google Scholar] [CrossRef]
- Dorris, G.M.; Gray, D.G. The surface analysis of paper and wood fibers by ESCA II. Cell. Chem. Technol. 1978, 12, 721–734. [Google Scholar]
- Dorris, G.M.; Gray, D.G. The surface analysis of paper and wood fibers by ESCA I. Cell. Chem. Technol. 1978, 12, 9–23. [Google Scholar]
- Rjiba, N.; Nardin, M.; Dréan, J.Y.; Frydrych, R. A study of the surface properties of cotton fibers by inverse gas chromatography. J. Colloid Interface Sci. 2007, 314, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Inari, G.N.; Pétrissans, M.; Dumarcay, S.; Lambert, J.; Ehrhardt, J.J.; Šernek, M.; Gérardin, P. Limitation of XPS for analysis of wood species containing high amounts of lipophilic extractives. Wood Sci. Technol. 2011, 45, 369–382. [Google Scholar] [CrossRef]
- Hua, X.; Kaliaguine, S.; Kokta, B.V.; Adnot, A. Surface analysis of explosion pulps by ESCA Part 1. Carbon (1s) spectra and oxygen-to-carbon ratios. Wood Sci. Technol. 1993, 27, 449–459. [Google Scholar] [CrossRef]
- Djajadi, D.T.; Jensen, M.M.; Oliveira, M.; Jensen, A.; Thygesen, L.G.; Pinelo, M.; Glasius, M.; Jørgensen, H.; Meyer, A.S. Lignin from hydrothermally pretreated grass biomass retards enzymatic cellulose degradation by acting as a physical barrier rather than by inducing nonproductive adsorption of enzymes. Biotechnol. Biofuels 2018, 11, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.; Wang, Y.; Wen, Y. Different micronization methods significantly improve the functionality of carrot insoluble fibre. Food Chem. 2007, 100, 1402–1408. [Google Scholar] [CrossRef]
- López, G.; Ros, G.; Rincón, F.; Periago, M.J.; Martínez, M.C.; Ortuño, J. Relationship between physical and hydration properties of soluble and insoluble fiber of artichoke. J. Agric. Food Chem. 1996, 44, 2773–2778. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Z.; Gai, G.; Yang, Y. Effect of superfine grinding on properties of ginger powder. J. Food Eng. 2009, 91, 217–222. [Google Scholar] [CrossRef]
- Farrés, M.; Platikanov, S.; Tsakovski, S.; Tauler, R. Comparison of the variable importance in projection (VIP) and of the selectivity ratio (SR) methods for variable selection and interpretation. J. Chemom. 2015, 29, 528–536. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | D10 (μm) | D50 (μm) | D90 (μm) | Span |
|---|---|---|---|---|
| BM0 | 172.7 ± 0.2 g | 369.7 ± 2.9 g | 593.0 ± 0.8 g | 1.14 ± 0.01 a |
| BM30 | 63.8 ± 0.5 f | 173.6 ± 0.9 f | 294.2 ± 1.6 f | 1.33 ± 0.00 a |
| BM60 | 33.7 ± 0.7 e | 118.3 ± 0.2 e | 241.9 ± 0.7 e | 1.76 ± 0.01 b |
| BM100 | 30.5 ± 0.8 d | 95.5 ± 0.4 d | 190.9 ± 2.4 d | 1.68 ± 0.01 b |
| BM160 | 24.7 ± 0.0 c | 60.2 ± 0.1 c | 134.7 ± 1.1 c | 1.83 ± 0.01 b |
| BM240 | 7.1 ± 0.1 b | 35.2 ± 0.1 b | 85.2 ± 1.9 b | 2.22 ± 0.05 c |
| BM400 | 3.2 ± 0.5 a | 16.6 ± 0.3 a | 45.8 ± 3.2 a | 2.58 ± 0.21 d |
| Sample | Glucose Diffusion Equation | R2 | Vmax (μmol/min) | GDRImax (%) |
|---|---|---|---|---|
| Control | y = −0.0191x2 + 8.13x + 248.6 | 0.953 | 8.13 | / |
| BM0 | y = −0.0186x2 + 8.05x + 206.7 | 0.964 | 8.05 | 9.35 |
| BM30 | y = −0.0170x2 + 7.88x + 187.1 | 0.951 | 7.88 | 12.89 |
| BM60 | y = −0.0163x2 + 7.71x + 129.3 | 0.971 | 7.71 | 14.03 |
| BM100 | y = −0.0166x2 + 7.81x + 123.6 | 0.973 | 7.81 | 15.77 |
| BM160 | y = −0.0164x2 + 7.75x + 99.5 | 0.975 | 7.75 | 19.48 |
| BM240 | y = −0.0143x2 + 7.00x + 106.7 | 0.965 | 7.00 | 30.69 |
| BM400 | y = −0.0134x2 + 6.58x + 96.7 | 0.964 | 6.58 | 36.15 |
| D50 | SSA | PV | CrI | O/C | GAC | GDRImax | |
|---|---|---|---|---|---|---|---|
| D50 | 1 | ||||||
| SSA | −0.903 ** | 1 | |||||
| PV | −0.977 ** | 0.967 ** | 1 | ||||
| CrI | 0.573 | −0.865 * | −0.711 | 1 | |||
| O/C | −0.979 ** | 0.965 ** | 0.997 ** | −0.711 | 1 | ||
| GAC | −0.642 | 0.902 ** | 0.774 * | −0.984 ** | 0.773 * | 1 | |
| GDRImax | −0.761 * | 0.942 ** | 0.860 * | −0.927 ** | 0.866 * | 0.974 ** | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Liu, J.; Zhang, Y.; Wang, Q.; Wang, J. Qualitative and Quantitative Correlation of Microstructural Properties and In Vitro Glucose Adsorption and Diffusion Behaviors of Pea Insoluble Dietary Fiber Induced by Ultrafine Grinding. Foods 2022, 11, 2814. https://doi.org/10.3390/foods11182814
Li L, Liu J, Zhang Y, Wang Q, Wang J. Qualitative and Quantitative Correlation of Microstructural Properties and In Vitro Glucose Adsorption and Diffusion Behaviors of Pea Insoluble Dietary Fiber Induced by Ultrafine Grinding. Foods. 2022; 11(18):2814. https://doi.org/10.3390/foods11182814
Chicago/Turabian StyleLi, Lingyi, Jianfu Liu, Yang Zhang, Qian Wang, and Jinrong Wang. 2022. "Qualitative and Quantitative Correlation of Microstructural Properties and In Vitro Glucose Adsorption and Diffusion Behaviors of Pea Insoluble Dietary Fiber Induced by Ultrafine Grinding" Foods 11, no. 18: 2814. https://doi.org/10.3390/foods11182814
APA StyleLi, L., Liu, J., Zhang, Y., Wang, Q., & Wang, J. (2022). Qualitative and Quantitative Correlation of Microstructural Properties and In Vitro Glucose Adsorption and Diffusion Behaviors of Pea Insoluble Dietary Fiber Induced by Ultrafine Grinding. Foods, 11(18), 2814. https://doi.org/10.3390/foods11182814