
Combined Profiling of Transcriptome and DNA Methylome Reveal Genes Involved in Accumulation of Soluble Sugars and Organic Acid in Apple Fruits

 , , ,
, , ,  and
and 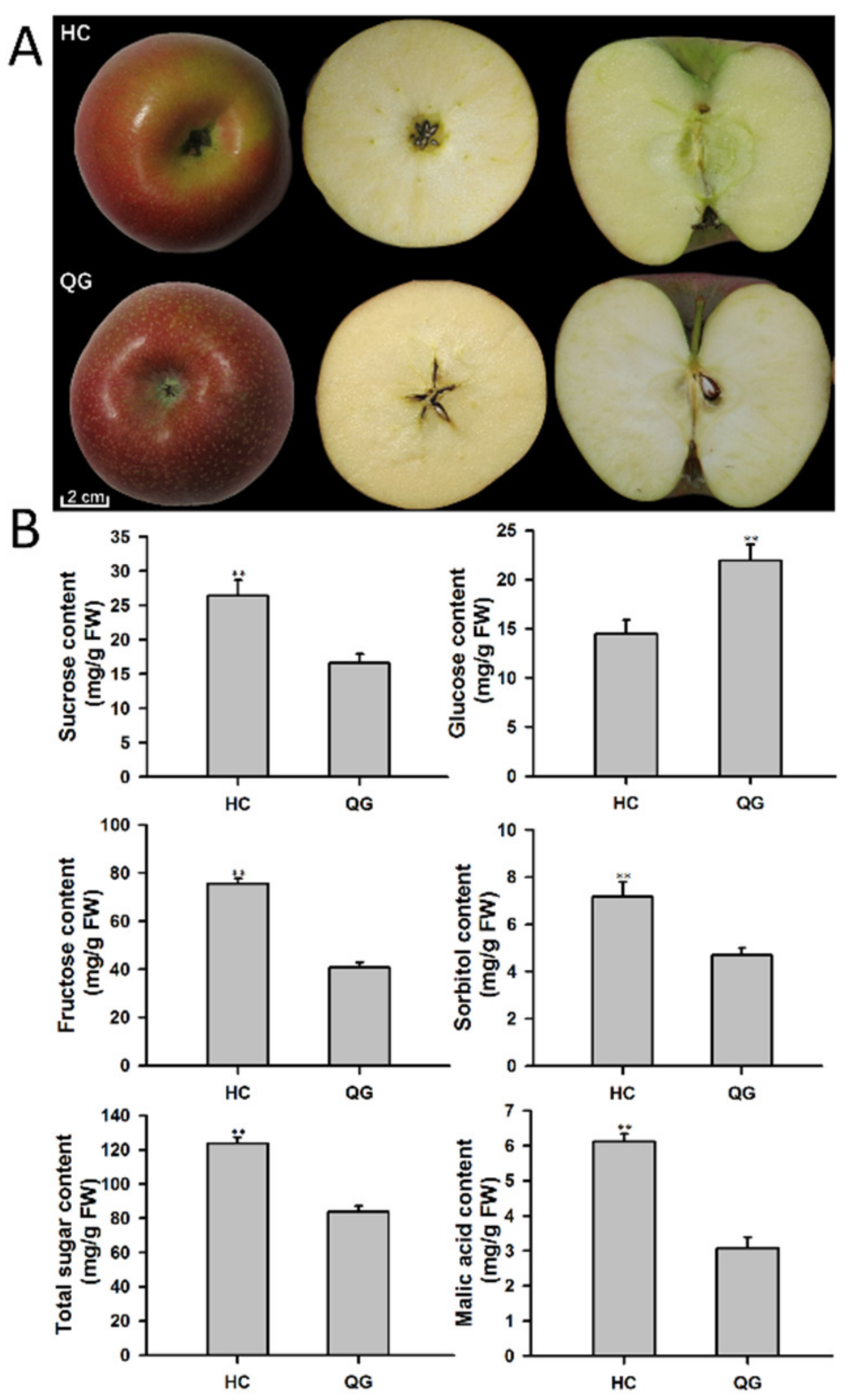{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatment
2.2. Measurement of Organic Acid and Soluble Sugar Contents
2.3. Bisulfite Sequencing (BS) Library Construction and Data Analysis
2.4. RNA Sequencing (RNA-Seq) and Identification of Differentially Expressed Genes (DEGs)
2.5. Subcellular Localization of MdTSTa and MdMa11 in N. benthamiana Leaf Epidermal Cells
2.6. Functional Analysis of MdTSTa and MdMa11
3. Results
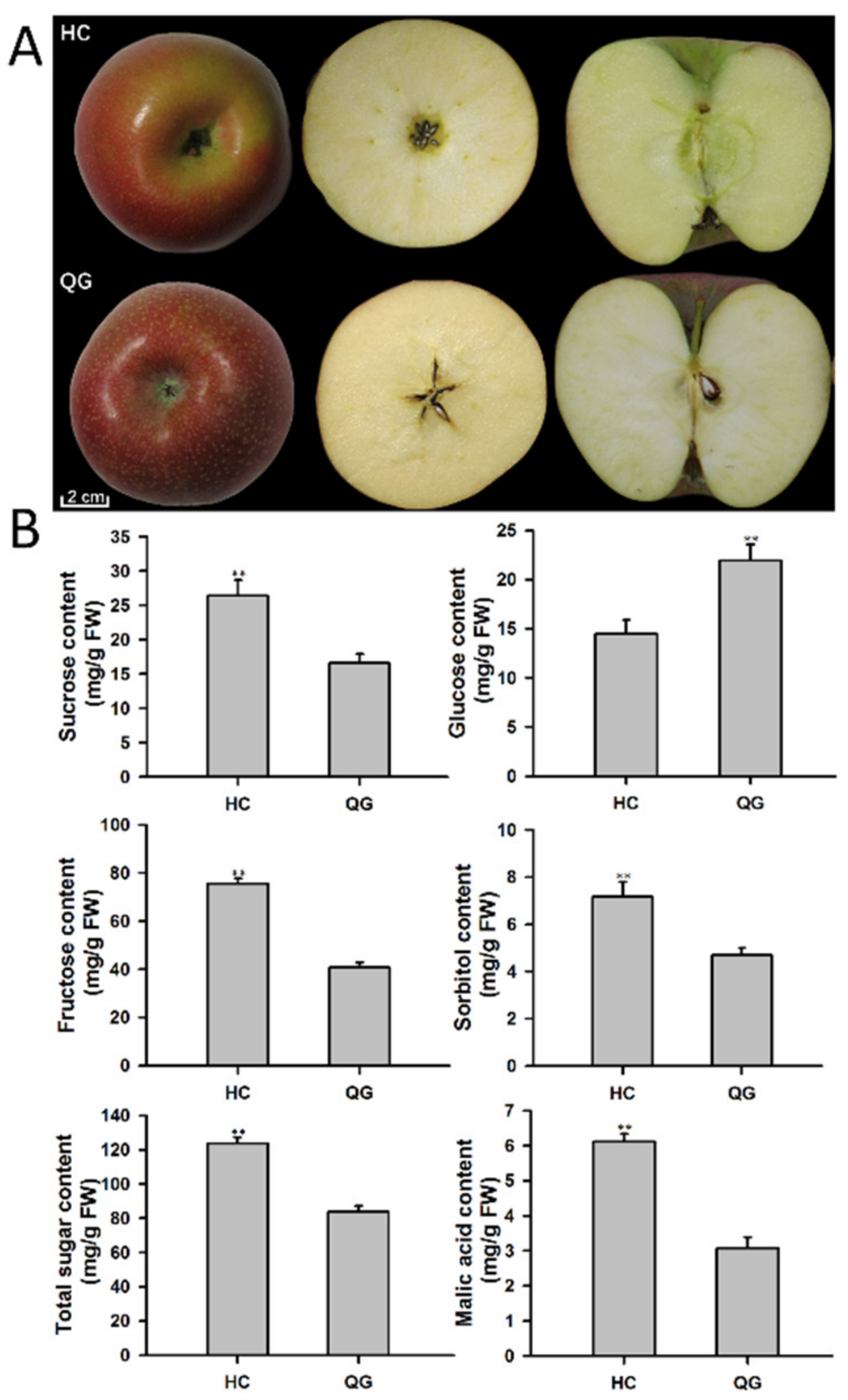
3.1. Phenotypic Difference between ‘HC’ and ‘QG’ Mature Apple Fruits
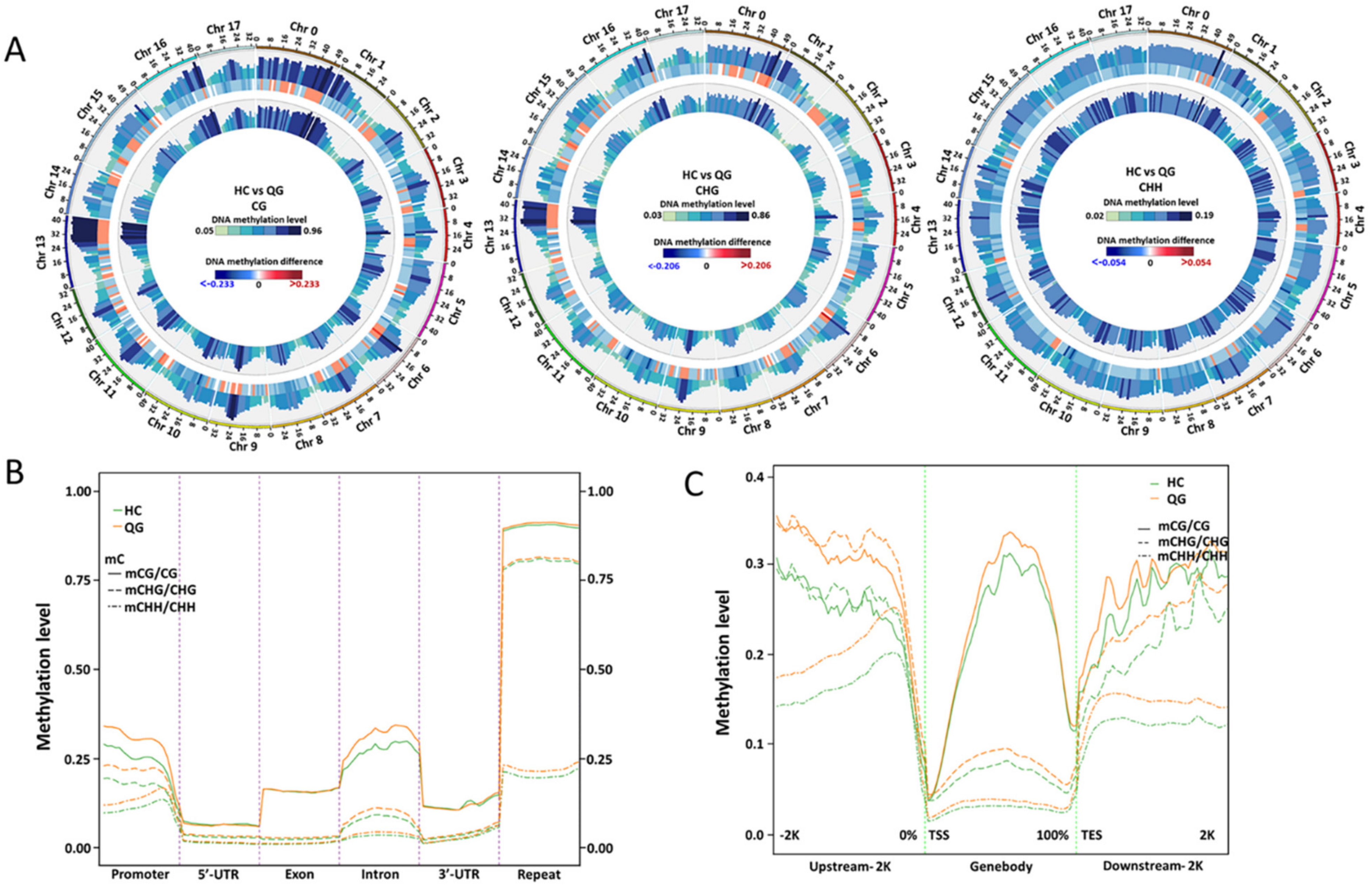
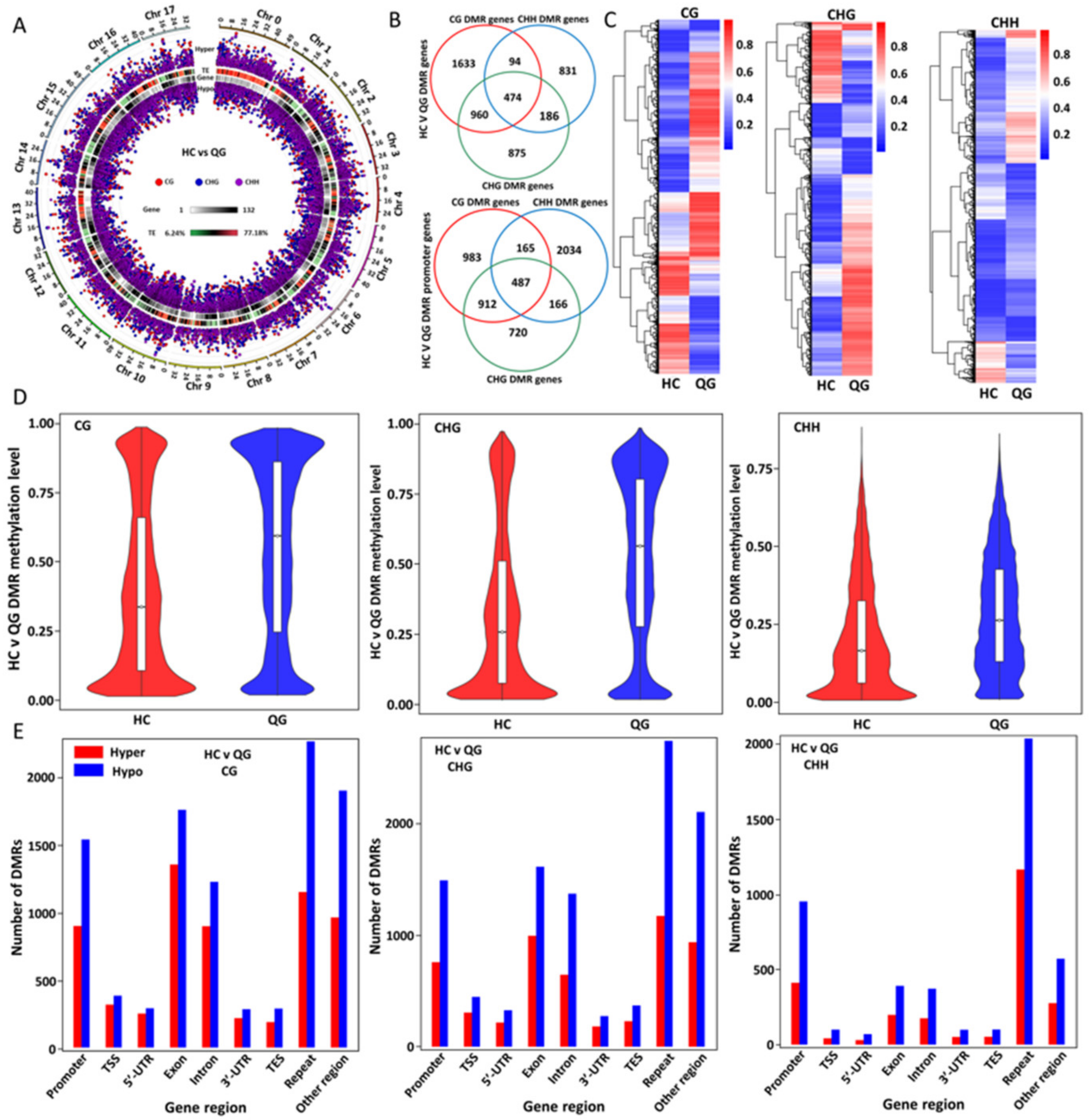
3.2. Genome-Wide Methylation Profiles in ‘HC’ and ‘QG’ Mature Fruits
3.3. Distribution of DNA Methylation among Gene Features and TEs
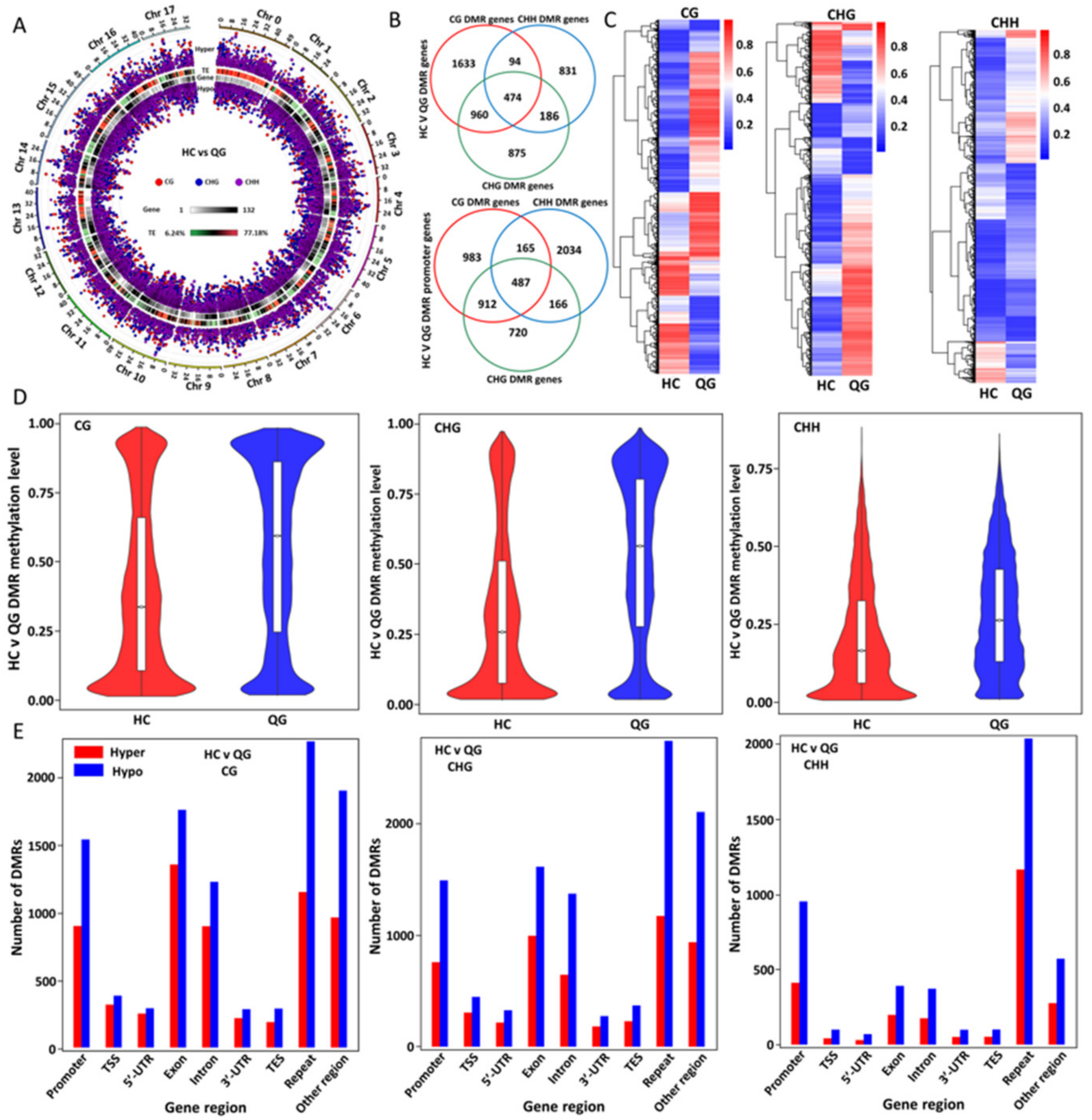
3.4. Identification and Analyses of the Distribution and Functions of the Differentially Methylated Regions (DMRs) between ‘HC’ and ‘QG’ Mature Fruits
3.5. Analysis of the Association between the Transcriptome and DNA Methylation
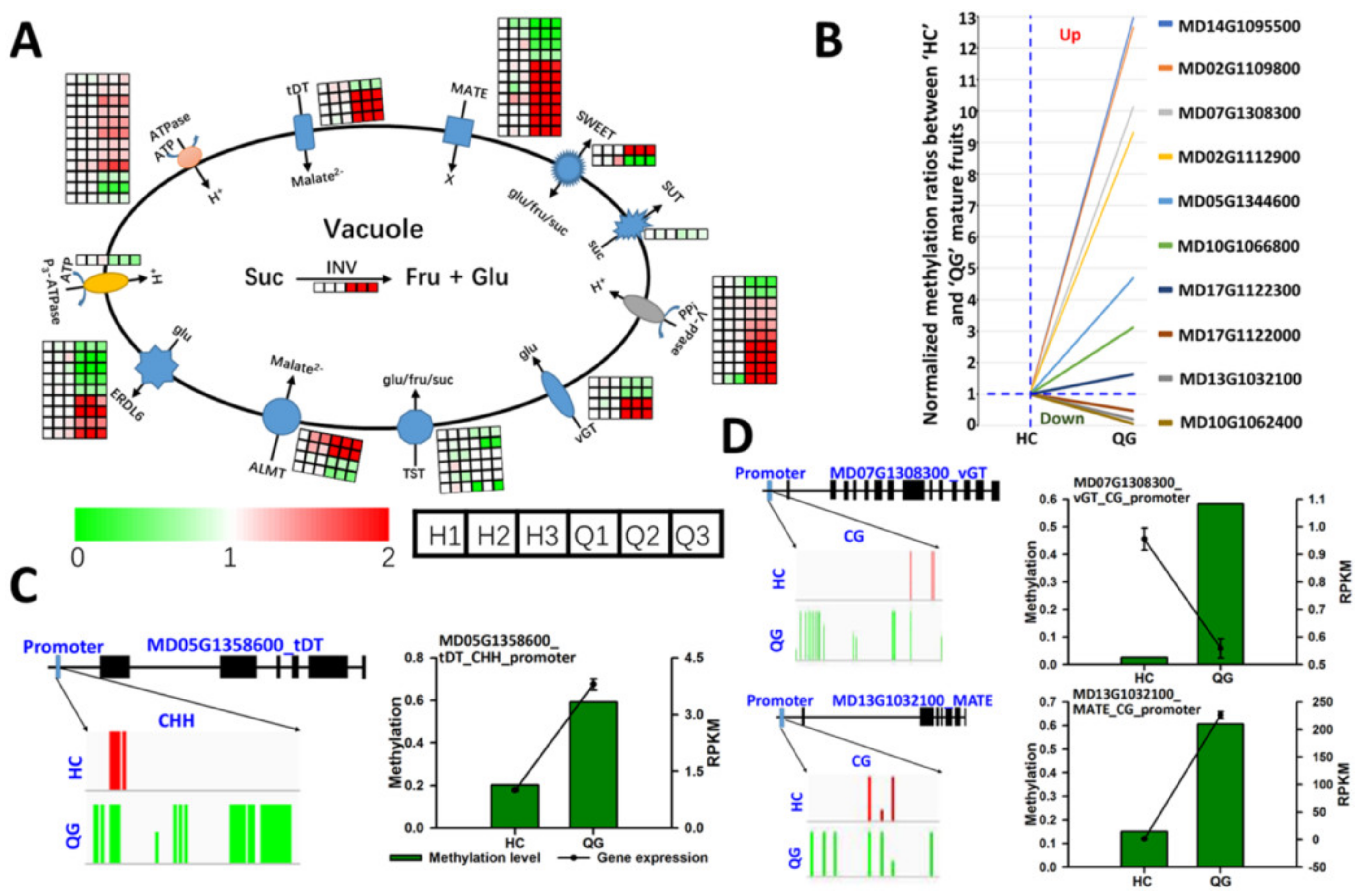
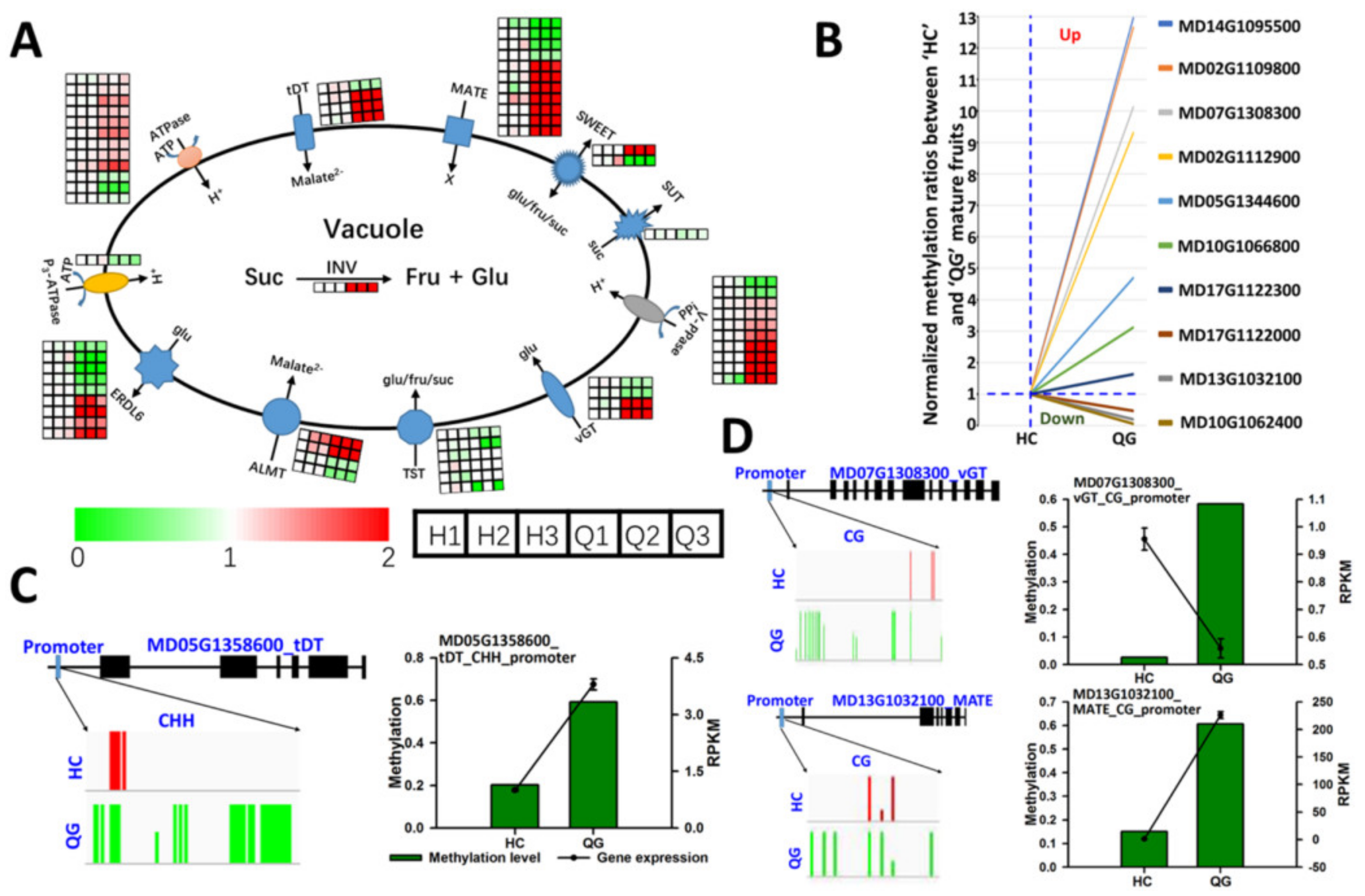
3.6. Differences in the Expression and Methylation Levels of Genes Involved in Soluble Sugar and Organic Acid Metabolism between ‘HC’ and ‘QG’ Mature Fruits
3.7. Vacuolar Transporter Genes: Differences in the Expression and Methylation Levels between ‘HC’ and ‘QG’ Mature Fruits
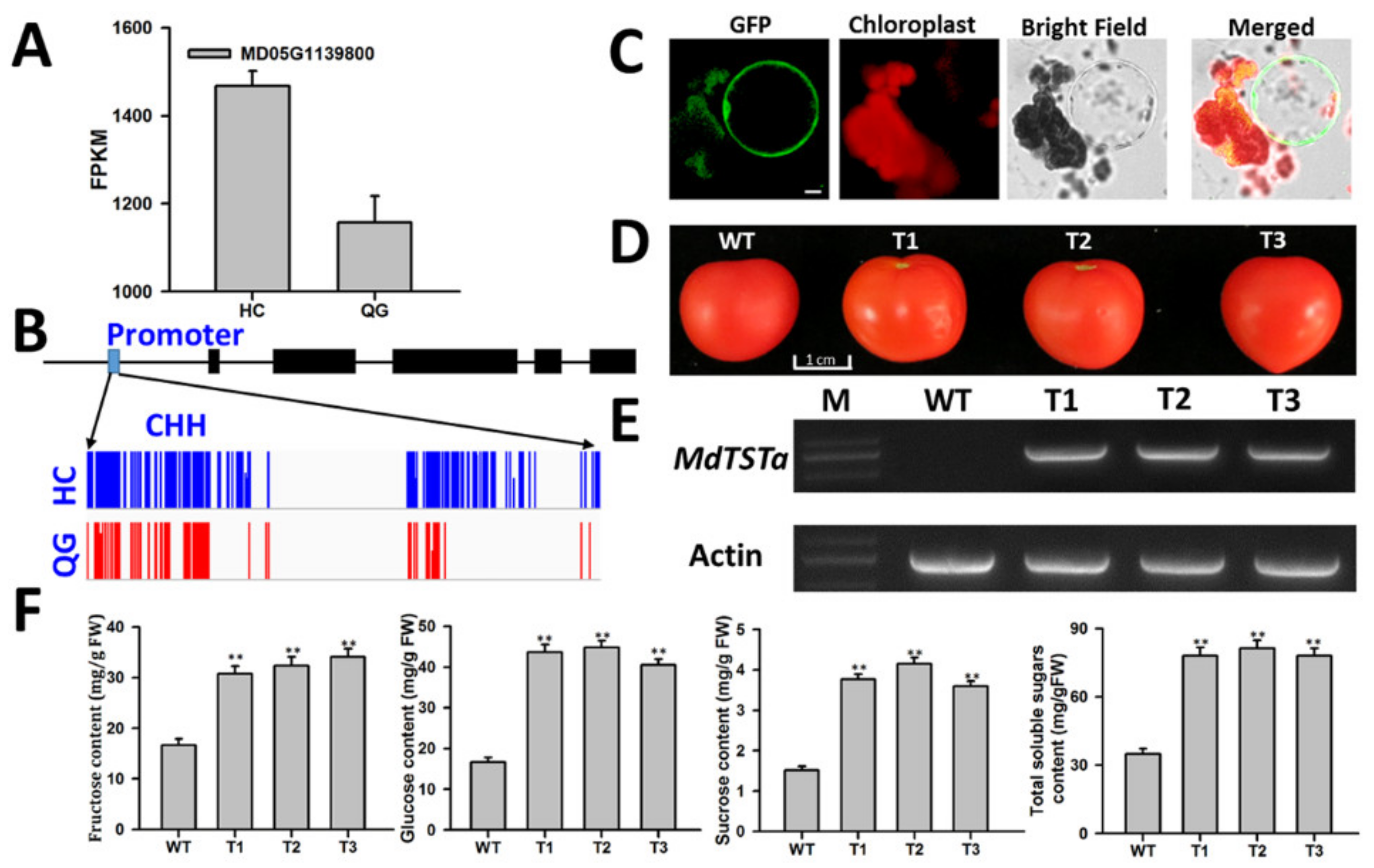
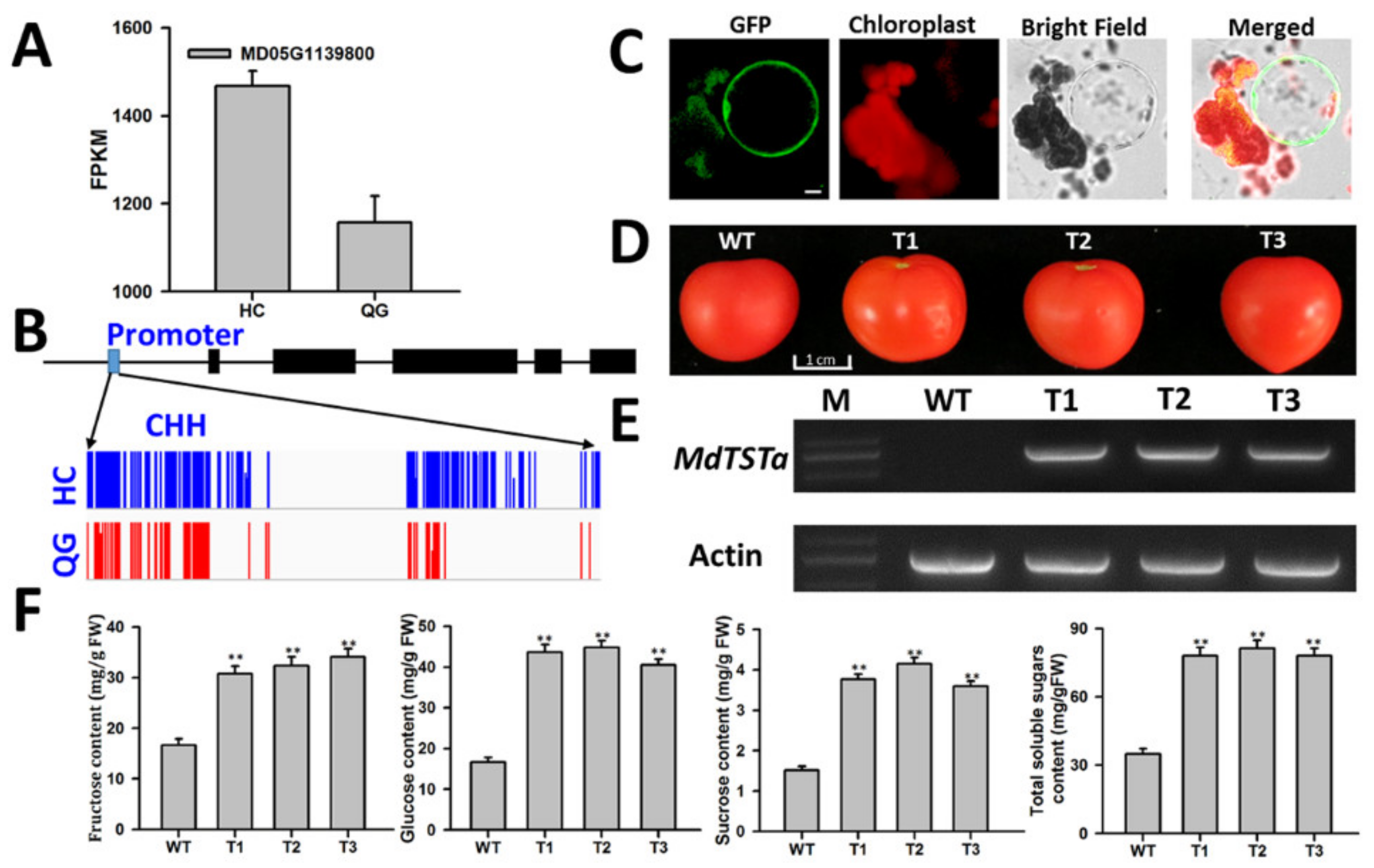
3.8. Identification and Functional Analysis of Genes Involved in Soluble Sugar Accumulation
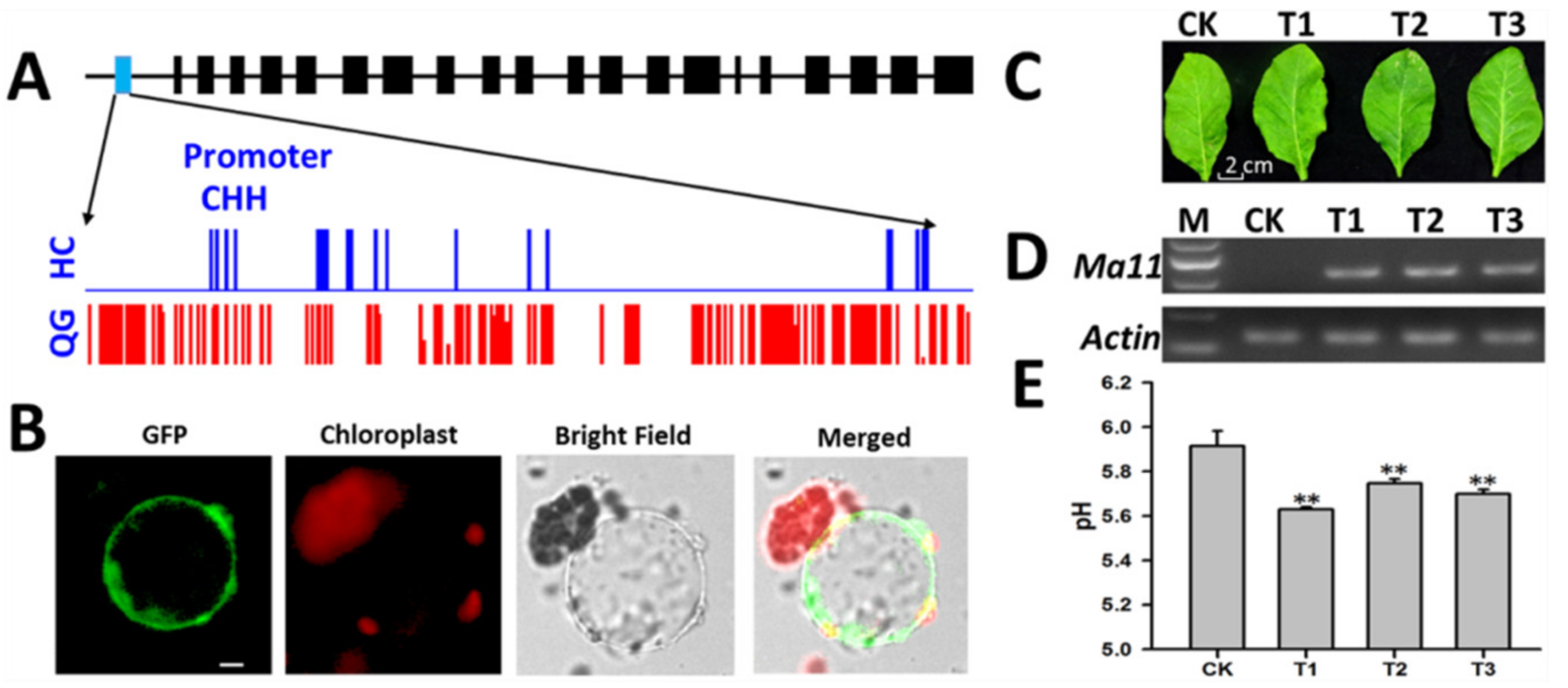
3.9. Identification of a Candidate Gene Affecting Vacuolar Acidity
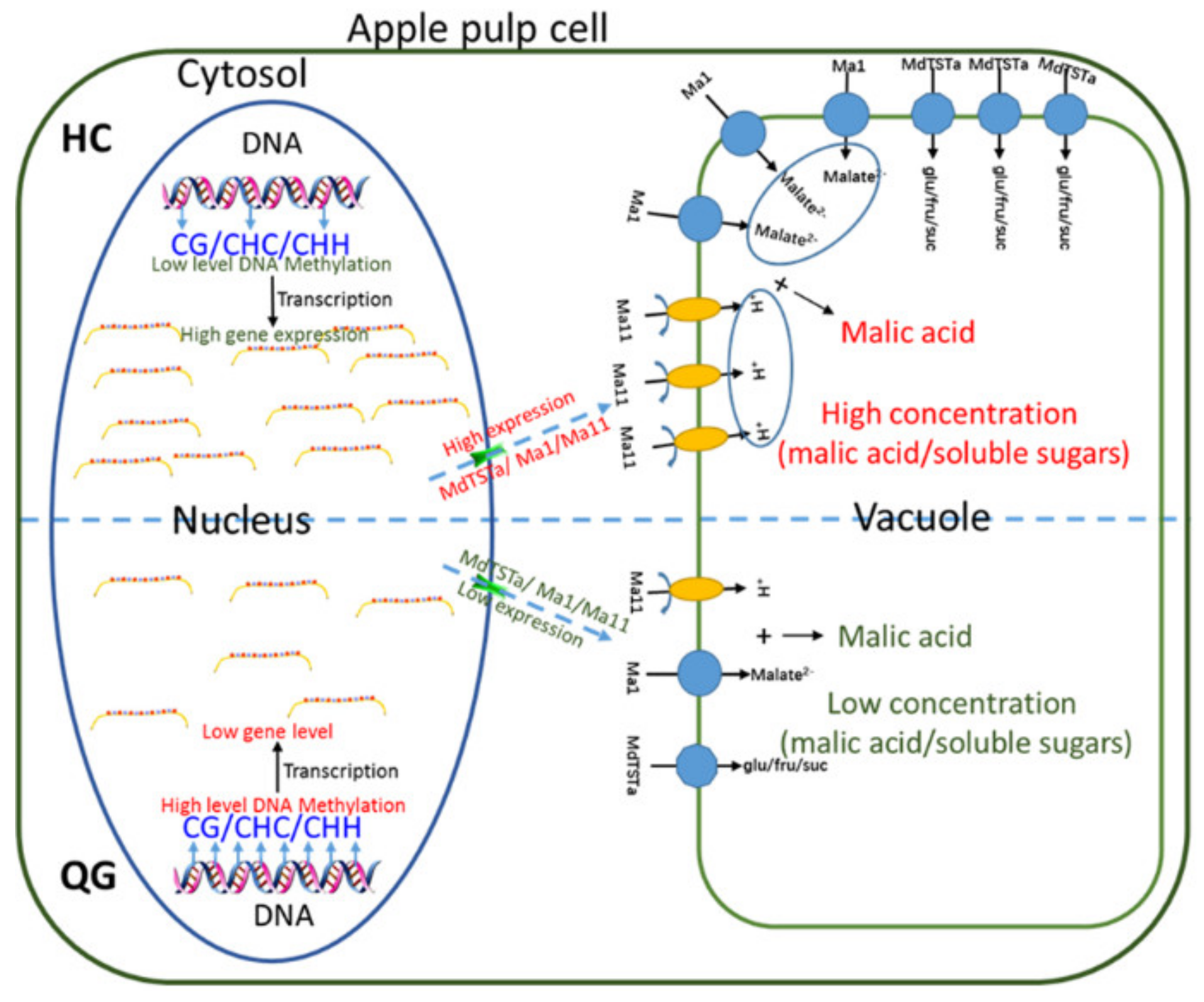
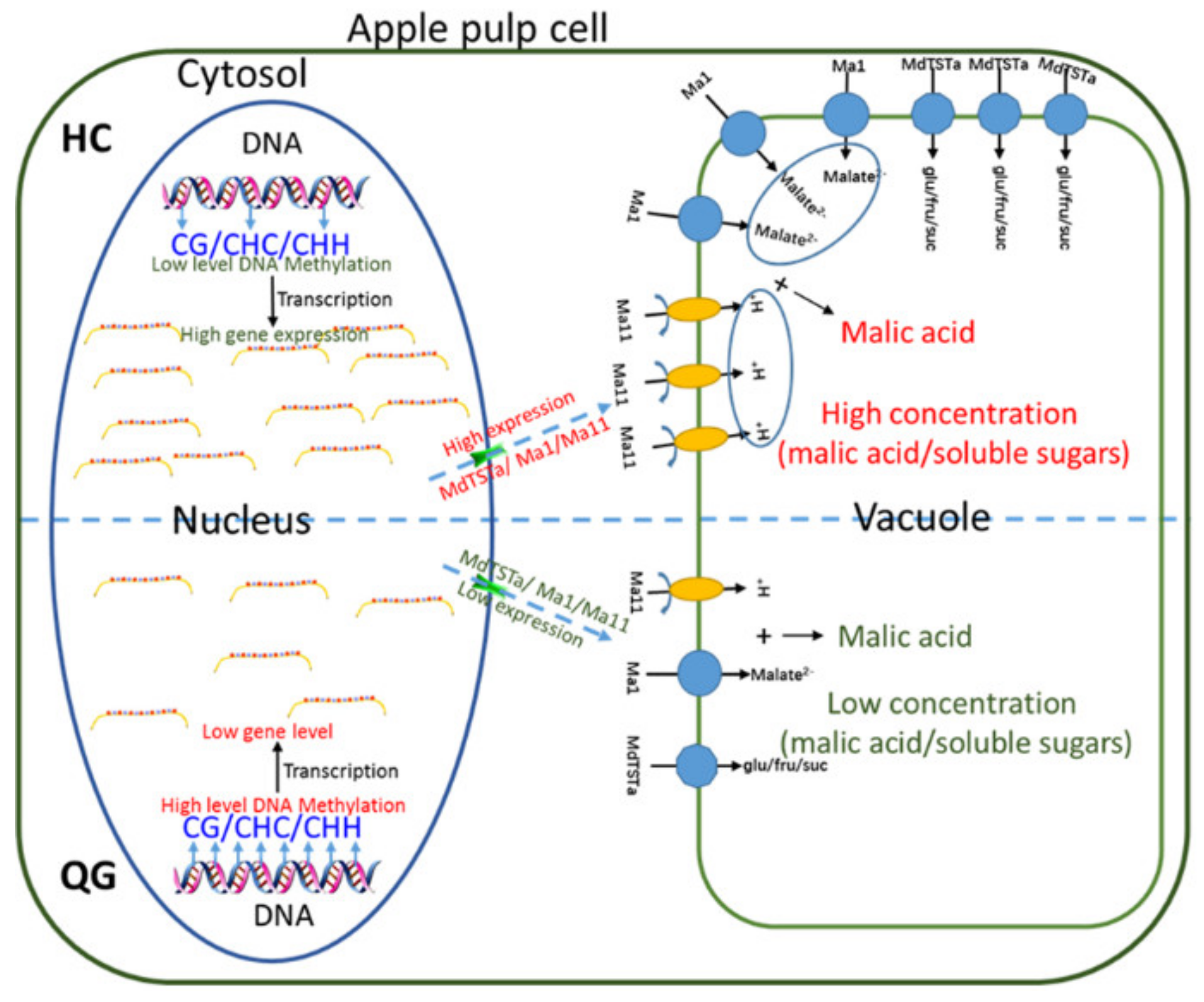
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borsani, J.; Budde, C.O.; Porrini, L.; Lauxmann, M.; Lombardo, V.A.; Murray, R.; Andreo, C.S.; Drincovich, M.F.; Lara, M.V. Carbon metabolism of peach fruit after harvest: Changes in enzymes involved in organic acid and sugar level modifications. J. Exp. Bot. 2009, 60, 1823–1837. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Chen, J.; Zheng, H.; Fang, T.; Ogutu, C.; Li, S.; Han, Y.; Wu, B. Comparative assessment of sugar and malic acid composition in cultivated and wild apples. Food Chem. 2015, 172, 86–91. [Google Scholar] [CrossRef]
- Doty, T.E. Fructose sweetness: A new dimension. Cereal Foods World 1976, 21, 62–63. [Google Scholar]
- Kulp, K.; Lorenz, K.; Stone, M. Functionality of carbohydrates ingredients in bakery products. Food Technol. 1991, 45, 136–142. [Google Scholar]
- Sheikh, M.; Vasanthaiah, H.K.; Kambiranda, D.; Easwaran, K.; Queeley, G. Genetic variation in sugar composition among muscadine, Florida hybrid bunch and bunch grape genotypes. Int. J. Wine Res. 2012, 4, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Patocka, J.; Bhardwaj, K.; Klimova, B.; Nepovimova, E.; Wu, Q.; Landi, M.; Kuca, K.; Valis, M.; Wu, W. Malus domestica: A Review on Nutritional Features, Chemical Composition, Traditional and Medicinal Value. Plants 2020, 9, 1408. [Google Scholar] [CrossRef]
- Li, M.; Li, P.; Ma, F.; Dandekar, A.M.; Cheng, L. Sugar metabolism and accumulation in thefruit of transgenic apple trees withdecreased sorbitol synthesis. Hortic. Res. 2018, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Cai, Y.; Lai, E.; Nakamura, M.; Liao, L.; Zheng, B.; Ogutu, C.; Cherono, S.; Han, Y. The sucrose transporter MdSUT4.1 participates in the regulation of fruit sugar accumulation in apple. BMC Plant Biol. 2020, 20, 1–14. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, X.; Yang, J.; Li, H.; Ma, B.; Zhang, K.; Zhang, Y.; Cheng, L.; Ma, F.; Li, M. Heterologous expression of the apple hexose transporter MdHT2.2 altered sugar concentration with increasing cell wall invertase activity in tomato fruit. Plant Biotechnol. J. 2019, 18, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Quoc, B.; Foyer, C. A role for ‘futile cycles’ involving invertase and sucrose synthase in sucrose metabolism of tomato fruit. J. Exp. Bot. 2001, 52, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Sweetman, C.; Deluc, L.G.; Cramer, G.R.; Ford, C.M.; Soole, K.L. Regulation of malate metabolism in grape berry and other developing fruits. Phytochemistry 2009, 70, 1329–1344. [Google Scholar] [CrossRef]
- Etienne, A.; Génard, M.; Lobit, P.; Mbeguié-A-Mbéguié, D.; Bugaud, C. What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J. Exp. Bot. 2013, 64, 1451–1469. [Google Scholar] [CrossRef] [Green Version]
- Etxeberria, E.; Pozueta-Romero, J.; Gonzalez, P. In and out of the plant storage vacuole. Plant Sci. 2012, 190, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Martinoia, E. Vacuolar Transporters—Companions on a Longtime Journey. Plant Physiol. 2018, 176, 1384–1407. [Google Scholar] [CrossRef]
- Peng, Q.; Wang, L.; Ogutu, C.; Liu, J.; Liu, L.; Mollah, M.D.A.; Han, Y. Functional Analysis Reveals the Regulatory Role of PpTST1 Encoding Tonoplast Sugar Transporter in Sugar Accumulation of Peach Fruit. Int. J. Mol. Sci. 2020, 21, 1112. [Google Scholar] [CrossRef] [Green Version]
- Poschet, G.; Hannich, B.; Raab, S.; Jungkunz, I.; Klemens, P.A.; Krueger, S.; Wic, S.; Neuhaus, H.E.; Büttner, M. A Novel Arabidopsis Vacuolar Glucose Exporter Is Involved in Cellular Sugar Homeostasis and Affects the Composition of Seed Storage Compounds. Plant Physiol. 2011, 157, 1664–1676. [Google Scholar] [CrossRef] [Green Version]
- Chardon, F.; Bedu, M.; Calenge, F.; Klemens, P.A.; Spinner, L.; Clement, G.; Chietera, G.; Léran, S.; Ferrand, M.; Lacombe, B.; et al. Leaf Fructose Content Is Controlled by the Vacuolar Transporter SWEET17 in Arabidopsis. Curr. Biol. 2013, 23, 697–702. [Google Scholar] [CrossRef]
- Zhu, L.; Li, B.; Wu, L.; Li, H.; Wang, Z.; Wei, X.; Ma, B.; Zhang, Y.; Ma, F.; Ruan, Y.-L.; et al. MdERDL6-mediated glucose efflux to the cytosol promotes sugar accumulation in the vacuole through up-regulating TSTs in apple and tomato. Proc. Natl. Acad. Sci. USA 2020, 118, e2022788118. [Google Scholar] [CrossRef]
- Ma, B.; Liao, L.; Zheng, H.; Chen, J.; Wu, B.; Ogutu, C.; Li, S.H.; Korban, S.S.; Han, Y. Genes encoding aluminum-activated malate transporter II and their association with fruit acidity in apple. Plant Genome 2015, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-L.; Dougherty, L.; Coluccio, A.E.; Meng, D.; El-Sharkawy, I.; Borejsza-Wysocka, E.; Liang, D.; Piñeros, M.A.; Xu, K.; Cheng, L. Apple ALMT9 Requires a Conserved C-Terminal Domain for Malate Transport Underlying Fruit Acidity. Plant Physiol. 2019, 182, 992–1006. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Shen, F.; Wang, Y.; Wu, T.; Xu, X.; Zhang, X.; Han, Z. Apple fruit acidity is genetically diversified by natural variations in three hierarchical epistatic genes: MdSAUR37, MdPP2CH and MdALMTII. Plant J. 2018, 95, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Liao, L.; Fang, T.; Peng, Q.; Ogutu, C.; Zhou, H.; Ma, F.; Han, Y. A Ma10 gene encoding P-type ATPase is involved in fruit organic acid accumulation in apple. Plant Biotechnol. J. 2019, 17, 674–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.G.; Sun, C.H.; Ma, Q.J.; You, C.Y.; Cheng, L.; Hao, Y.J. MdMYB1 Regulates Anthocyanin and Malate Accumulation by Directly Facilitating Their Transport into Vacuoles in Apples. Plant Physiol. 2016, 170, 1315–1330. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.-G.; Li, Y.-Y.; Zhang, Q.-Y.; Li, M.; Sun, C.-H.; Yu, J.-Q.; Hao, Y.-J. The R2R3-MYB transcription factor MdMYB73 is involved in malate accumulation and vacuolar acidification in apple. Plant J. 2017, 91, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shi, Y.; Chang, X.; Jing, S.; Zhang, Q.; You, C.; Yuan, H.; Wang, H. DNA methylome analysis provides evidence that the expansion of the tea genome is linked to TE bursts. Plant Biotechnol. J. 2019, 17, 826–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schob, H.; Grossniklaus, U. The first high-resolution DNA “methylome”. Cell 2006, 126, 1025–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.W.-L.; Henderson, I.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Stroud, H.; Greenberg, M.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive Analysis of Silencing Mutants Reveals Complex Regulation of the Arabidopsis Methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.M.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2013, 21, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzke, M.A.; Mosher, R. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Qi, S.; Zhou, H.; Zhang, W.; Zhang, C.; Ma, W.; Zhang, Q.; Shah, K.; Han, M.; Zhao, J. Epigenomic Regulatory Mechanism in Vegetative Phase Transitionof Malus hupehensis. J. Agric. Food Chem. 2020, 68, 4812–4829. [Google Scholar] [CrossRef]
- Li, J.; Wang, M.; Li, Y.; Zhang, Q.; Lindsey, K.; Daniell, H.; Jin, S.; Zhang, X. Multi-omics analyses reveal epigenomics basis for cotton somatic embryogenesis through successive regeneration acclimation process. Plant Biotechnol. J. 2018, 17, 435–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.-F.; Ning, G.-X.; Mao, J.; Guo, Z.-G.; Zhou, Q.; Chen, B.-H. Whole-genome DNA methylation patterns and complex associations with gene expression associated with anthocyanin biosynthesis in apple fruit skin. Planta 2019, 250, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Sun, Q.-G.; Chen, M.; Wang, N.; Xu, H.-F.; Fang, H.-C.; Wang, Y.-C.; Zhang, Z.-Y.; Chen, X.-S. Methylome and transcriptome analyses of apple fruit somatic mutations reveal the difference of red phenotype. BMC Genom. 2019, 20, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.B.; Lougheed, E.C.; Franklin, E.W.; McMillan, I. The starch iodine test for determining stage of maturation in apples. Can. J. Plant Sci. 1979, 59, 725–735. [Google Scholar] [CrossRef]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jiang, X.; Zhao, L.; Wang, F.; Liu, Y.; Zhou, H.; He, H.; Han, Y. A candidate PpRPH gene of the D locus con-trolling fruit acidity in peach. Plant Mol. Biol. 2021, 105, 321–332. [Google Scholar] [CrossRef]
- Bugaud, C.; Deverge, E.; Daribo, M.O.; Ribeyre, F.; Fils-Lycaon, B.; Mbéguié-A-Mbéguié, D. Sensory characterisation enabled the first classification of dessert bananas. J. Sci. Food Agric. 2011, 91, 992–1000. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; Van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2017, 16, 672–687. [Google Scholar] [CrossRef]
- Fan, S.; Gao, X.; Gao, C.; Yang, Y.; Zhu, X.; Feng, W.; Li, R.; Tahir, M.M.; Zhang, D.; Han, M.; et al. Dynamic Cytosine DNA Methylation Patterns Associated with mRNA and siRNA Expression Profiles in Alternate Bearing Apple Trees. J. Agric. Food Chem. 2019, 67, 5250–5264. [Google Scholar] [CrossRef]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Domcke, S.; Bardet, A.F.; Ginno, P.A.; Hartl, D.; Burger, L.; Schübeler, D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 2015, 528, 575–579. [Google Scholar] [CrossRef]
- Takuno, S.; Gaut, B.S. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl. Acad. Sci. USA 2013, 110, 1797–1802. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Yang, X.; Sun, W.; Miao, Y.; He, L.; Zhang, X. The Calcium Sensor CBL2 and Its Interacting Kinase CIPK6 Are Involved in Plant Sugar Homeostasis via Interacting with Tonoplast Sugar Transporter TST2. Plant Physiol. 2020, 183, 236–249. [Google Scholar] [CrossRef]
- Ma, B.; Yuan, Y.; Gao, M.; Li, C.; Ogutu, C.; Li, M.; Ma, F. Determination of Predominant Organic Acid Components in Malus Species: Correlation with Apple Domestication. Metabolites 2018, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Chandler, V.L.; Stam, M. Chromatin conversations: Mechanisms and implications of paramutation. Nat. Rev. Genet. 2004, 5, 532–544. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Li, B.; Zheng, L.; Peng, Y.; Tian, R.; Yuan, Y.; Zhu, L.; Su, J.; Ma, F.; Li, M.; et al. Combined Profiling of Transcriptome and DNA Methylome Reveal Genes Involved in Accumulation of Soluble Sugars and Organic Acid in Apple Fruits. Foods 2021, 10, 2198. https://doi.org/10.3390/foods10092198
Ma W, Li B, Zheng L, Peng Y, Tian R, Yuan Y, Zhu L, Su J, Ma F, Li M, et al. Combined Profiling of Transcriptome and DNA Methylome Reveal Genes Involved in Accumulation of Soluble Sugars and Organic Acid in Apple Fruits. Foods. 2021; 10(9):2198. https://doi.org/10.3390/foods10092198
Chicago/Turabian StyleMa, Wenfang, Baiyun Li, Litong Zheng, Yunjing Peng, Rui Tian, Yangyang Yuan, Lingcheng Zhu, Jing Su, Fengwang Ma, Mingjun Li, and et al. 2021. "Combined Profiling of Transcriptome and DNA Methylome Reveal Genes Involved in Accumulation of Soluble Sugars and Organic Acid in Apple Fruits" Foods 10, no. 9: 2198. https://doi.org/10.3390/foods10092198
APA StyleMa, W., Li, B., Zheng, L., Peng, Y., Tian, R., Yuan, Y., Zhu, L., Su, J., Ma, F., Li, M., & Ma, B. (2021). Combined Profiling of Transcriptome and DNA Methylome Reveal Genes Involved in Accumulation of Soluble Sugars and Organic Acid in Apple Fruits. Foods, 10(9), 2198. https://doi.org/10.3390/foods10092198