
Phase II Metabolism of Asarone Isomers In Vitro and in Humans Using HPLC-MS/MS and HPLC-qToF/MS


Abstract
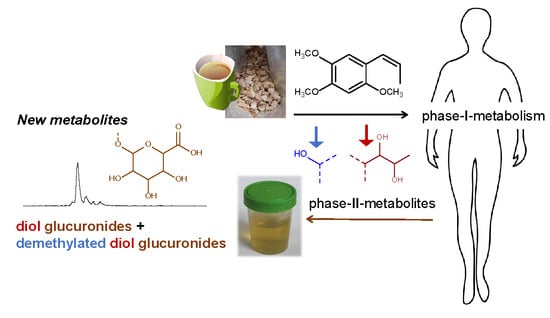
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. In Vitro Experiments
Sample Preparation
2.3. Human Study
2.3.1. Study Conditions and Subjects
2.3.2. Study Design
2.3.3. Calamus Tea Selection
2.3.4. Urine Sample Preparation
2.4. Method Validation
2.5. HPLC-MS/MS and HPLC-qTOF-MS Settings
3. Results
3.1. Microsome Experiments
3.2. Method Validation
3.3. Human Study
3.3.1. Analysis of the Consumed Tea Infusion
3.3.2. HPLC-MS/MS and qTOF-MS Analysis of Urine Samples
3.3.3. Kinetic Studies and Excretion Rate Determination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuo, H.L.; Yang, F.Q.; Zhang, X.M.; Xia, Z.N. Separation of cis- and trans-asarone from Acorus tatarinowii by preparative gas chromatography. J. Anal. Methods Chem. 2012, 2012, 402081. [Google Scholar] [CrossRef]
- Rajput, S.B.; Tonge, M.B.; Karuppayil, S.M. An overview on traditional uses and pharmacological profile of Acorus calamus Linn. (Sweet flag) and other Acorus species. Phytomedicine 2014, 21, 268–276. [Google Scholar] [CrossRef]
- Varshney, V.K.; Song, B.H.; Ginwal, H.S.; Mittal, N. High Levels of diversity in the phytochemistry, ploidy and genetics of the medicinal plant Acorus calamus L. J. Med. Aromat. Plants 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Rana, T.S.; Mahar, K.S.; Pandey, M.M.; Srivastava, S.K.; Rawat, A.K.S. Molecular and chemical profiling of ‘sweet flag’ (Acorus calamus L.) germplasm from India. Physiol. Mol. Biol. Plants 2013, 19, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Scientific Committee on Food; European Commission. Opinion of the Scientific Committee on Food on the Presence of Beta-asarone in Flavourings and Other Food Ingredients with Flavouring Properties (SCF/CS/FLAV/FLAVOUR/9 ADD1 Final). 2002. Available online: https://ec.europa.eu/food/system/files/2016-10/fs_food-improvement-agents_flavourings-out111.pdf (accessed on 27 August 2021).
- Das, B.K.; Swamy, A.V.; Koti, B.C.; Gadad, P.C. Experimental evidence for use of Acorus calamus (asarone) for cancer chemoprevention. Heliyon 2019, 5, e01585. [Google Scholar] [CrossRef] [PubMed]
- Chellian, R.; Pandy, V.; Mohamed, Z. Pharmacology and toxicology of α- and β-asarone: A review of preclinical evidence. Phytomedicine 2017, 32, 41–58. [Google Scholar] [CrossRef]
- Haupenthal, S.; Berg, K.; Gründken, M.; Vallicotti, S.; Hemgesberg, M.; Sak, K.; Schrenk, D.; Esselen, M. In vitro genotoxicity of carcinogenic asarone isomers. Food Func. 2017, 8, 1227–1234. [Google Scholar] [CrossRef]
- Berg, K.; Bischoff, R.; Stegmüller, S.; Cartus, A.; Schrenk, D. Comparative investigation of the mutagenicity of propenylic and allylic asarone isomers in the Ames fluctuation assay. Mutagenesis 2016, 31, 443–451. [Google Scholar] [CrossRef]
- Hermes, L.; Haupenthal, S.; Uebel, T.; Esselen, M. DNA double strand break repair as cellular response to genotoxic asarone isomers considering phase I metabolism. Food Chem. Toxicol. 2020, 142, 111484. [Google Scholar] [CrossRef]
- Wiseman, R.W.; Miller, E.C.; Miller, J.A.; Liem, A. Structure-activity studies of the hepatocarcinogenicities of alkenylbenzene derivatives related to estragole and safrole on administration to preweanling male C57BL/6J x C3H/HeJ F1 mice. Cancer Res. 1987, 47, 2275–2283. [Google Scholar] [CrossRef]
- Cartus, A.T.; Schrenk, D. Metabolism of carcinogenic alpha-asarone by human cytochrome P450 enzymes. Naunyn-Schmiedebergs Arch. Pharmacol. 2020, 393, 213–223. [Google Scholar] [CrossRef]
- Cartus, A.T.; Stegmüller, S.; Simson, N.; Wahl, A.; Neef, S.; Kelm, H.; Schrenk, D. Hepatic metabolism of carcinogenic β-asarone. Chem. Res. Toxicol. 2015, 28, 1760–1773. [Google Scholar] [CrossRef]
- Cartus, A.T.; Schrenk, D. Metabolism of the carcinogen alpha-asarone in liver microsomes. Food Chem. Toxicol. 2016, 87, 103–112. [Google Scholar] [CrossRef]
- Björnstad, K.; Helander, A.; Hultén, P.; Beck, O. Bioanalytical investigation of asarone in connection with Acorus calamus oil intoxications. J. Anal. Toxicol. 2009, 33, 604–609. [Google Scholar] [CrossRef][Green Version]
- Morales-Ramírez, P.; Madrigal-Bujaidar, E.; Mercader-Martínez, J.; Cassani, M.; González, G.; Chamorro-Cevallos, G.; Salazar-Jacobo, M. Sister-chromatid exchange in-duction produced by in vivo and in vitro exposure to alpha-asarone. Mutat. Res. Genet. Toxicol. Environ. Mutagen 1992, 279, 269–273. [Google Scholar] [CrossRef]
- Yang, Q.; Deng, Z.; Zhang, F.; Sun, P.; Li, J.; Zheng, W. Development of an LC–MS/MS method for quantification of two isomeric phenylpropenes and the application to pharmacokinetic studies in rats. Biomed. Chromatogr. 2018, 32, e4115. [Google Scholar] [CrossRef]
- Lu, J.; Fu, T.; Qian, Y.; Zhang, Q.; Zhu, H.; Pan, L.; Guo, L.; Zhang, M. Distribution of α-asarone in brain following three different routes of administration in rats. Eur. J. Pharm. Sci. 2014, 63, 63–70. [Google Scholar] [CrossRef]
- Meng, X.; Zhao, X.; Wang, S.; Jia, P.; Bai, Y.; Liao, S.; Zheng, X. Simultaneous determination of volatile constituents from Acorus tatarinowii Schott in rat plasma by gas chromatography-mass spectrometry with selective ion monitoring and application in pharmacokinetic study. J. Anal. Methods Chem. 2013, 2013, 949830. [Google Scholar] [CrossRef]
- Fang, Y.Q.; Shi, C.; Liu, L.; Fang, R.M. Pharmacokinetics of beta-asarone in rabbit blood, hippocampus, cortex, brain stem, thalamus and cerebellum. Pharmazie 2012, 67, 120–123. [Google Scholar] [CrossRef]
- Regulation (EC) No 1334/2008 of the European Parliament and of the Council of 16 December 2008 on Flavourings and Certain food Ingredients with Flavouring Properties for use in and on Foods and Amending Council Regulation (EEC) No 1601/91, Regulations (EC) No 2232/96 and (EC) No 110/2008 and Directive 2000/13/EC. 2008. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32008R1334&from=en (accessed on 27 August 2021).
- European Medicines Agency, Commitee on Herbal Medicinal Products. Public Statement on the Use of Herbal Medicinal Products Containing Asarone. 2005. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/public-statement-use-herbal-medicinal-products-containing-asarone_en.pdf (accessed on 27 August 2021).
- Van den Berg, S.J.P.L.; Restani, P.; Boersma, M.G.; Delmulle, L.; Rietjens, I.M.C.M. Levels of genotoxic and carcinogenic ingredients in plant food supplements and associated risk assessment. Food Nutr. Sci. 2011, 2, 989–1010. [Google Scholar] [CrossRef]
- Lake, B.G. Preparation and characterisation of microsomal fractions for studies of xenobiotic metabolism. In Biochemical Toxicology—A Practical Approach; Snell, K., Mullock, B., Eds.; IRL Press: Oxford, UK, 1987; pp. 183–215. [Google Scholar]
- Wu, X.; Murphy, P.; Cunnick, J.; Hendrich, S. Synthesis and characterization of deoxynivalenol glucuronide: Its comparative immunotoxicity with deoxynivalenol. Food Chem. Toxicol. 2007, 45, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Hermes, L.; Römermann, J.; Cramer, B.; Esselen, M. Quantitative analysis of β-asarone derivatives in Acorus calamus and herbal food products by HPLC-MS/MS. J. Agric. Food Chem. 2021, 69, 776–782. [Google Scholar] [CrossRef]
- US Food & Drug Administration Office of Foods and Veterinary Medicine. Guidelines for the Validation of Chemical Methods for the FDA FVM Program, 3rd ed.; Regulatory Science Steering Committee (RSSC): Silver Spring, MD, USA, 2019. Available online: https://www.fda.gov/media/81810/download (accessed on 27 August 2021).
- Stegmüller, S.; Schrenk, D.; Cartus, A.T. Formation and fate of DNA adducts of alpha- and beta-asarone in rat hepatocytes. Food Chem. Toxicol. 2018, 116, 138–146. [Google Scholar] [CrossRef]
- Kim, S.G.; Liem, A.; Stewart, B.C.; Miller, J.A. New studies on trans-anethole oxide and trans-asarone oxide. Carcinogenesis 1999, 20, 1303–1307. [Google Scholar] [CrossRef]
- Luo, G.; Guenthner, T.M. Metabolism of allylbenzene 2′,3′-oxide and estragole 2′,3′-oxide in the isolated perfused rat liver. J. Pharm. Exp. Ther. 1995, 272, 588–596. [Google Scholar]
- Beyer, J.; Ehlers, D.; Maurer, H.H. Abuse of nutmeg (Myristica fragrans Houtt.): Studies on the metabolism and the toxicologic detection of its ingredients elemicin, myristicin, and safrole in rat and human urine using gas chromatography/mass spec-trometry. Ther. Drug. Monit. 2006, 28, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.U.; von Unruh, G.E.; Dengler, H.J. The metabolism of eugenol in man. Xenobiotica 1990, 20, 209–222. [Google Scholar] [CrossRef]
- Sangster, S.A.; Caldwell, J.; Hutt, A.J.; Anthony, A.; Smith, R.L. The metabolic disposition of methoxy-14C-labelled trans-anethole, estragole and p-propylanisole in human volunteers. Xenobiotica 1987, 17, 1223–1232. [Google Scholar] [CrossRef]
- Punt, A.; Delatour, T.; Scholz, G.; Schilter, B.; van Bladeren, P.J.; Rietjens, I.M.C.M. Tandem mass spectrometry analysis of N2-(trans-Isoestragol-3′-yl)-2′-deoxyguanosine as a strategy to study species differences in sulfotransferase conversion of the proximate carcinogen 1′-hydroxyestragole. Chem. Res. Toxicol. 2007, 20, 991–998. [Google Scholar] [CrossRef]
- Hasheminejad, G.; Caldwell, J. Genotoxicity of the alkenylbenzenes alpha- and beta-asarone, myristicin and elimicin as determined by the UDS assay in cultured rat hepatocytes. Food Chem. Toxicol. 1994, 32, 223–231. [Google Scholar] [CrossRef]
- Zeller, A.; Horst, K.; Rychlik, M. Study of the metabolism of estragole in humans consuming fennel tea. Chem. Res. Toxicol. 2009, 22, 1929–1937. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Linear Range [ng/mL] | LOQ [ng/mL] | LOQ [ng/mL] | Interday Repeatability [%] | Intraday Repeatability [%] | Recovery [%] |
|---|---|---|---|---|---|---|
| erythro-asarone diols | 0.25–50 | 0.09 | 0.30 | 12.3 | 3.4 | 103 |
| threo-asarone diols | 0.25–50 | 0.06 | 0.25 | 8.5 | 8.3 | 83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hermes, L.; Römermann, J.; Cramer, B.; Esselen, M. Phase II Metabolism of Asarone Isomers In Vitro and in Humans Using HPLC-MS/MS and HPLC-qToF/MS. Foods 2021, 10, 2032. https://doi.org/10.3390/foods10092032
Hermes L, Römermann J, Cramer B, Esselen M. Phase II Metabolism of Asarone Isomers In Vitro and in Humans Using HPLC-MS/MS and HPLC-qToF/MS. Foods. 2021; 10(9):2032. https://doi.org/10.3390/foods10092032
Chicago/Turabian StyleHermes, Lena, Janis Römermann, Benedikt Cramer, and Melanie Esselen. 2021. "Phase II Metabolism of Asarone Isomers In Vitro and in Humans Using HPLC-MS/MS and HPLC-qToF/MS" Foods 10, no. 9: 2032. https://doi.org/10.3390/foods10092032
APA StyleHermes, L., Römermann, J., Cramer, B., & Esselen, M. (2021). Phase II Metabolism of Asarone Isomers In Vitro and in Humans Using HPLC-MS/MS and HPLC-qToF/MS. Foods, 10(9), 2032. https://doi.org/10.3390/foods10092032