
Proton Low Field NMR Relaxation Time Domain Sensor for Monitoring of Oxidation Stability of PUFA-Rich Oils and Emulsion Products



Abstract
1. Introduction
1.1. 1H LF NMR Relaxation Sensor Technology
1.2. Determination of Chemical and Physical Structure of Oils and Emulsions by the 1H LF NMR TD Sensor
1.3. Oxidation of PUFA-Rich Oils and O/W Emulsions Food Products
1.4. Comparison of 1H LF NMR with Common Methods of Characterizing Oil Oxidation
1.5. Applications of 1H LF NMR Relaxation in Food Industry
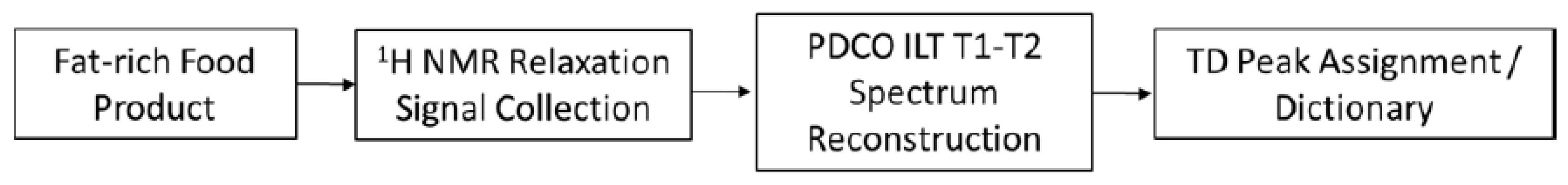
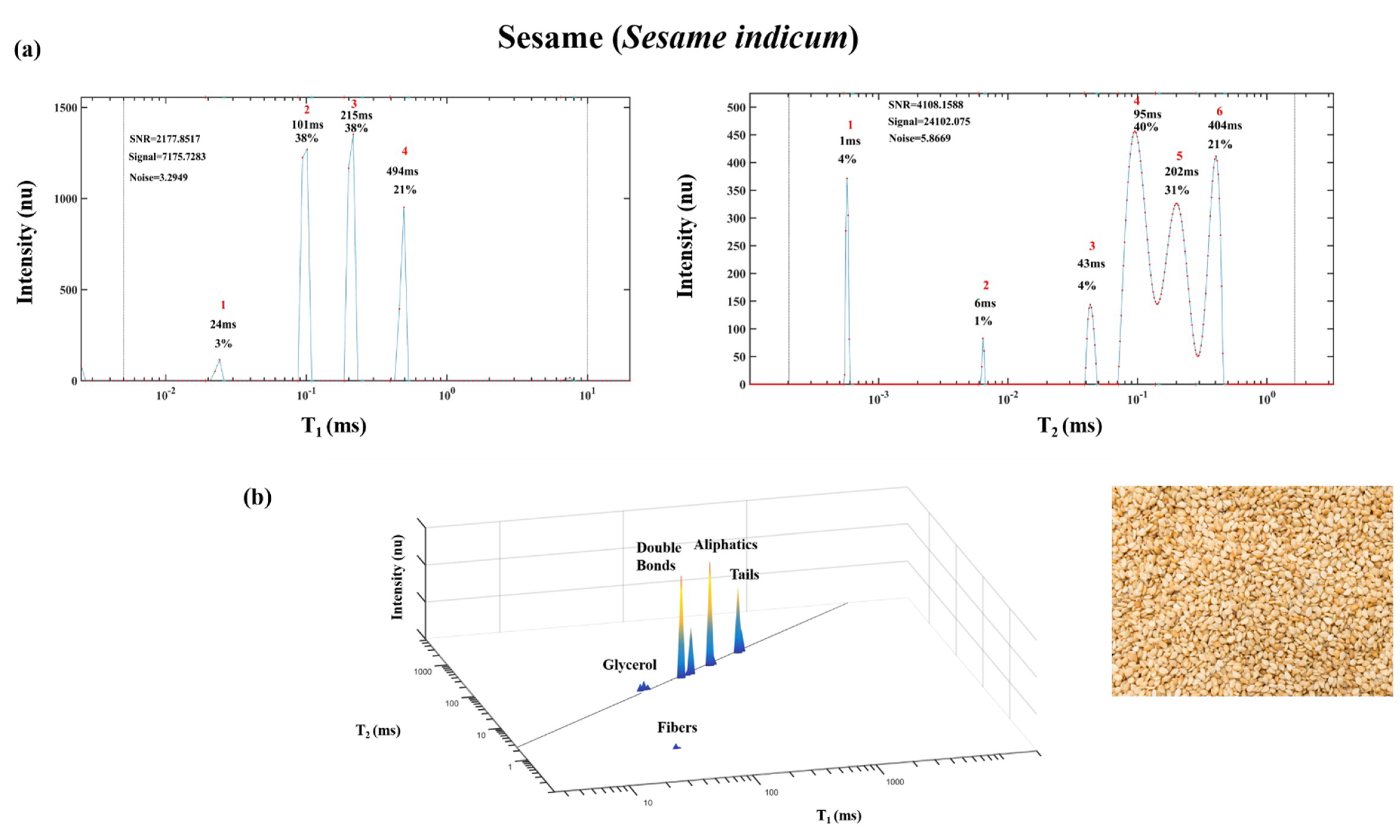
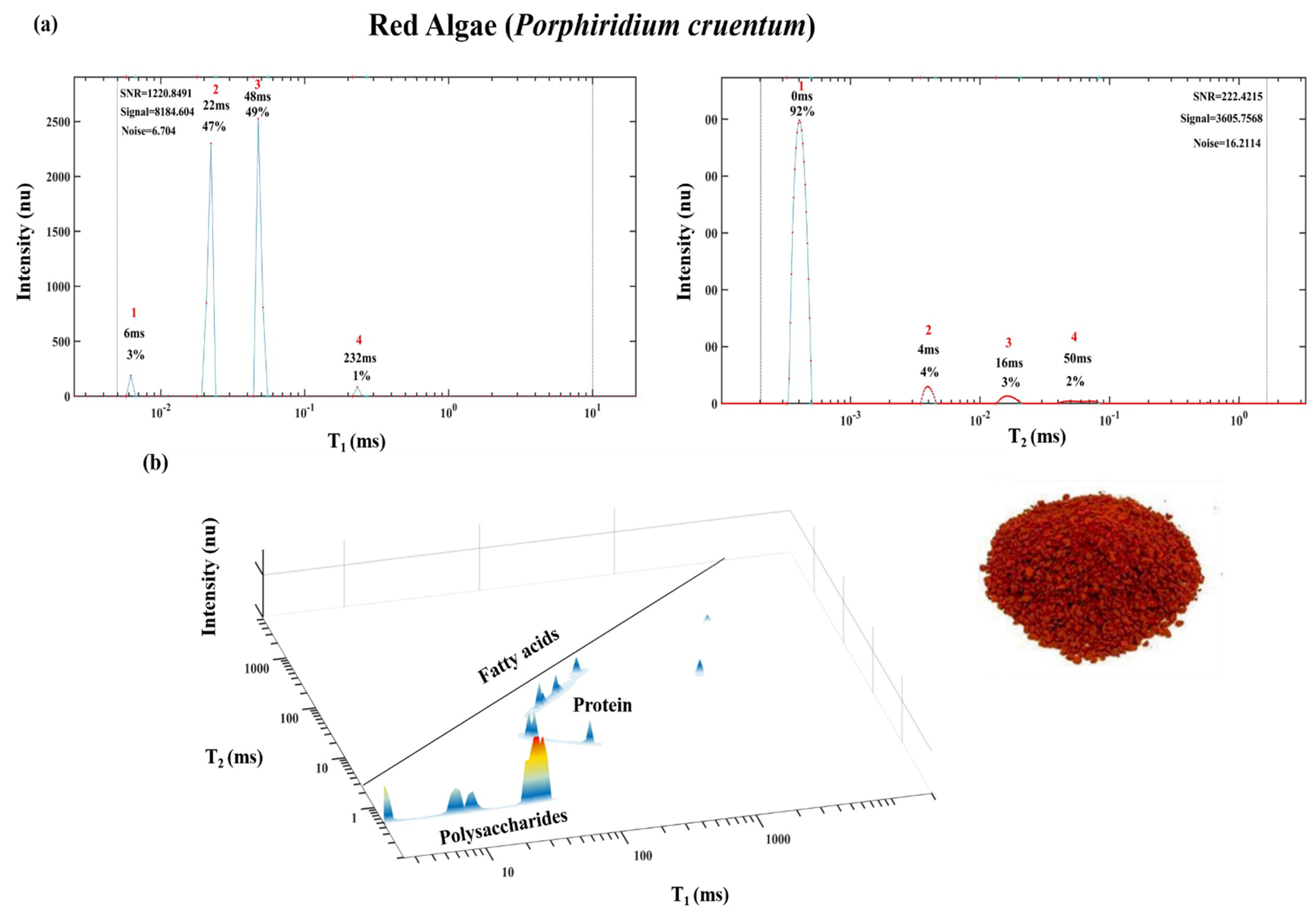
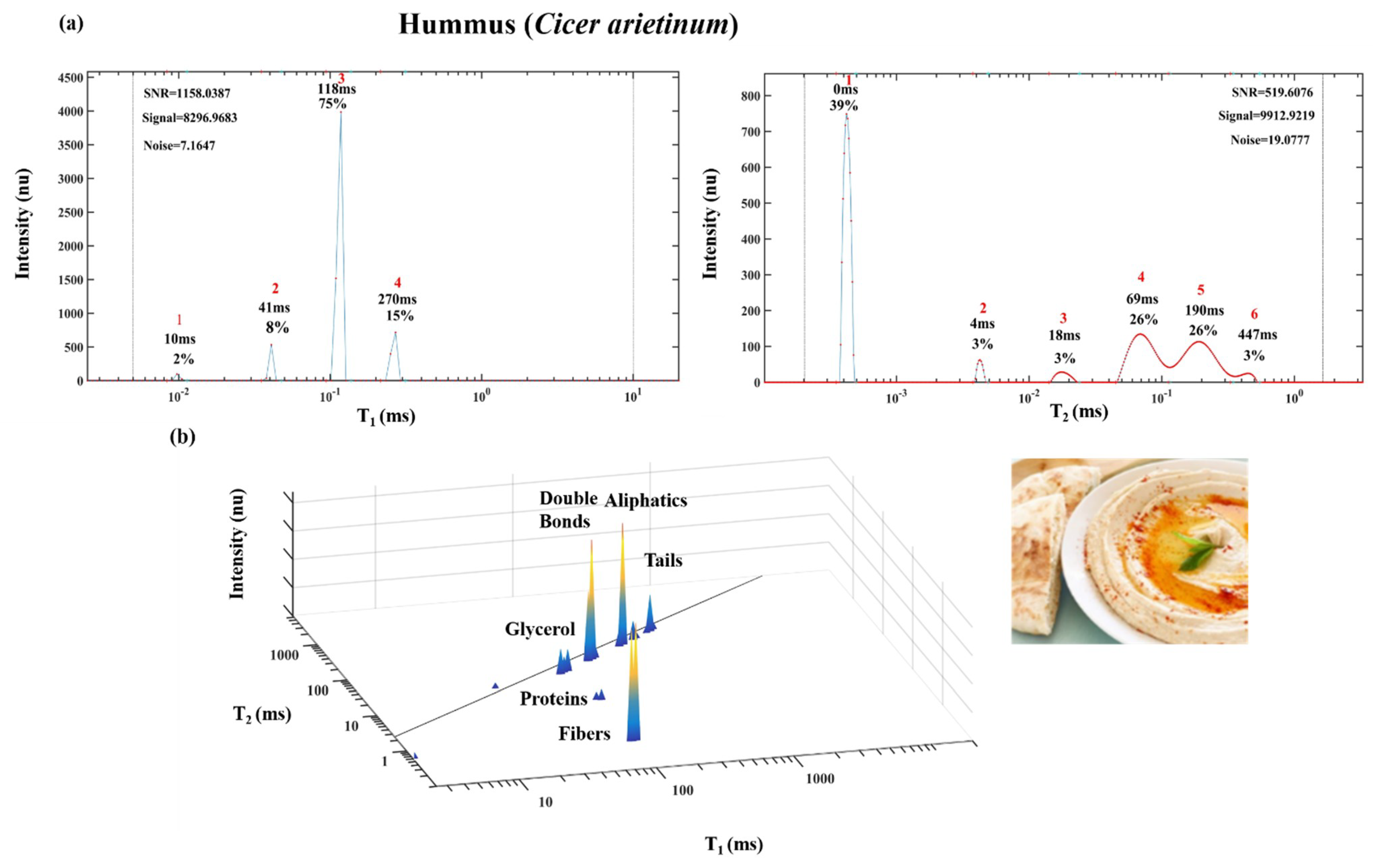
2. Demonstration of TD Sensor Fingerprint Mapping of FAs, Oils, Seeds and Paste Products
2.1. TD Fingerprint Mapping of FAs, Oils, Seeds and Paste Products
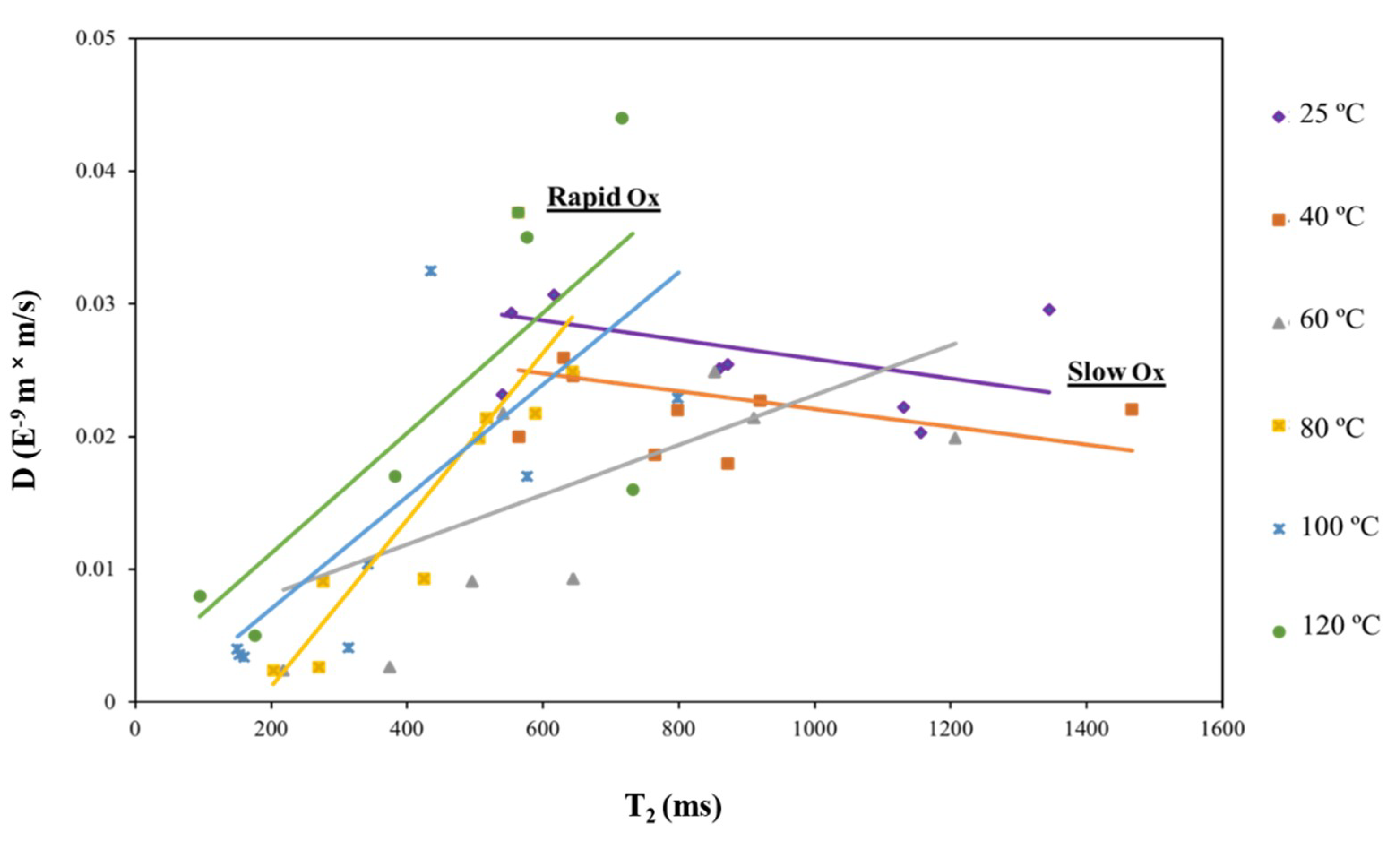
2.2. Demonstration of TD Sensor Fingerprint Mapping of FA Oil Oxidation
2.3. Demonstration of TD Sensor Fingerprint Mapping of Food O/W Emulsion Products and Oxidation
2.4. Demonstration of TD Sensor Fingerprint Mapping of Oleosome Oil Bodies Encapsulating of External Oils
3. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health Implications of High Dietary Omega-6 Polyunsaturated Fatty Acids. J. Nutr. Metab. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Ruxton, C.H.; Reed, S.C.; Simpson, M.J.; Millington, K.J. The health benefits of omega-3 polyunsaturated fatty acids: A review of the evidence. J. Hum. Nutr. Diet. 2007, 20, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, R.S.; Chouinard-Watkins, R.; Bazinet, R.P. Brain docosahexaenoic acid uptake and metabolism. Mol. Asp. Med. 2018, 64, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Burdge, G. Long-chain n−3 PUFA: Plant v. marine sources. Proc. Nutr. Soc. 2006, 65, 42–50. [Google Scholar] [CrossRef]
- Yehuda, S.; Rabinovitz, S.; Mostofsky, D.I. Essential fatty acids are mediators of brain biochemistry and cognitive functions. J. Neurosci. Res. 1999, 56, 565–570. [Google Scholar] [CrossRef]
- Nikiforidis, C.V.; Matsakidou, A.; Kiosseoglou, V. Composition, properties and potential food applications of natural emulsions and cream materials based on oil bodies. RSC Adv. 2014, 4, 25067–25078. [Google Scholar] [CrossRef]
- Hwang, H.-S. Advances in NMR Spectroscopy for Lipid Oxidation Assessment, 1st ed.; Springer Briefs in Foods, Health and Nutrition: Cham, Switzerland, 2017. [Google Scholar]
- Wiesman, Z.; Linder, C.; Ayalon, N.; Resende, T.M.; Levi, O.; Bernardinelli, O.D.; Nascimento Mitre, C.I. 2D and 3D Spectrum graphics of the Chemical-Morphological Domains of Complex Biomass by Low Field Proton NMR Energy Relaxation Signal Analysis. Energy Fuels 2018, 32, 5090–5102. [Google Scholar] [CrossRef]
- Wiesman, Z.; Linder, C. Facile NMR Relaxation Sensor for Monitoring of Biomass Degradation Products during Conversion to Biogas. In Nuclear Magnetic Resonance, 1st ed.; Khaneja, N., Ed.; IntechOpen: Rijeka, Croatia, 2020; pp. 1–20. [Google Scholar] [CrossRef]
- Barbosa, L.L.; Kock, F.V.C.; Silva, R.C.; Freitas, J.C.C.; Lacerda, V., Jr.; Castro, E.V.R. Application of Low-Field NMR for the Determination of Physical Properties of Petroleum Fractions. Energy Fuels 2013, 27, 673–679. [Google Scholar] [CrossRef]
- Guo, J.-C.; Zhou, H.-Y.; Zeng, J.; Wang, K.-J.; Lai, J.; Liu, Y.-X. Advances in low-field nuclear magnetic resonance (NMR) technologies applied for characterization of pore space inside rocks: A critical review. Pet. Sci. 2020, 17, 1281–1297. [Google Scholar] [CrossRef]
- Berman, P.; Leshem, A.; Etziony, O.; Levi, O.; Parmat, Y.; Saunders, M.; Wiesman, Z. Novel 1H Low Resolution (LR)-NMR Applications for the Biodiesel Industry. Biotechnol. Biofuels 2013, 6, 55–75. [Google Scholar] [CrossRef]
- Song, Y.-Q.; Venkataramanan, L.; Hürlimann, M.; Flaum, M.; Frulla, P.; Straley, C. T1-T2 Correlation Spectra Obtained Using a Fast Two-Dimensional Laplace Inversion. J. Magn. Reson. 2002, 154, 261–268. [Google Scholar] [CrossRef]
- Kroeker, R.M.; Henkelman, R.M. Analysis of biological NMR relaxation data with continuous distributions of relaxation times. J. Magn. Reason. 1986, 69, 218–235. [Google Scholar] [CrossRef]
- Hahn, E.L. Spin Echoes. Phys. Rev. 1950, 80, 580–594. [Google Scholar] [CrossRef]
- Carr, H.Y.; Purcell, E.M. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D.R. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Oxford University Press: Oxford, UK, 1961. [Google Scholar]
- McDonald, P.; Korb, J.-P.; Mitchell, J.; Monteilhet, L. Surface relaxation and chemical exchange in hydrating cement pastes: A two-dimensional NMR relaxation study. Phys. Rev. E 2005, 72, 011409. [Google Scholar] [CrossRef]
- Callaghan, P.; Godefroy, S.; Ryland, B. Diffusion–relaxation correlation in simple pore structures. J. Magn. Reson. 2003, 162, 320–327. [Google Scholar] [CrossRef]
- Berman, P.; Levi, O.; Parmat, Y.; Saunders, M.; Wiesman, Z. Laplace Inversion of LR-NMR Relaxometry Data using Sparse Representation Methods. Concept. Magn. Reson. A 2013, 42, 72–88. [Google Scholar] [CrossRef]
- Campisi-Pinto, S.; Levi, O.; Benson, D.; Cohen, M.; Resende, M.T.; Saunders, M.; Linder, C.; Wiesman, Z. Analysis of the Regularization Parameters of Primal–Dual Interior Method for Convex Objectives Applied to 1H Low Field Nuclear Magnetic Resonance Data Processing. Appl. Magn. Reson. 2018, 49, 1129–1150. [Google Scholar] [CrossRef]
- Campisi-Pinto, S.; Resende, M.T.; Levi, O.; Benson, D.; Cohen, M.; Saunders, M.; Linder, C.; Wiesman, Z. Simula-tion-Based Sensitivity Analysis of Regularization Parameters for Robust Reconstruction of Complex Material’s T1-T2 1H LF-NMR Energy Relaxation Signals. Appl. NMR 2020, 51, 41–58. [Google Scholar]
- Resende, M.T.; Campisi-Pinto, S.; Linder, C.; Wiesman, Z. Multidimensional proton nuclear magnetic resonance relaxation morphological and chemical spectrum graphics for monitoring and characterization of polyunsaturated fatty acid oxidation. J. Am. Oil Chem. Soc. 2019, 96, 125–135. [Google Scholar] [CrossRef]
- Berman, P.; Meiri, N.; Colnago, L.A.; Moraes, T.B.; Linder, C.; Levi, O.; Parmet, Y.; Saunders, M.; Wiesman, Z. Study of liquid-phasemolecular packing interactions and morphology of fatty acid methyl esters (biodiesel). Biotechnol. Biofuels 2015, 8, 12. [Google Scholar] [CrossRef]
- Iwahashi, M.; Kasahara, Y. Dynamic molecular movements and aggregation structures of lipids in a liquid state. Curr. Opin. Colloid Interface Sci. 2011, 16, 359–366. [Google Scholar] [CrossRef]
- Resende, M.T.; Linder, C.; Wiesman, Z. 1H LF-NMR energy relaxation time characterization of the chemical and mor-phological structure of PUFA-rich linseed oil during oxidation with and without antioxidants. Eur. J. Lipid Sci. Technol. 2019, 121, 1800339–1800347. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, L.; Xie, L.; Lu, X.; Liu, Q.; He, J.; Mantilla, C.A.; Berg, F.G.A.V.D.; Zeng, H. Surface Interaction of Water-in-Oil Emulsion Droplets with Interfacially Active Asphaltenes. Langmuir 2017, 33, 1265–1274. [Google Scholar] [CrossRef]
- Nilsen-Nygaard, J.; Sletmoen, M.; Draget, K.I. Stability and interaction forces of oil-in-water emulsions as observed by optical tweezers—A proof-of-concept study. RSC Adv. 2014, 4, 52220–52229. [Google Scholar] [CrossRef]
- Khalil, A.; Puel, F.; Chevalier, Y.; Galvan, J.-M.; Rivoire, A.; Klein, J.-P. Study of droplet size distribution during emulsi-fication process using in situ video probe coupled with an automatic image analysis. Chem. Eng. J. 2010, 165, 946–957. [Google Scholar] [CrossRef]
- McClements, D.J. Critical Review of Techniques and Methodologies for Characterization of Emulsion Stability. Crit. Rev. Food Sci. Nutr. 2007, 47, 611–649. [Google Scholar] [CrossRef]
- Chen, G.; Tao, D. An experimental study of stability of oil–water emulsion. Fuel Process. Technol. 2005, 86, 499–508. [Google Scholar] [CrossRef]
- Waschatko, G.; Junghans, A.; Vilgis, T.A. Soy milk oleosome behaviour at the air–water interface. Faraday Discuss. 2012, 158, 157–169. [Google Scholar] [CrossRef]
- Resende, M.; Linder, C.; Wiesman, Z. Alkyl Tail Segments Mobility as a Marker for Omega-3 PUFA-rich Linseed Oil Oxidative Aging. J. Am. Oil Chem. Soc. 2020, 97, 1283–1297. [Google Scholar] [CrossRef]
- Jacobsen, C. Some strategies for the stabilization of long chain n-3 PUFA-enriched foods: A review. Eur. J. Lipid Sci. Technol. 2015, 117, 1853–1866. [Google Scholar] [CrossRef]
- Choe, E.; Min, D.B. Chemistry of Deep-Fat Frying Oils. J. Food Sci. 2007, 72, R77–R86. [Google Scholar] [CrossRef] [PubMed]
- Barriuso, B.; Astiasarán, I.; Ansorena, D. A review of analytical methods measuring lipid oxidation status in foods: A challenging task. Eur. Food Res. Technol. 2013, 236, 1–15. [Google Scholar] [CrossRef]
- Berton-Carabin, C.; Ropers, M.-H.; Genot, C. Lipid Oxidation in Oil-in-Water Emulsions: Involvement of the Interfacial Layer. Compr. Rev. Food Sci. Food Saf. 2014, 13, 945–977. [Google Scholar] [CrossRef]
- Merkx, D.W.H.; Hong, G.T.S.; Ermacora, A.; Van Duynhoven, J.P.M. Rapid Quantitative Profiling of Lipid Oxidation Products in a Food Emulsion by 1H NMR. Anal. Chem. 2018, 90, 4863–4870. [Google Scholar] [CrossRef]
- Gromadzka, J.; Wardencki, W. Trends in edible vegetable oils analysis. Part A Determination of different components of edible oilS-a Review. Pol. J. Food Nutr. Sci. 2011, 61, 33–43. [Google Scholar] [CrossRef]
- Resende, M.T.; Linder, C.; Wiesman, Z. Low-Field Nuclear Magnetic Resonance Time Domain Characterization of Polyunsaturated Fatty Acid—Rich Linseed and Fish Oil Emulsions during Thermal Air Oxidation. J. Am. Oil Chem. Soc. 2021. [Google Scholar] [CrossRef]
- Colnago, L.A.; Wiesman, Z.; Pages, G.; Musse, M.; Monaretto, T.; Windt, C.W.; Rondeau-Mouro, C. Low field, time domain NMR in the agriculture and agrifood sectors: An overview of applications in plants, foods and biofuels. J. Magn. Reson. 2021, 323, 106899. [Google Scholar] [CrossRef]
- Hills, B.; Benamira, S.; Marigheto, N.; Wright, K. T1-T2 correlation analysis of complex foods. Appl. Magn. Reson. 2004, 26, 543–560. [Google Scholar] [CrossRef]
- Corrêa, C.C.; Forato, L.A.; Colnago, L.A. High-throughput non-destructive nuclear magnetic resonance method to measure intramuscular fat content in beef. Anal. Bioanal. Chem. 2008, 393, 1357–1360. [Google Scholar] [CrossRef]
- Bertram, H.C.; Dønstrup, S.; Karlsson, A.H.; Andersen, H.J. Continuous distribution analysis of T2 relaxation in meat—an approach in the determination of water-holding capacity. Meat Sci. 2002, 60, 279–285. [Google Scholar] [CrossRef]
- Rutledge, D.N. Characterization of water in agro-food products by time domain-NMR. Food Control 2001, 12, 437–445. [Google Scholar] [CrossRef]
- Choi, S.-G.; Kerr, W.L. 1H NMR studies of molecular mobility in wheat starch. Food Res. Int. 2003, 36, 341–348. [Google Scholar] [CrossRef]
- Goetz, J.; Koehler, P. Study of the thermal denaturation of selected proteins of whey and egg by low resolution NMR. LWT Food Sci. Technol. 2005, 38, 501–512. [Google Scholar] [CrossRef]
- Lucas, T.; Le Ray, D.; Barey, P.; Mariette, F. NMR assessment of ice cream: Effect of formulation on liquid and solid fat. Int. Dairy J. 2005, 15, 1225–1233. [Google Scholar] [CrossRef]
- Marigheto, N.; Duarte, S.; Hills, B.P. NMR relaxation study of avocado quality. Appl. Magn. Reson. 2005, 29, 687–701. [Google Scholar] [CrossRef]
- Hills, B. Applications of Low-Field NMR to Food Science. Annu. Rep. NMR Spectrosc. 2006, 58, 177–230. [Google Scholar] [CrossRef]
- Geresh, S.; Arad, M.S. The extracellular polysaccharide of the red microalgae: Chemistry and Rheology. Biores. Technol. 1991, 38, 195–201. [Google Scholar] [CrossRef]
- Besghini, D.; Mauri, M.; Simonutti, R. Time Domain NMR in Polymer Science: From the Laboratory to the Industry. Appl. Sci. 2019, 9, 1801. [Google Scholar] [CrossRef]
- Bonsegna, S.; Bettini, S.; Pagano, R.; Zacheo, A.; Vergaro, V.; Giovinazzo, G.; Caminati, G.; Leporatti, S.; Valli, L.; Santino, A. Plant Oil Bodies: Novel Carriers to Deliver Lipophilic Molecules. Appl. Biochem. Biotechnol. 2010, 163, 792–802. [Google Scholar] [CrossRef]
- Goyal, A.; Sharma, V.; Upadhyay, N.; Singh, A.K.; Arora, S.; Lal, D.; Sabikhi, L. Development of stable flaxseed oil emulsions as a potential delivery system of ω-3 fatty acids. J. Food Sci. Technol. 2014, 52, 4256–4265. [Google Scholar] [CrossRef]
- Nikovska, K. Study of olive oil-in-water emulsions with protein emulsifiers. Emir. J. Food Agric. 2012, 24, 17–24. [Google Scholar] [CrossRef]
- Fabre, J.-F.; Lacroux, E.; Cerny, M.; Mouloungui, Z. Barriers to the release of flaxseed oil bodies and ways of overcoming them. Oilseeds Fats Crop. Lipids 2015, 22, D607. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | Linoleic Acid | Linolenic Acid | ||
|---|---|---|---|---|
| T1 (ms) | T2 (ms) | T1 (ms) | T2 (ms) | |
| 1 | 320 ± 10 | 292 ± 10 | 309 ± 29 | 302 ± 33 |
| 2 | 845 ± 35 | 512 ± 29 | 442 ± 54 | 444 ± 35 |
| 3 | 1111 ± 59 | 852 ± 34 | 1099 ± 207 | 927 ± 103 |
| 4 | 1267 ± 68 | 950 ± 22 | 2288 ± 192 | 1948 ± 113 |
| Peak | T1 (ms) | T2 (ms) | Dictionary |
|---|---|---|---|
| 1 | 94 | 53 | Glycerol |
| 2 | 191 | 135 | Double Bonds |
| 3 | 437 | 344 | Aliphatic Chain |
| 4 | 1003 | 766 | Tail |
| Peak | T-0 | T-96 | 1H TD Assignment | ||
|---|---|---|---|---|---|
| T1 (ms) | T2 (ms) | T1 (ms) | T2 (ms) | ||
| 1 | 2941 | 1287 | - | - | Surface (small OB) |
| 2 | 2732 | 1206 | 1756 | 615 | Surface (medium OB) |
| 3 | 905 | 577 | 374 | 307 | FA-Aliphatic chains |
| 4 | 92 | 55 | 80 | 57 | FA-D. Bonds |
| 5 | 59 | 18 | 64 | 25 | Glycerol |
| Peak | T-0 h | T-96 h | 1H TD Assignment | ||
|---|---|---|---|---|---|
| T1 (ms) | T2 (ms) | T1 (ms) | T2 (ms) | ||
| 1 | 64 | 29 | 55 | 21 | Glycerol |
| 2 | 115 | 84 | 92 | 57 | FA D. Bonds |
| 3 | 726 | 282 | 583 | 416 | FA Aliphatic chains |
| 4 | 3948 | 1902 | 2538 | 971 | OB surface |
| T-0 | T-96 | |
|---|---|---|
| T1/T2 | 2.07 * | 2.61 * |
| Self-diffusion (10−9 m*m/s) | 2.902 ± 0.020 | 2.7338 ± 0.025 |
| DLS (nm) | 1374 ± 81 | 1951 ± 89 |
| Z Potential (mV) | −27.3 ± 6.6 | −25.1 ± 8.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Resende, M.T.; Osheter, T.; Linder, C.; Wiesman, Z. Proton Low Field NMR Relaxation Time Domain Sensor for Monitoring of Oxidation Stability of PUFA-Rich Oils and Emulsion Products. Foods 2021, 10, 1385. https://doi.org/10.3390/foods10061385
Resende MT, Osheter T, Linder C, Wiesman Z. Proton Low Field NMR Relaxation Time Domain Sensor for Monitoring of Oxidation Stability of PUFA-Rich Oils and Emulsion Products. Foods. 2021; 10(6):1385. https://doi.org/10.3390/foods10061385
Chicago/Turabian StyleResende, Maysa T., Tatiana Osheter, Charles Linder, and Zeev Wiesman. 2021. "Proton Low Field NMR Relaxation Time Domain Sensor for Monitoring of Oxidation Stability of PUFA-Rich Oils and Emulsion Products" Foods 10, no. 6: 1385. https://doi.org/10.3390/foods10061385
APA StyleResende, M. T., Osheter, T., Linder, C., & Wiesman, Z. (2021). Proton Low Field NMR Relaxation Time Domain Sensor for Monitoring of Oxidation Stability of PUFA-Rich Oils and Emulsion Products. Foods, 10(6), 1385. https://doi.org/10.3390/foods10061385