
Application of Whole Genome Sequencing to Understand Diversity and Presence of Genes Associated with Sanitizer Tolerance in Listeria monocytogenes from Produce Handling Sources

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Whole Genome Sequencing and Data Assembly
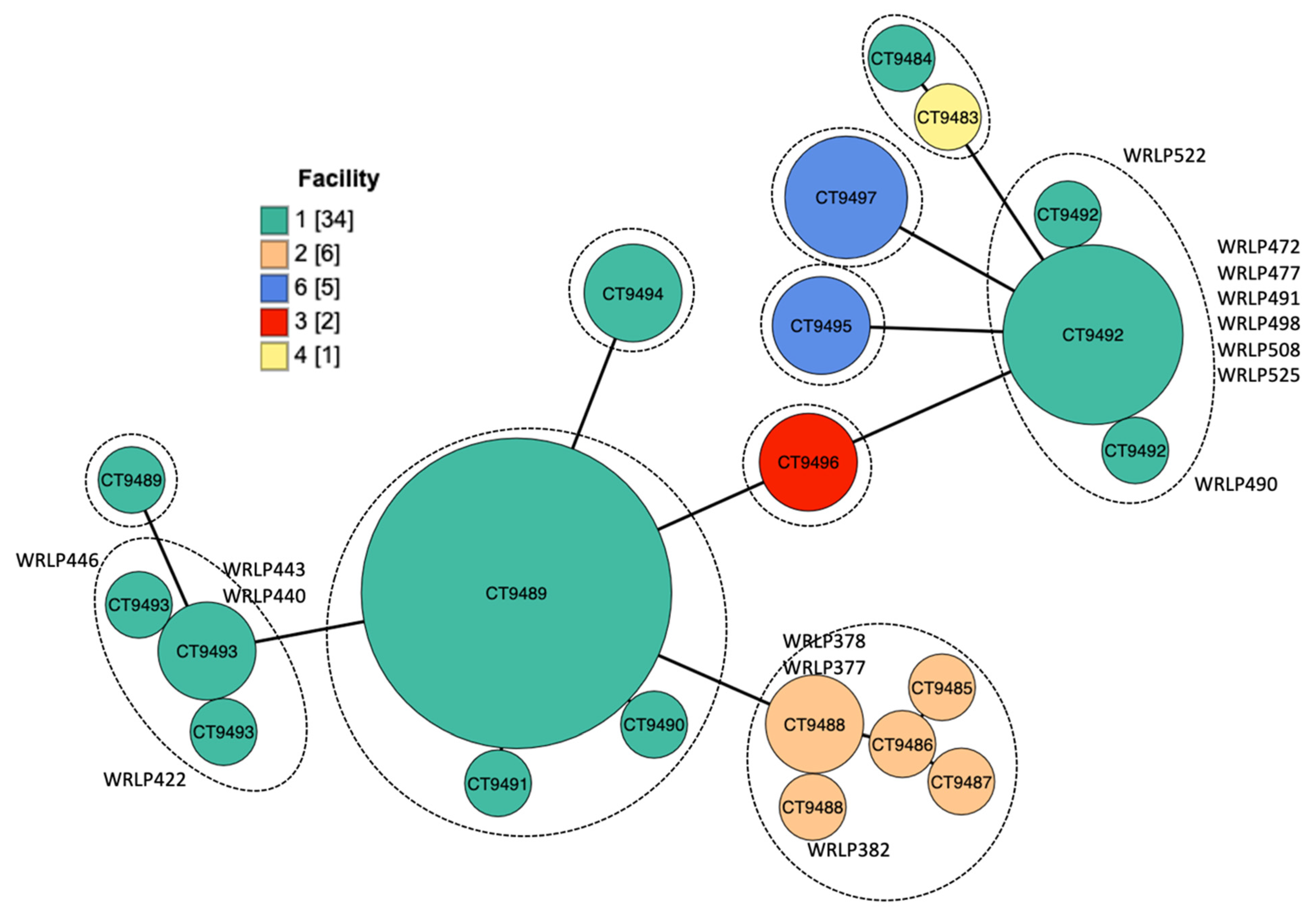
2.2. Core Genome Multilocus Sequence Typing (cgMLST)
2.3. Detection and Assessment of Antimicrobial Resistance, Stress Survival, and Pathogenicity Genes
2.4. Whole Genome SNP and Pangenome Analyses
2.5. Growth in the Presence of Quaternary Ammonium Compound (QAC) Sanitizer
3. Results
3.1. General Genome Characteristics
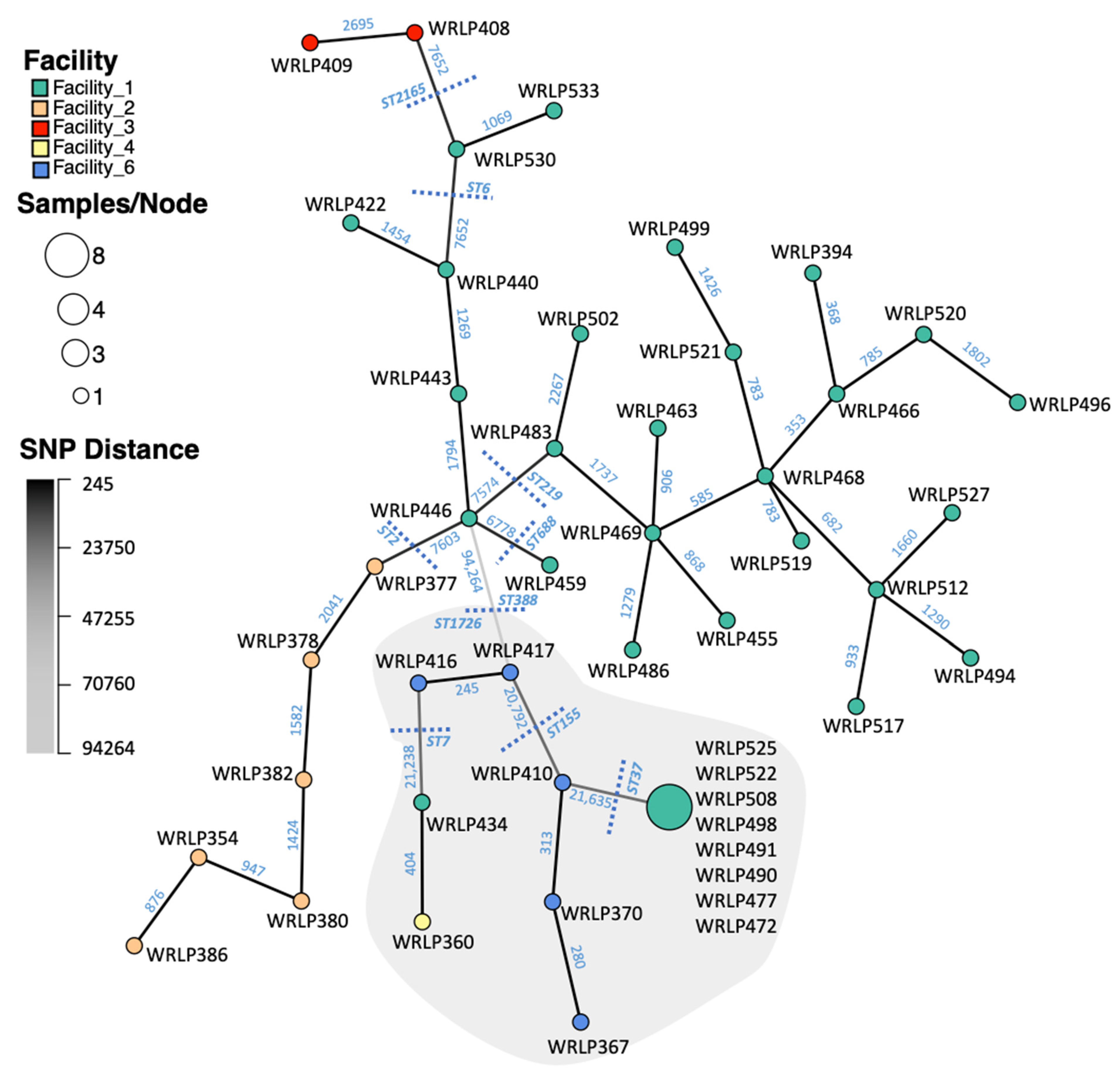
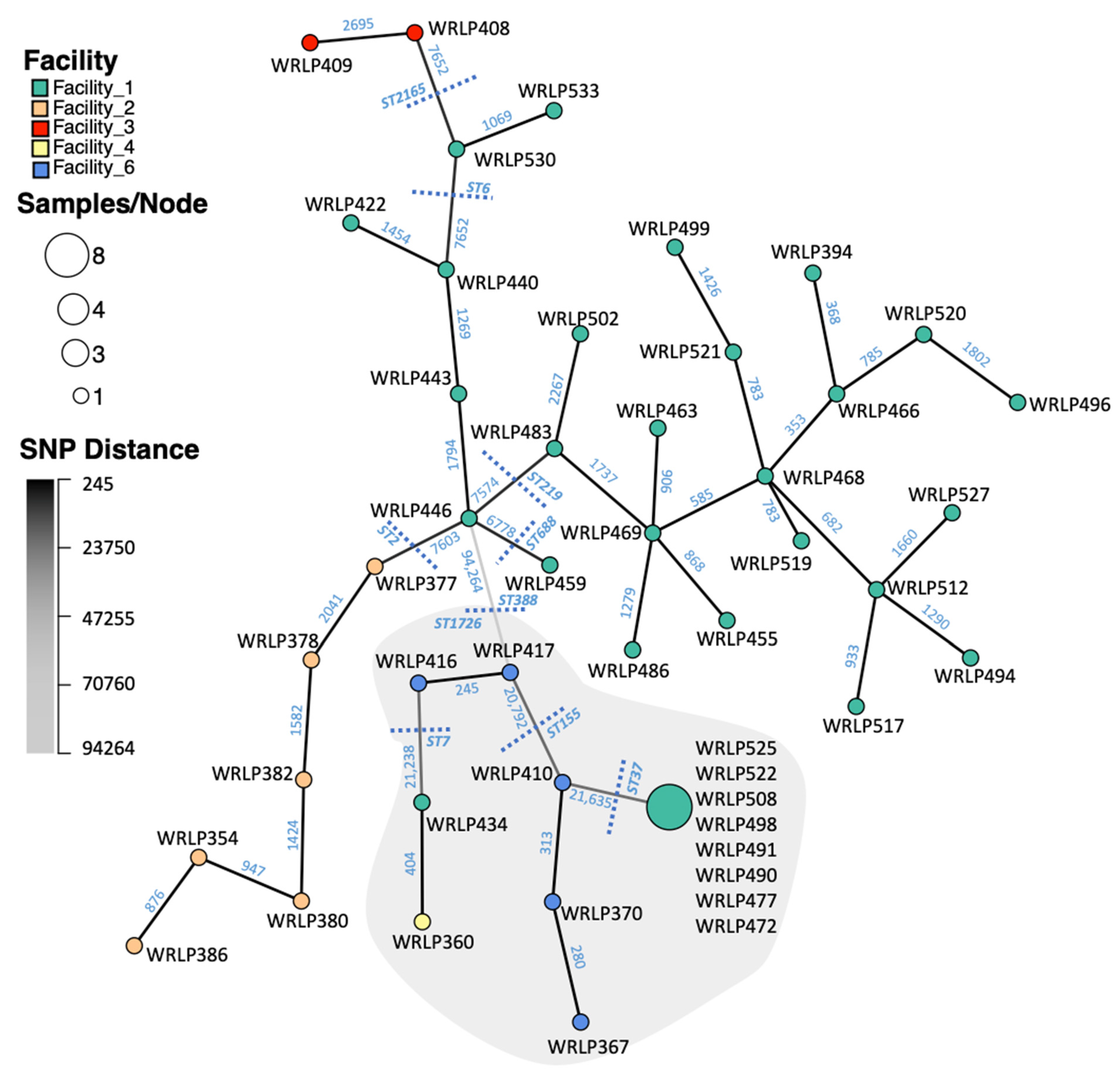
3.2. SNP Analyses
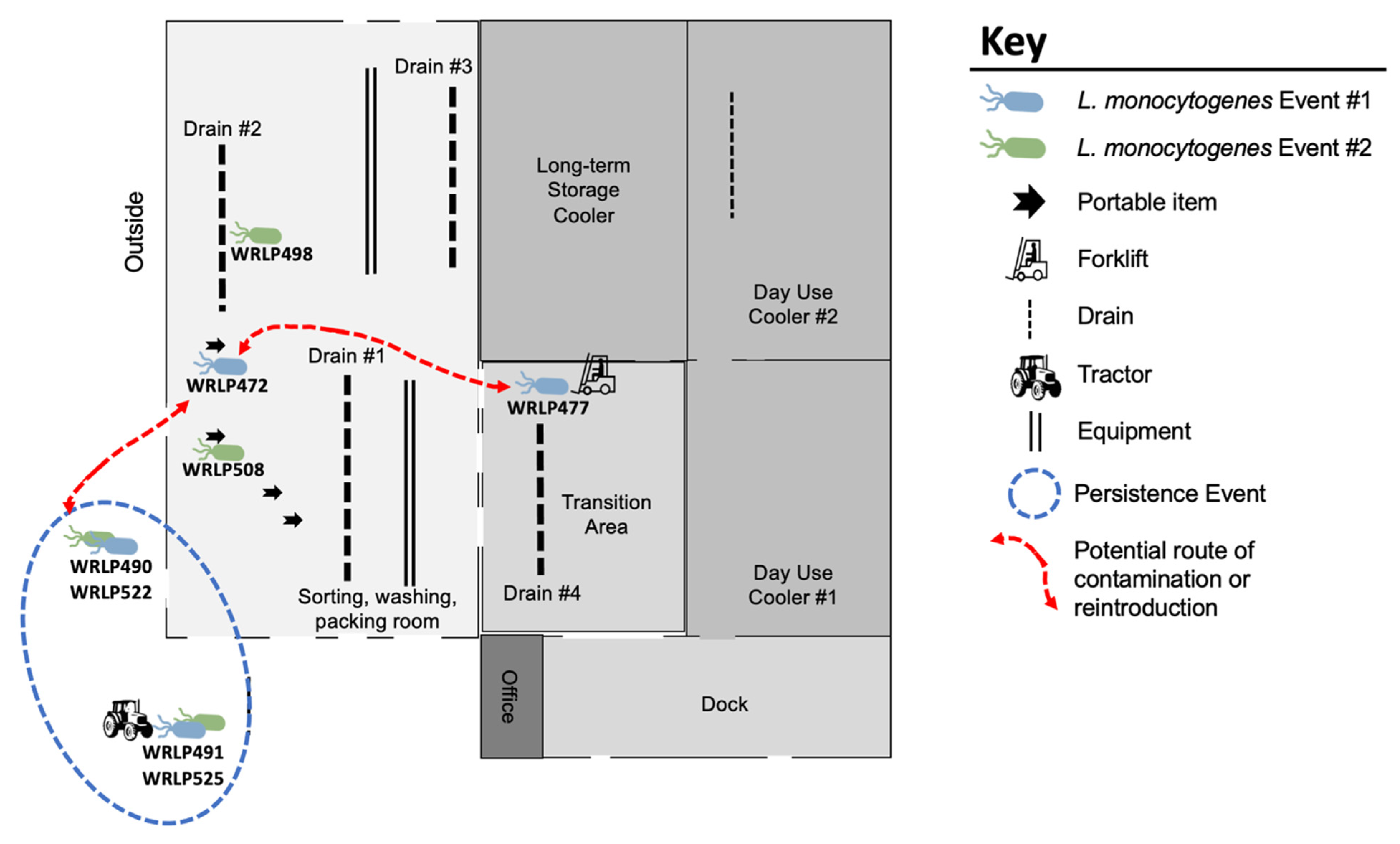
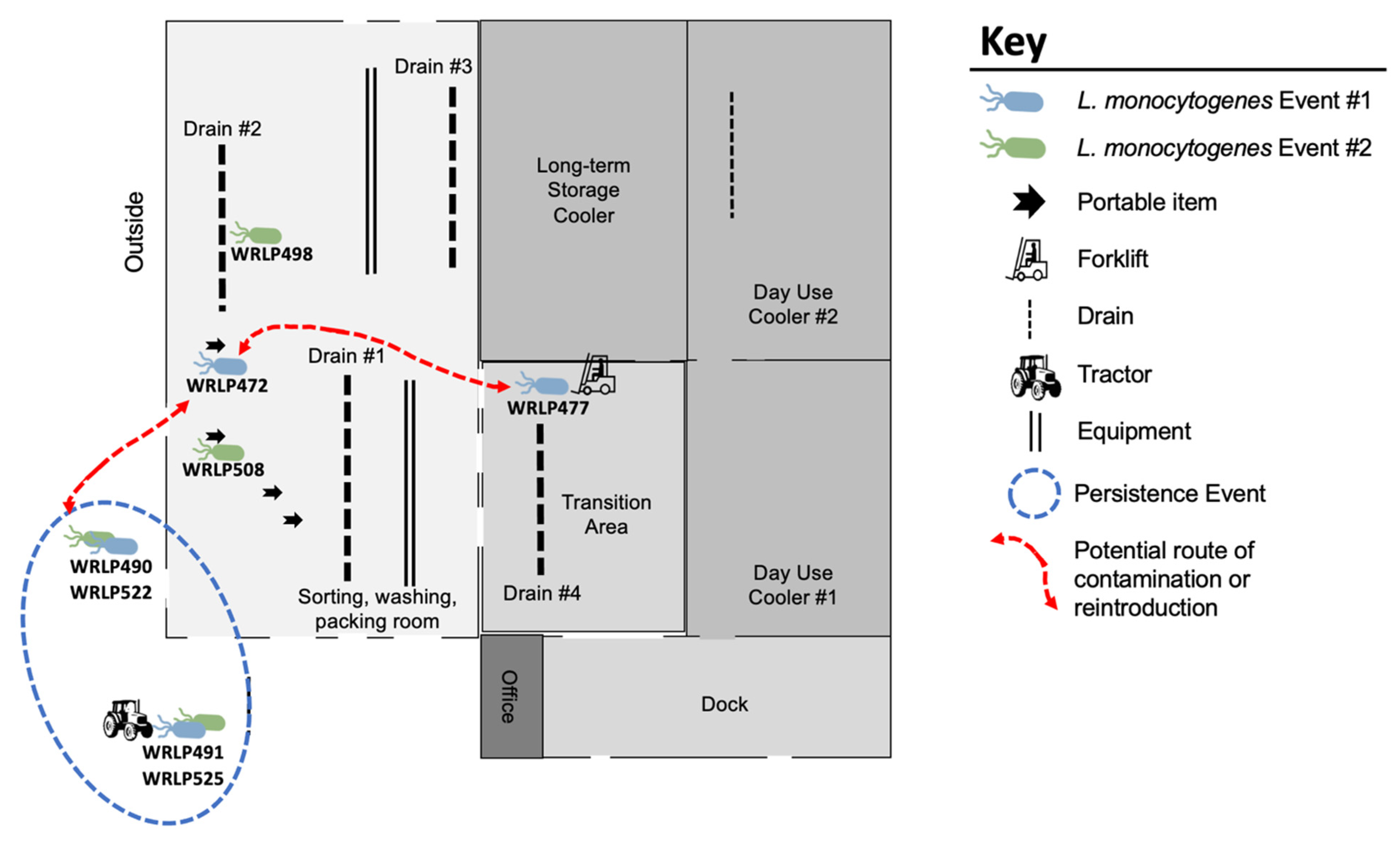
3.3. Highly Related CC37 Cluster in Facility 1
3.4. Accessory Genome Analysis
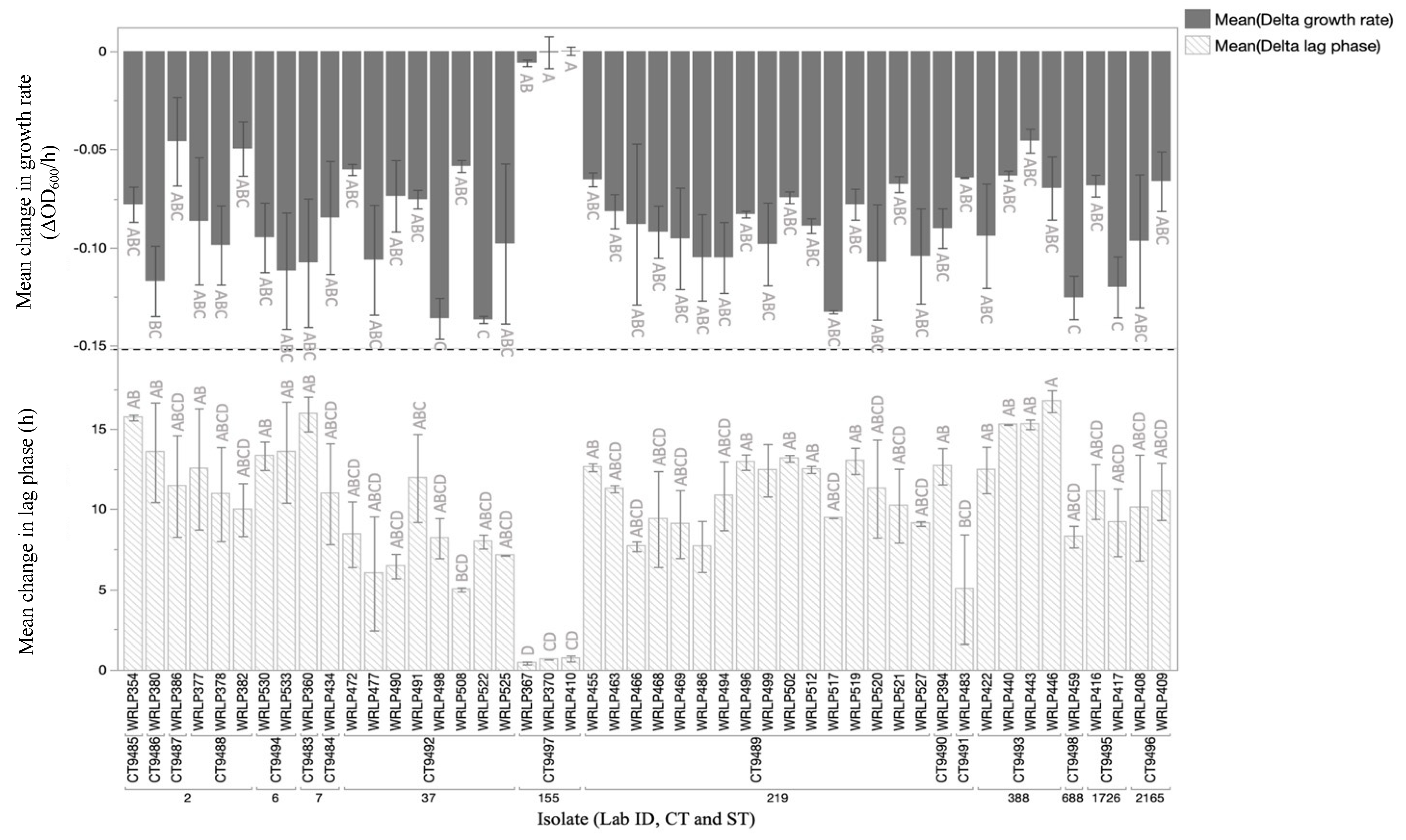
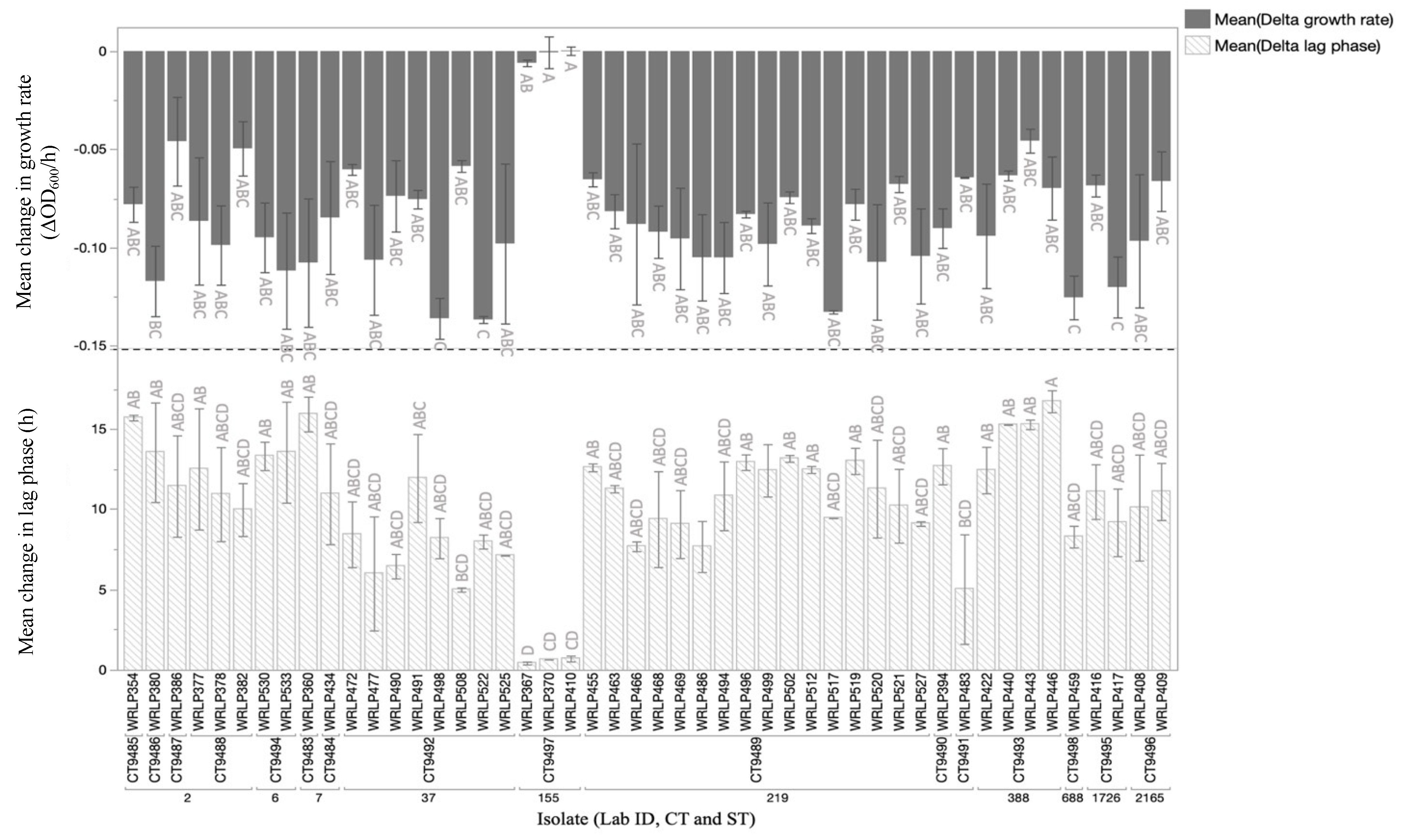
3.5. Phenotypic Analyses and Presence of Genetic Elements Assocaited with QAC Tolerance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tompkin, R. Control of Listeria monocytogenes in the food-processing environment. J. Food Prot. 2002, 65, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.; Luque-Sastre, L.; Parker, C.T.; Huynh, S.; Eshwar, A.K.; Nguyen, S.V.; Andrews, N.; Moura, A.; Fox, E.M.; Jordan, K. Whole-genome sequencing-based characterization of 100 Listeria monocytogenes isolates collected from food processing environments over a four-year period. mSphere 2019, 4, e00252-19. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, Y.; Pouillot, R.; Dennis, S.; Xian, Z.; Luchansky, J.B.; Porto-Fett, A.C.S.; Lindsay, J.A.; Hammack, T.S.; Allard, M.; et al. Genetic diversity and profiles of genes associated with virulence and stress resistance among isolates from the 2010–2013 interagency Listeria monocytogenes market basket survey. PLoS ONE 2020, 15, e0231393. [Google Scholar] [CrossRef]
- Angelo, K.; Conrad, A.; Saupe, A.; Dragoo, H.; West, N.; Sorenson, A.; Barnes, A.; Doyle, M.; Beal, J.; Jackson, K. Multistate outbreak of Listeria monocytogenes infections linked to whole apples used in commercially produced, prepackaged caramel apples: United States, 2014–2015. Epidemiol. Infect. 2017, 145, 848–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC. Multistate Outbreak of Listeriosis Linked to Frozen Vegetables (Final Update). Available online: https://www.cdc.gov/listeria/outbreaks/frozen-vegetables-05-16/index.html (accessed on 10 September 2021).
- CDC. Multistate Outbreak of Listeriosis Linked to Packaged Salads Produced at Springfield, Ohio Dole Processing Facility (Final Update). Available online: https://www.cdc.gov/listeria/outbreaks/bagged-salads-01-16/index.html (accessed on 10 September 2021).
- CDC. Outbreak of Listeria Infections Linked to Enoki Mushrooms (Final Update). Available online: https://www.cdc.gov/listeria/outbreaks/enoki-mushrooms-03-20/index.html (accessed on 10 September 2021).
- Gaul, L.K.; Farag, N.H.; Shim, T.; Kingsley, M.A.; Silk, B.J.; Hyytia-Trees, E. Hospital-acquired listeriosis outbreak caused by contaminated diced celery—Texas, 2010. Clin. Infect. Dis. 2013, 56, 20–26. [Google Scholar] [CrossRef] [PubMed]
- FDA. Memorandum to the File on the Environmental Assessment; FDA: Silver Spring, MD, USA, 2012. [Google Scholar]
- Zhu, Q.; Gooneratne, R.; Hussain, M.A. Listeria monocytogenes in fresh produce: Outbreaks, prevalence and contamination levels. Foods 2017, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Freitag, N.E.; Port, G.C.; Miner, M.D. Listeria monocytogenes—From saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 2009, 7, 623–628. [Google Scholar] [CrossRef]
- Ferreira, V.; Wiedmann, M.; Teixeira, P.; Stasiewicz, M.J. Listeria monocytogenes persistence in food-associated environments: Epidemiology, strain characteristics, and implications for public health. J. Food Prot. 2014, 77, 150–170. [Google Scholar] [CrossRef]
- Wendorf, M.; Feldpausch, E.; Pinkava, L.; Luplow, K.; Hosking, E.; Norton, P.; Biswas, P.; Mozola, M.; Rice, J. Validation of the ANSR® Listeria Method for Detection of Listeria spp. in Environmental Samples. J. AOAC Int. 2019, 96, 1414–1424. [Google Scholar] [CrossRef]
- Benesh, D.L.; Crowley, E.S.; Bird, P.M. 3M™ Tecra™ Listeria Visual Immunoassay: AOAC Official MethodsSM 995.22 and 2002.09. J. AOAC Int. 2019, 96, 218–224. [Google Scholar] [CrossRef]
- Benesh, D.L.; Crowley, E.S.; Bird, P.M. 3M™ Petrifilm™ Environmental Listeria Plate. J. AOAC Int. 2019, 96, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Rapid Microbiology. Listeria Detection and Identification Methods in Foods. Available online: https://www.rapidmicrobiology.com/test-method/listeria-detection-and-identification-methods (accessed on 8 October 2021).
- International Organization for Standardization (ISO). Microbiology of the food chain—Horizontal Method for the Detection and Enumeration of Listeria monocytogenes and of Listeria spp.—Part 1: Detection Method. Available online: https://www.iso.org/standard/60313.html/ (accessed on 8 October 2021).
- Hitchins, A.D.; Jinneman, K.; Chen, Y. BAM Chapter 10: Detection of Listeria monocytogenes in Foods and Environmental Samples, and Enumeration of Listeria monocytogenes in Foods. In Bacteriological Analytical Manual (BAM); U.S. Food and Drug Administration (FDA): Silver Spring, MD, USA, 2017. [Google Scholar]
- Jorgensen, J.; Waite-Cusic, J.; Kovacevic, J. Prevalence of Listeria spp. in produce handling and processing facilities in the Pacific Northwest. Food Microbiol. 2020, 90, 103468. [Google Scholar]
- Jorgensen, J.; Bland, R.; Waite-Cusic, J.; Kovacevic, J. Diversity and antimicrobial resistance of Listeria spp. and L. monocytogenes clones from produce handling and processing facilities in the Pacific Northwest. Food Control 2021, 123, 107665. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Moura, A.; Criscuolo, A.; Pouseele, H.; Maury, M.M.; Leclercq, A.; Tarr, C.; Björkman, J.T.; Dallman, T.; Reimer, A.; Enouf, V. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2016, 2, 16185. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). Basic Local Alignment Search Tool (BLAST). Standard Nucleotide BLAST (blastn). Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 8 October 2021).
- Weisberg, A.J.; Davis, E.W., 2nd; Tabima, J.; Belcher, M.S.; Miller, M.; Kuo, C.H.; Loper, J.E.; Grunwald, N.J.; Putnam, M.L.; Chang, J.H. Unexpected conservation and global transmission of agrobacterial virulence plasmids. Science 2020, 368, eaba5256. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Broad Institute. Picard Toolkit. Available online: http://broadinstitute.github.io/picard (accessed on 8 October 2021).
- Eggertsson, H.P.; Jonsson, H.; Kristmundsdottir, S.; Hjartarson, E.; Kehr, B.; Masson, G.; Zink, F.; Hjorleifsson, K.E.; Jonasdottir, A.; Jonasdottir, A.; et al. Graphtyper enables population-scale genotyping using pangenome graphs. Nat. Genet. 2017, 49, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Kronenberg, Z.N.; Dawson, E.T.; Pedersen, B.S.; Prins, P. Vcflib and tools for processing the VCF variant call format. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A.; et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 2020, 21, 180. [Google Scholar] [CrossRef]
- Baranyi, J.; Roberts, T.A. A dynamic approach to predicting bacterial growth in food. Int. J. Food Microbiol. 1994, 23, 277–294. [Google Scholar] [CrossRef]
- Ragon, M.; Wirth, T.; Hollandt, F.; Lavenir, R.; Lecuit, M.; Le Monnier, A.; Brisse, S. A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 2008, 4, e1000146. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.L.; Xu, J.J.; Chen, N.; Shuai, J.B.; Fang, W.H. Virulence phenotyping and molecular characterization of a low-pathogenicity isolate of Listeria monocytogenes from cow’s milk. Acta Biochim. Biophys. Sin. 2006, 38, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.; Zaiser, A.; Leitner, R.; Quijada, N.M.; Pracser, N.; Pietzka, A.; Ruppitsch, W.; Schmitz-Esser, S.; Wagner, M.; Rychli, K. Virulence characterization and comparative genomics of Listeria monocytogenes sequence type 155 strains. BMC Genom. 2020, 21, 847. [Google Scholar] [CrossRef]
- Cotter, P.D.; Draper, L.A.; Lawton, E.M.; Daly, K.M.; Groeger, D.S.; Casey, P.G.; Ross, R.P.; Hill, C. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 2008, 4, e1000144. [Google Scholar] [CrossRef] [Green Version]
- Maury, M.M.; Bracq-Dieye, H.; Huang, L.; Vales, G.; Lavina, M.; Thouvenot, P.; Disson, O.; Leclercq, A.; Brisse, S.; Lecuit, M. Hypervirulent Listeria monocytogenes clones’ adaption to mammalian gut accounts for their association with dairy products. Nat. Commun. 2019, 10, 2488. [Google Scholar] [CrossRef] [Green Version]
- Raschle, S.; Stephan, R.; Stevens, M.J.A.; Cernela, N.; Zurfluh, K.; Muchaamba, F.; Nuesch-Inderbinen, M. Environmental dissemination of pathogenic Listeria monocytogenes in flowing surface waters in Switzerland. Sci. Rep. 2021, 11, 9066. [Google Scholar] [CrossRef]
- Pirone-Davies, C.; Chen, Y.; Pightling, A.; Ryan, G.; Wang, Y.; Yao, K.; Hoffmann, M.; Allard, M.W. Genes significantly associated with lineage II food isolates of Listeria monocytogenes. BMC Genom. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Den Bakker, H.C.; Desjardins, C.A.; Griggs, A.D.; Peters, J.E.; Zeng, Q.; Young, S.K.; Kodira, C.D.; Yandava, C.; Hepburn, T.A.; Haas, B.J. Evolutionary dynamics of the accessory genome of Listeria monocytogenes. PLoS ONE 2013, 8, e67511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Guo, X.; Weller, D.L.; Pollak, S.; Buckley, D.H.; Wiedmann, M.; Cordero, O.X. Nationwide genomic atlas of soil-dwelling Listeria reveals effects of selection and population ecology on pangenome evolution. Nat. Microbiol. 2021, 6, 1021–1030. [Google Scholar] [CrossRef]
- Hilliard, A.; Leong, D.; O’Callaghan, A.; Culligan, E.P.; Morgan, C.A.; DeLappe, N.; Hill, C.; Jordan, K.; Cormican, M.; Gahan, C.G. Genomic characterization of Listeria monocytogenes isolates associated with clinical listeriosis and the food production environment in Ireland. Genes 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullapudi, S.; Siletzky, R.M.; Kathariou, S. Diverse cadmium resistance determinants in Listeria monocytogenes isolates from the turkey processing plant environment. Appl. Environ. Microbiol. 2010, 76, 627–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelbicova, T.; Florianova, M.; Hluchanova, L.; Kalova, A.; Korena, K.; Strakova, N.; Karpiskova, R. Comparative analysis of genetic determinants encoding cadmium, arsenic, and benzalkonium chloride resistance in Listeria monocytogenes of human, food, and environmental origin. Front. Microbiol. 2020, 11, 599882. [Google Scholar] [CrossRef]
- Dutta, V.; Elhanafi, D.; Kathariou, S. Conservation and distribution of the benzalkonium chloride resistance cassette bcrABC in Listeria monocytogenes. Appl. Environ. Microbiol. 2013, 79, 6067–6074. [Google Scholar] [CrossRef] [Green Version]
- Katharios-Lanwermeyer, S.; Rakic-Martinez, M.; Elhanafi, D.; Ratani, S.; Tiedje, J.; Kathariou, S. Coselection of cadmium and benzalkonium chloride resistance in conjugative transfers from nonpathogenic Listeria spp. to other Listeriae. Appl. Environ. Microbiol. 2012, 78, 7549–7556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhanafi, D.; Dutta, V.; Kathariou, S. Genetic characterization of plasmid-associated benzalkonium chloride resistance determinants in a Listeria monocytogenes strain from the 1998–1999 outbreak. Appl. Environ. Microbiol. 2010, 76, 8231–8238. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Yu, T.; Liang, Y.; Ji, S.; Guo, X.; Ma, J.; Zhou, L. Efflux pump-mediated benzalkonium chloride resistance in Listeria monocytogenes isolated from retail food. Int. J. Food Microbiol. 2016, 217, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.B.; Guldimann, C.; Markkula, A.; Pontinen, A.; Korkeala, H.; Tasara, T. Comparative phenotypic and genotypic analysis of Swiss and Finnish Listeria monocytogenes isolates with respect to benzalkonium chloride resistance. Front. Microbiol. 2017, 8, 397. [Google Scholar] [CrossRef]
- Cherifi, T.; Carrillo, C.; Lambert, D.; Miniai, I.; Quessy, S.; Lariviere-Gauthier, G.; Blais, B.; Fravalo, P. Genomic characterization of Listeria monocytogenes isolates reveals that their persistence in a pig slaughterhouse is linked to the presence of benzalkonium chloride resistance genes. BMC Microbiol. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.L.; Carrillo, C.D.; DeschEnes, M.; Blais, B.W. Genomic markers for quaternary ammonium compound resistance as a persistence indicator for Listeria monocytogenes contamination in food manufacturing environments. J. Food Prot. 2021, 84, 389–398. [Google Scholar] [CrossRef]
- Ebner, R.; Stephan, R.; Althaus, D.; Brisse, S.; Maury, M.; Tasara, T. Phenotypic and genotypic characteristics of Listeria monocytogenes strains isolated during 2011–2014 from different food matrices in Switzerland. Food Control. 2015, 57, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, S.; Lopez, V.; Martinez-Suarez, J.V. The influence of subminimal inhibitory concentrations of benzalkonium chloride on biofilm formation by Listeria monocytogenes. Int. J. Food Microbiol. 2014, 189, 106–112. [Google Scholar] [CrossRef]
- Jiang, X.; Yu, T.; Xu, Y.; Wang, H.; Korkeala, H.; Shi, L. MdrL, a major facilitator superfamily efflux pump of Listeria monocytogenes involved in tolerance to benzalkonium chloride. Appl. Microbiol. Biotechnol. 2019, 103, 1339–1350. [Google Scholar] [CrossRef]
- Rakic-Martinez, M.; Drevets, D.A.; Dutta, V.; Katic, V.; Kathariou, S. Listeria monocytogenes strains selected on ciprofloxacin or the disinfectant benzalkonium chloride exhibit reduced susceptibility to ciprofloxacin, gentamicin, benzalkonium chloride, and other toxic compounds. Appl. Environ. Microbiol. 2011, 77, 8714–8721. [Google Scholar] [CrossRef] [Green Version]
- Romanova, N.A.; Wolffs, P.F.; Brovko, L.Y.; Griffiths, M.W. Role of efflux pumps in adaptation and resistance of Listeria monocytogenes to benzalkonium chloride. Appl. Environ. Microbiol. 2006, 72, 3498–3503. [Google Scholar] [CrossRef] [Green Version]
- To, M.S.; Favrin, S.; Romanova, N.; Griffiths, M.W. Postadaptational resistance to benzalkonium chloride and subsequent physicochemical modifications of Listeria monocytogenes. Appl. Environ. Microbiol. 2002, 68, 5258–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, S.; Begley, M.; Hill, C.; Gahan, C.G. A five-gene stress survival islet (SSI-1) that contributes to the growth of Listeria monocytogenes in suboptimal conditions. J. Appl. Microbiol. 2010, 109, 984–995. [Google Scholar] [CrossRef]
- Keeney, K.; Trmcic, A.; Zhu, Z.; Delaquis, P.; Wang, S. Stress survival islet 1 contributes to serotype-specific differences in biofilm formation in Listeria monocytogenes. Lett. Appl. Microbiol. 2018, 67, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.D.; Fletcher, G.C. Assessing manufacturers’ recommended concentrations of commercial sanitizers on inactivation of Listeria monocytogenes. Food Control 2012, 26, 194–199. [Google Scholar] [CrossRef]
- Pang, X.; Wong, C.; Chung, H.-J.; Yuk, H.-G. Biofilm formation of Listeria monocytogenes and its resistance to quaternary ammonium compounds in a simulated salmon processing environment. Food Control 2019, 98, 200–208. [Google Scholar] [CrossRef]
- Orsi, R.H.; den Bakker, H.C.; Wiedmann, M. Listeria monocytogenes lineages: Genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 2011, 301, 79–96. [Google Scholar] [CrossRef]
- Van Stelten, A.; Simpson, J.; Ward, T.; Nightingale, K. Revelation by single-nucleotide polymorphism genotyping that mutations leading to a premature stop codon in inlA are common among Listeria monocytogenes isolates from ready-to-eat foods but not human listeriosis cases. Appl. Environ. Microbiol. 2010, 76, 2783–2790. [Google Scholar] [CrossRef] [Green Version]
- Nightingale, K.; Ivy, R.; Ho, A.; Fortes, E.; Njaa, B.L.; Peters, R.; Wiedmann, M. inlA premature stop codons are common among Listeria monocytogenes isolates from foods and yield virulence-attenuated strains that confer protection against fully virulent strains. Appl. Environ. Microbiol. 2008, 74, 6570–6583. [Google Scholar] [CrossRef] [Green Version]
- Maury, M.M.; Tsai, Y.-H.; Charlier, C.; Touchon, M.; Chenal-Francisque, V.; Leclercq, A.; Criscuolo, A.; Gaultier, C.; Roussel, S.; Brisabois, A. Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 2016, 48, 308. [Google Scholar] [CrossRef] [Green Version]
- Kovacevic, J.; Arguedas-Villa, C.; Wozniak, A.; Tasara, T.; Allen, K.J. Examination of food chain-derived Listeria monocytogenes strains of different serotypes reveals considerable diversity in inlA genotypes, mutability, and adaptation to cold temperatures. Appl. Environ. Microbiol. 2013, 79, 1915–1922. [Google Scholar] [CrossRef] [Green Version]
- Kanki, M.; Naruse, H.; Taguchi, M.; Kumeda, Y. Characterization of specific alleles in InlA and PrfA of Listeria monocytogenes isolated from foods in Osaka, Japan and their ability to invade Caco-2 cells. Int. J. Food Microbiol. 2015, 211, 18–22. [Google Scholar] [CrossRef]
- Smith, A.; Hearn, J.; Taylor, C.; Wheelhouse, N.; Kaczmarek, M.; Moorhouse, E.; Singleton, I. Listeria monocytogenes isolates from ready to eat plant produce are diverse and have virulence potential. Int. J. Food Microbiol. 2019, 299, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.A.; Theriot, J.A.; Portnoy, D.A. The tandem repeat domain in the Listeria monocytogenes ActA protein controls the rate of actin-based motility, the percentage of moving bacteria, and the localization of vasodilator-stimulated phosphoprotein and profilin. J. Cell Biol. 1996, 135, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travier, L.; Guadagnini, S.; Gouin, E.; Dufour, A.; Chenal-Francisque, V.; Cossart, P.; Olivo-Marin, J.C.; Ghigo, J.M.; Disson, O.; Lecuit, M. ActA promotes Listeria monocytogenes aggregation, intestinal colonization and carriage. PLoS Pathog. 2013, 9, e1003131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holch, A.; Gottlieb, C.T.; Larsen, M.H.; Ingmer, H.; Gram, L. Poor invasion of trophoblastic cells but normal plaque formation in fibroblastic cells despite actA deletion in a group of Listeria monocytogenes strains persisting in some food processing environments. Appl. Environ. Microbiol. 2010, 76, 3391–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poimenidou, S.V.; Dalmasso, M.; Papadimitriou, K.; Fox, E.M.; Skandamis, P.N.; Jordan, K. Virulence gene sequencing highlights similarities and differences in sequences in Listeria monocytogenes serotype 1/2a and 4b strains of clinical and food origin from 3 different geographic locations. Front. Microbiol. 2018, 9, 1103. [Google Scholar] [CrossRef] [PubMed]
- Vilchis-Rangel, R.E.; Espinoza-Mellado, M.D.R.; Salinas-Jaramillo, I.J.; Martinez-Pena, M.D.; Rodas-Suarez, O.R. Association of Listeria monocytogenes LIPI-1 and LIPI-3 marker llsX with invasiveness. Curr. Microbiol. 2019, 76, 637–643. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate ID | Lineage a | ST | CC | cgMLST | Fac.# | bcrABC | SSI-1 b | cadA Type | LIPI1 c | LIPI3 | LIPI4 | inlAd | AMR Profile e |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WRLP360 | 2 | 7 | 7 | CT9483 | 4 | - | + | - | + | - | - | + | CLI |
| WRLP434 | 2 | 7 | 7 | CT9484 | 1 | - | - | - | + | - | - | + | CLI |
| WRLP472 | 2 | 37 | 37 | CT9492 | 1 | - | - | - | + | - | - | + | PEN |
| WRLP477 | 2 | 37 | 37 | CT9492 | 1 | - | - | - | + | - | - | + | PEN |
| WRLP490 | 2 | 37 | 37 | CT9492 | 1 | - | - | - | + | - | - | + | CLI, PEN |
| WRLP491 | 2 | 37 | 37 | CT9492 | 1 | - | - | - | + | - | - | + | CLI, PEN |
| WRLP498 | 2 | 37 | 37 | CT9492 | 1 | - | - | - | + | - | - | + | |
| WRLP508 | 2 | 37 | 37 | CT9492 | 1 | - | - | - | + | - | - | + | |
| WRLP522 | 2 | 37 | 37 | CT9492 | 1 | - | - | - | + | - | - | + | |
| WRLP525 | 2 | 37 | 37 | CT9492 | 1 | - | - | - | + | - | - | + | |
| WRLP367 | 2 | 155 | 155 | CT9497 | 6 | - | +Δ | - | + | - | - | + | CLI |
| WRLP370 | 2 | 155 | 155 | CT9497 | 6 | + | +Δ | - | + | - | - | + | CLI |
| WRLP410 | 2 | 155 | 155 | CT9497 | 6 | + | +Δ | - | + | - | - | + | CIP, CLI |
| WRLP417 | 2 | 1726 | 452 | CT9495 | 6 | - | - | - | + | - | - | + | CHL, CIP, NOV |
| WRLP416 | 2 | 1726 | 452 | CT9495 | 6 | - | - | - | + | - | - | + | CHL, CIP, PEN |
| WRLP354 | 1 | 2 | 2 | CT9485 | 2 | - | - | A1, A2 | + | - | - | + | CLI |
| WRLP380 | 1 | 2 | 2 | CT9486 | 2 | - | - | A1, A2 | + | - | - | + | AMP, CIP, PEN |
| WRLP386 | 1 | 2 | 2 | CT9487 | 2 | - | - | A1, A2 | + | - | - | + | CLI, PEN |
| WRLP377 | 1 | 2 | 2 | CT9488 | 2 | - | - | A1, A2 | + | - | - | + | |
| WRLP378 | 1 | 2 | 2 | CT9488 | 2 | - | - | A1, A2 | + | - | - | + | CLI |
| WRLP382 | 1 | 2 | 2 | CT9488 | 2 | - | - | A1, A2 | + | - | - | + | CIP, CLI |
| WRLP530 | 1 | 6 | 6 | CT9494 | 1 | - | - | A2 | + | + | - | Δ | CLI |
| WRLP533 | 1 | 6 | 6 | CT9494 | 1 | - | - | A2 | + | + | - | Δ | CLI |
| WRLP499 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | |
| WRLP521 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI |
| WRLP455 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI, PEN |
| WRLP463 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI, PEN |
| WRLP466 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI, PEN |
| WRLP468 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI, PEN, NOV |
| WRLP469 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI, PEN, NOV |
| WRLP486 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI, PEN |
| WRLP494 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI, PEN |
| WRLP496 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | |
| WRLP502 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | |
| WRLP512 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | |
| WRLP517 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI |
| WRLP519 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI |
| WRLP520 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI, PEN |
| WRLP527 | 1 | 219 | 4 | CT9489 | 1 | - | - | A2 | +Δ | + | + | + | CLI |
| WRLP394 | 1 | 219 | 4 | CT9490 | 1 | - | - | A2 | +Δ | + | + | + | CLI |
| WRLP483 | 1 | 219 | 4 | CT9491 | 1 | - | - | A2 | +Δ | + | + | + | PEN, NOV |
| WRLP422 | 1 | 388 | 388 | CT9493 | 1 | - | - | A2 | + | - | + | + | CLI |
| WRLP440 | 1 | 388 | 388 | CT9493 | 1 | - | - | A2 | + | - | + | + | CLI |
| WRLP443 | 1 | 388 | 388 | CT9493 | 1 | - | - | A2 | + | - | + | + | AMP, CLI |
| WRLP446 | 1 | 388 | 388 | CT9493 | 1 | - | - | A2 | + | - | + | + | CLI |
| WRLP459 | 1 | 688 | 688 | CT9498 | 1 | - | A2 | + | + | + | + | CLI, NOV | |
| WRLP408 | 1 | 2165 | 345 | CT9496 | 3 | - | - | A1, A2 | +Δ | - | - | + | CIP, CLI, NOV |
| WRLP409 | 1 | 2165 | 345 | CT9496 | 3 | - | - | A1, A2 | +Δ | - | - | + | CIP, CLI |
| Isolate Grouping (Lineage/Serogroup; Sequence Type; Strain ID) | No. Unique Genes a |
|---|---|
| Lineage I/Serogroup 4b, 4d, 4e | 11 |
| ST7 | 20 |
| WRLP360 | 62 |
| WRLP434 | 16 |
| ST37 | 49 |
| WRLP472 | 118 |
| WRLP477 | 143 |
| WRLP490 | 108 |
| WRLP491 | 141 |
| WRLP498 | 96 |
| WRLP508 | 94 |
| WRLP522 | 139 |
| WRLP525 | 210 |
| ST155 | 39 |
| WRLP367 | 103 |
| WRLP370 | 239 |
| WRLP410 | 536 |
| ST1726 | 47 |
| WRLP416 | 406 |
| WRLP417 | 46 |
| Lineage II/Serogroup 1/2a or 3a | 104 |
| ST2 | 36 |
| WRLP354 | 12 |
| WRLP377 | 1 |
| WRLP378 | 7 |
| WRLP380 | 1 |
| WRLP382 | 4 |
| WRLP386 | 13 |
| ST6 | 21 |
| WRLP530 | 509 |
| WRLP533 | 308 |
| ST219 | 3 |
| WRLP394 | 22 |
| WRLP455 | 41 |
| WRLP463 | 72 |
| WRLP466 | 26 |
| WRLP468 | 30 |
| WRLP469 | 26 |
| WRLP483 | 110 |
| WRLP486 | 75 |
| WRLP494 | 101 |
| WRLP496 | 70 |
| WRLP499 | 64 |
| WRLP502 | 1 |
| WRLP512 | 52 |
| WRLP517 | 67 |
| WRLP519 | 83 |
| WRLP520 | 56 |
| WRLP521 | 46 |
| WRLP527 | 51 |
| ST388 | 9 |
| WRLP422 | 212 |
| WRLP440 | 129 |
| WRLP443 | 173 |
| WRLP446 | 683 |
| ST688 | 19 |
| WRLP459 | nd b |
| ST2165 | 103 |
| WRLP408 | 157 |
| WRLP409 | 530 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bland, R.N.; Johnson, J.D.; Waite-Cusic, J.G.; Weisberg, A.J.; Riutta, E.R.; Chang, J.H.; Kovacevic, J. Application of Whole Genome Sequencing to Understand Diversity and Presence of Genes Associated with Sanitizer Tolerance in Listeria monocytogenes from Produce Handling Sources. Foods 2021, 10, 2454. https://doi.org/10.3390/foods10102454
Bland RN, Johnson JD, Waite-Cusic JG, Weisberg AJ, Riutta ER, Chang JH, Kovacevic J. Application of Whole Genome Sequencing to Understand Diversity and Presence of Genes Associated with Sanitizer Tolerance in Listeria monocytogenes from Produce Handling Sources. Foods. 2021; 10(10):2454. https://doi.org/10.3390/foods10102454
Chicago/Turabian StyleBland, Rebecca N., Jared D. Johnson, Joy G. Waite-Cusic, Alexandra J. Weisberg, Elizabeth R. Riutta, Jeff H. Chang, and Jovana Kovacevic. 2021. "Application of Whole Genome Sequencing to Understand Diversity and Presence of Genes Associated with Sanitizer Tolerance in Listeria monocytogenes from Produce Handling Sources" Foods 10, no. 10: 2454. https://doi.org/10.3390/foods10102454
APA StyleBland, R. N., Johnson, J. D., Waite-Cusic, J. G., Weisberg, A. J., Riutta, E. R., Chang, J. H., & Kovacevic, J. (2021). Application of Whole Genome Sequencing to Understand Diversity and Presence of Genes Associated with Sanitizer Tolerance in Listeria monocytogenes from Produce Handling Sources. Foods, 10(10), 2454. https://doi.org/10.3390/foods10102454