Role of Nickel in Microbial Pathogenesis

1
Department of Microbiology, University of Georgia, Athens, GA 30602, USA
2
Center for Metalloenzyme Studies, University of Georgia, Athens, GA 30602, USA
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(7), 80; https://doi.org/10.3390/inorganics7070080
Submission received: 21 May 2019
/
Revised: 14 June 2019
/
Accepted: 20 June 2019
/
Published: 26 June 2019
(This article belongs to the Special Issue Bioinorganic Chemistry of Nickel)
Abstract
:Nickel is an essential cofactor for some pathogen virulence factors. Due to its low availability in hosts, pathogens must efficiently transport the metal and then balance its ready intracellular availability for enzyme maturation with metal toxicity concerns. The most notable virulence-associated components are the Ni-enzymes hydrogenase and urease. Both enzymes, along with their associated nickel transporters, storage reservoirs, and maturation enzymes have been best-studied in the gastric pathogen Helicobacter pylori, a bacterium which depends heavily on nickel. Molecular hydrogen utilization is associated with efficient host colonization by the Helicobacters, which include both gastric and liver pathogens. Translocation of a H. pylori carcinogenic toxin into host epithelial cells is powered by H2 use. The multiple [NiFe] hydrogenases of Salmonella enterica Typhimurium are important in host colonization, while ureases play important roles in both prokaryotic (Proteus mirabilis and Staphylococcus spp.) and eukaryotic (Cryptoccoccus genus) pathogens associated with urinary tract infections. Other Ni-requiring enzymes, such as Ni-acireductone dioxygenase (ARD), Ni-superoxide dismutase (SOD), and Ni-glyoxalase I (GloI) play important metabolic or detoxifying roles in other pathogens. Nickel-requiring enzymes are likely important for virulence of at least 40 prokaryotic and nine eukaryotic pathogenic species, as described herein. The potential for pathogenic roles of many new Ni-binding components exists, based on recent experimental data and on the key roles that Ni enzymes play in a diverse array of pathogens.
1. Introduction
Nickel (Ni) is well established as an essential cofactor for some pathogen virulence factors. While the majority of studies on pathogens’ Ni enzymes relate to human pathogens, a sizable portion of animal pathogens (Helicobacter hepaticus, Helicobacter mustelae, Ureaplasma diversum, Brucella species, Campylobacter species) also use Ni-containing virulence factors (Table 1). In contrast, the literature on Ni-dependent plant pathogens is scarce. This discrepancy reflects a fundamental difference between plants and mammals: plants use nickel as a cofactor for urease, which they oftentimes make in abundance, therefore reducing its availability to pathogens. On the other hand, mammals do not synthesize any (known) Ni-requiring protein(s), hence (host) nickel is likely to be more available for Ni-utilizing pathogens. Still, the intestinal microflora of mammals is comprised of many Ni-utilizing members, such as urease-producing lactobacilli and Bifidobacterium species, or gut methanogens relying on nickel-dependent coenzyme M reductase [1]; those are likely to compete for nickel with pathogens. Interestingly, there is a strong link between nickel pools in plants (e.g., urease-bound) and mammals nickel pools, as plants are one of the main dietary sources of nickel for mammals.
The most notable virulence-associated components are the Ni-enzymes hydrogenase and urease, both of which have been shown to be important for pathogen virulence in various organisms [2,3,4]. Both hydrogenase and urease, along with their associated nickel transporters, storage reservoirs, maturation enzymes, and nickel-dependent regulators have been best-studied in the gastric pathogen H. pylori, so much of our review will discuss the nickel-metabolism factors in the gastric pathogen. Nevertheless, significant progress has been made towards understanding the role of [NiFe] hydrogenases in enteric pathogens, especially in S. Typhimurium; this microbe will thus be covered in our review as well. Also, urease enzymes play important roles in eukaryotic pathogens belonging to the Cryptoccoccus genus, as well as in prokaryotic pathogens such as P. mirabilis and Staphylococcus spp., which are causative agents of urinary tract infections (UTIs). Since the synthesis, structure, and catalytic activity of ureases and hydrogenases have been recently presented and discussed in comprehensive reviews [5,6,7,8,9], these aspects will not be covered in the present review. Likewise, H. pylori hydrogenase and urease maturation, as well as NikR-mediated gene regulation, have been extensively reviewed by our group and others [3,10,11,12], hence they will not be discussed herein.
Other Ni-requiring enzymes, such as Ni-acireductone dioxygenase (ARD) [6,13], Ni-superoxide dismutase (SOD) [14,15], and Ni-glyoxalase I (GloI) [6,16] play important metabolic or detoxifying roles in a few pathogens; little is known about their contribution to pathogenicity; nevertheless, their role will be briefly discussed. The hypothetical or demonstrated role of all Ni-enzymes in pathogens is summarized in Table 1.
2. Nickel Availability to Pathogens and Host-Mediated Influences
Nickel, more so than for other required metals, presents both a difficult acquisition and a homeostasis problem for pathogens, ultimately due to its low availability within the host. Indeed, nickel is found at less than 5 ppm (μg/g of ash) in most human organs [100]. Compared to other metals, such as zinc, nickel is far less prevalent in organs: for instance it is found at a level of less than 1% of the amount of zinc in the brain, heart, lung, or muscle, and the amount of nickel is less than 0.1% of that of zinc measured in both the human liver and kidney [101].
To limit pathogen growth, animal hosts have developed metal sequestering strategies to abrogate the invading pathogen [102]. Indeed, this is thought to be a key antibacterial mechanism used to inhibit initial infection as well as to combat tissue-established ones. Metals are bound by the mammalian host mucosa (a process termed “nutritional immunity”), and this is known to involve mucosal-associated metal binding proteins [102,103]. Although we have significant knowledge of iron, zinc, and manganese sequestering by pathogenic bacteria, and the competition between host and pathogen for these metals is beginning to be understood [102,104,105], much less is known about these aspects with respect to cobalt, nickel, and copper [105]. Recruited neutrophils at inflammation sites express metal-binding proteins, such as calprotectin, lipocalin and lactoferrin. The role of the host defense protein calprotectin in zinc binding and subsequent pathogen inhibition is well-established [105]; however, a recent study from Nakashige and colleagues showed that coordination of Ni(II) at the hexahistidine site of calprotectin is preferred over that for Zn(II) [106]. In agreement with this finding, calprotectin was shown to sequester nickel away from two pathogens, S. aureus and K. pneumoniae, subsequently inhibiting their respective urease activity in bacterial culture [106].
Although the antibacterial effects of the multifunctional globular protein lactoferrin are attributed in part to its iron-binding capacity [107,108], the histidine and tyrosine ligands can bind other metals, including nickel; therefore, a nickel-sequestering effect of lactoferrin towards pathogens should not be ruled out. The same thinking might also apply to the peptide hormone hepcidin, a regulator of host iron homeostasis, which binds both Fe and Ni (II) [109]. While the role of hepcidin in starving pathogens of nickel has not been investigated, its antimicrobial activity via iron sequestering has been well established [110]. The list of siderophilic pathogens affected by hepcidin-driven iron chelation includes E. coli, Staphylococcus spp., including S. epidermidis and S aureus, group B Streptococcus bacteria, Y. enterocolitica, and Candida albicans. In particular, one demonstrated role of hepcidin is to affect metals levels within macrophages [111], so nickel availability may also be expected to be impacted for immune cell-engulfed pathogens. It is well known that macrophages use metal (iron, copper, zinc) sequestration to starve pathogens of essential metals [112]. Of relevance here is that one of the [Ni–Fe] H2-uptake hydrogenases of S. Typhimurium (Hya or Hyd-1) is needed for survival within macrophages [64], an environment in which this hydrogenase, as well as another one, Hyd-5, are greatly up-expressed. Likewise, the hyc operon in Mycobacterium tuberculosis (encoding for a putative [Ni–Fe] H2-evolving complex) is upregulated in human macrophage-like cells, as well as in resting or activated murine bone marrow macrophages [33,34]. Similarly, urease has been shown to be important for survival in macrophages of several pathogens, for instance that of A. pleuropneumoniae [51,52] or C. neoformans [19]. Thus, nickel starvation would be expected to be an immune cell strategy to attenuate pathogen growth.
Many more aspects of nickel restriction to pathogens by host metabolites need to be studied. For example, although not specifically studied with regard to Ni(II), the divalent metal ion transporter NRAMP1 can export metals out of the macrophage phagolysosome, thus restricting the metal availability to the engulfed or intracellular pathogen [113]. It seems likely that some of the antipathogen affects attributed to host iron restriction (or other metals starvation) actually employ nickel starvation as a goal as well. However, host-mediated metal restriction aimed at exacerbating pathogens also complicates metabolism for the host, as host processes are often metal-dependent as well. For example, calprotectin-mediated zinc and manganese starvation attenuates S. aureus abscess infection [114], but at the same time, the metal is needed for host enzymes and for normal immune processes [102].
Regarding nickel restriction, no major effect on the host metabolism is expected, as the mammalian hosts do not contain (known) Ni-dependent enzymes. This has caused several research groups to suggest nickel sequestrations as a possible therapeutic approach to combat Ni-requiring pathogens [10,115,116]. For instance, targeting nickel trafficking pathways to prevent proper maturation of both the H2-uptake [Ni–Fe] hydrogenase and the urease in the gastric pathogen H. pylori has been proposed by several groups, including ours [10,116]. Similarly, a recent study identified the nickel requirement for C. neoformans’s urease as the fungus’s “Achilles’ heels” [117]. However, one has to keep in mind that hosts also contain Ni-requiring prokaryotic and eukaryotic microorganisms as part of their (healthy) microbiota [1], thus multiple aspects of host physiology would be affected. Indeed, disruption of nickel homeostasis in these microflora would be expected to lead to dysbiosis and subsequent health consequences for the host. In this particular case, Ni-enzymes do not play a direct role in pathogenesis, but rather they can be considered a “health-related factor” [118].
3. Ureases
Ureases, which catalyze the hydrolytic decomposition of urea into bicarbonate and ammonia, play a dual role: (i) the ammonia produced by the enzyme is an important source of nitrogen for microorganisms and (ii) both ammonia and bicarbonate can be used to neutralize the pH, thus allowing urease-containing organisms to survive and even thrive in acidic environments [7]. Indeed, mammalian pathogens encounter acid in transit through the stomach, on skin, within abscesses, and inside host cells; the nickel-containing urease activity is thus an enzyme that is critical to many bacteria for surviving acidic environments, for instance, H. pylori in the stomach. The acidic environment the pathogen encounters may be while in transit (such as when temporarily residing in the gastric milieu) whereby the pathogen is “surviving and seeking” a more hospitable host area, or may be encountered while inside the (acidified) phagolysosome. Before its concentration in the kidneys, urea is present in the bloodstream, thus in blood-rich organs. It is abundant in the blood, with levels estimated to range between 2.5 and 7.1 mM; urea is also found in sweat, saliva, and gastric juices [38,119]. Saliva levels in healthy individuals vary from 3 to 10 mM, albeit they can reach up to 15 mM in patients with renal disease [120]. Urea is also present at high levels in the lungs (2–4 mM) and can be used by urease-positive lung pathogens (e.g., H. influenzae, M. tuberculosis, K. pneumoniae, or C. neoformans) as a nitrogen source and/or as a way to neutralize acidic pH; in fact, urease can be used as breath test diagnosis of lung pathogens [121].
As stated above, in addition to its acid-neutralizing properties, urease hydrolyzes urea into ammonium, an important nitrogen source for many pathogens, especially urinary tract pathogens such as P. stuartii, Morganella spp., Pseudomonas spp., U. urealyticum, Klebsiella spp., P. mirabilis, and others (see Table 1) [16]. Urea is extremely abundant in human urine; although urea levels fluctuate widely, the average concentration is around 400 mM [119]. The formation of urinary stones is a direct result of alkalinization of the urinary tract by these pathogens’ urease activity [63]. In this case, Ni-related pathogenesis can be viewed as having several notable outcomes. While exacerbating the host excretory system, the crystalline stones also provide a surface for the pathogen(s) to build biofilms and augment its (their) growth [63]. Furthermore, synergistic induction of urease activity in polymicrobial populations (belonging to the species listed above) leads to an increased incidence of urolithiasis and bacteremia [59]. S. salivarius uses salivary urea both as a source of nitrogen and to combat acid stress [41]. In S. aureus, a pathogen causing significant morbidity due to both acute and chronic infections, the transcription of urease-associated (e.g., structural and accessory/maturation) genes is up-expressed during bacterial biofilm growth [37]. This up-expression is considered to be one component of its acid response network [37,122].
While most ureases are Ni-enzymes, there are a few exceptions. Interestingly, some gastric Helicobacter species, such as H. mustelae (ferret), H. felis (big cats) and H. acinonychis (cheetah), possess two distinct urease gene loci, ureABIEFGH and ureA2B2 [123,124]. The former encodes for a nickel-containing urease (similar to that found in H. pylori) while the latter encodes for a nickel-free, iron-containing isoform [125]. Transcription of the ureABIEFGH operon is induced by the addition of nickel, whereas transcription of ureA2B2 is upregulated by iron and downregulated by nickel; the nickel-responsive transcriptional regulator NikR is involved in this dual Ni-dependent control [124]. The role of the iron-containing urease is not clear; however, the fact that it is only present in species inhabiting the stomach of carnivores may reflect an evolutionary adaptation, according to the authors of these studies [123,124,125]. Carnivores encounter an iron-rich, nickel-scarce diet [126] so infection by these Helicobacter species could be limited if they were to possess only Ni-ureases. The flexibility to produce either a Ni- or Fe-urease allows these Helicobacter species to colonize the gastric mucosa regardless of their host’s diets.
We review here the role in pathogenesis of some of the best characterized microbial urease systems, emphasizing perspectives on the most recently published findings.
3.1. H. pylori
H. pylori must first survive the harsh environment of the human stomach and then survive a prolonged immune response that includes bombardment with oxidative radicals and oxidizing acids. The most severe H. pylori-mediated disease is due to long term infection. Urease, which comprises up to 10% of the total H. pylori proteome [127], is essential for the in vivo survival of H. pylori, as the buffering molecules from urea hydrolysis are essential to maintain the pathogen’s cytoplasmic pH close to neutral. The constant production of ammonia in H. pylori-infected patients with cirrhosis can lead to blood hyperammonemia, which has been linked to a condition named minimal hepatic encephalopathy (MHE). As expected, anti-H. pylori therapy led to a reduction in blood ammonia levels, with subsequent improvement in MHE [128]. Besides the well-established acid-combatting role, many additional roles have been attributed to H. pylori urease over the last 25 years (see Table 1). The Ni-containing active form of urease is clearly required for initial colonization; however, several studies suggest that non-neutralizing roles also exist for urease, and those may not even require the Ni-form. For instance, a urease negative strain was unable to colonize a pH neutral pig stomach [85]. A recent study from Debowski et al. found that urease is needed for persistence in the mouse gastric mucosa, where pH approaches neutrality [96]. This is at first puzzling, since the main role of urease (to survive the low pH) requires considerable expense in terms of number of maturation/accessory enzymes and energy (in the form of GTP hydrolysis); this expenditure and Ni-drain should not occur (i.e., should not be needed) when the bacterium occupies neutral pH environments. However, urease-expressing bacteria are favored for survival/colonization in vivo over long time periods, while the pathogen resides in the mucosa, so urease seems to be needed for chronic and persistent infection [96]. In addition, urease is linked to gastric carcinoma incidence via its ability to promote angiogenesis [97]; the enzyme has been shown to induce proinflammatory cytokines, stimulate chemotaxis of neutrophils and monocytes, and to induce apoptosis in gastric endothelial cells [88,98].
As previously stated, urease (UreAB) is the most abundant enzyme synthesized in H. pylori; however, it appears most of it is never active as a ureolytic enzyme; this is probably due to limiting extracellular (host) and intracellular nickel levels. Indeed, a study from Stingl and De Reuse estimated that the fraction of nickel-activated urease ranged from 2% to 25% depending on growth conditions [129]. Therefore, this observation raises a question: why does H. pylori synthesize so much urease, if the bulk of it is not activated and therefore is not useful to combat acidity? Perhaps much of the above can be explained at the molecular level by a study indicating inactive urease plays a large role in H. pylori survival, due simply to its amino acid residue composition. Indeed, a new role for urease as an antioxidant or reactive oxygen-combating protein has been recently unveiled [99] (Figure 1). Catalytically inactive urease was able to protect the pathogen from oxidative damage, via a Met residue oxidant quenching mechanism. This mechanism does not require nickel, but requires surface Met residues, that cycle between oxidized and reduced forms, yet it would seem that nickel is important to facilitate the overall process, as the nickel containing version would be more stable to proteolysis within the cell, and urease synthesis is up-expressed by nickel [130,131]. The UreAB heterodimer contains 25 Met residues; 11 of these are subject to oxidation and subsequent methionine sulfoxide reductase (MSR) repair through a Met/Met-sulfoxide cycle [99]. In summary, both the catalytic and the noncatalytic role(s) of urease are important for H. pylori initial infection and long-term persistence in the host, as depicted in Figure 1.
3.2. S. aureus
S. aureus is both a commensal bacterium and a human pathogen. It colonizes approximately 30% of the population asymptomatically; however, it can also cause infections ranging from mild skin and soft tissue infections to invasive infections, including sepsis and pneumonia [132]. A recent study by Zhou and coworkers examined the S. aureus urease response and its roles in host persistence [38]. The authors note that S. aureus combats many different host environments, and urease may have very different roles in different pH conditions, in part due to the largely underappreciated differing mode of action of strong versus weak acids on the cells. For example, weak acids such as acetate enter the cytoplasm more easily than strong acids that fully dissociate in water. Therefore, the weak acids cause macromolecular damage via intracellular proton release. Using a mutant strain analysis approach, it was concluded that urease activity is important to cell viability under weak acid stress conditions [38]. Kidney colonization was compared in a mouse bacteremia model; kidneys infected with the S. aureus Δure mutant strain had significantly lower bacterial burden in the longer term (12 and 19-day) infections than did the wild type strain. The host immune response, as assessed by leukocyte populations, did not differ between the two infected groups, so the absence of urease did not seem to influence the immune response (i.e., to augment clearance). These authors postulate that urease is important for host skin survival, where S. aureus resides; indeed, human skin is a major reservoir of this pathogen (for instance in sweat glands and hair follicles) and the authors reported that sweat contains 22 mM urea and the skin pH ranges from 4 to 6.
Biofilms are a significant contributor to host colonization and subsequent virulence by many pathogens, including S. aureus. Interestingly, a study from Resch et al. found increased expression of some of the urease structural and accessory genes (in correlation with increased urease activity) in biofilm-embedded S. aureus cells compared to planktonic cells of the same strain [37]. Connections between staphylococcal biofilms, urease production, and antibiotic resistance were further analyzed in a recent study [39]. When mixed biofilms composed of two species of Staphyloccoccus (S. aureus and S. epidermidis) were compared to monospecies biofilms, the urease subunit genes as well as the accessory protein genes were downexpressed in the mixed source. Since each species did not influence survival of the other, and the initial ratios used in biofilm formation were maintained, it was therefore determined that the cospecies influence on urease gene expression was specific to biofilm cultures [39]. According to the authors, S. epidermidis inhibits metabolic activity of S. aureus, leading to less acid production. As a consequence, less urease activity is required to compensate for low pH. Importantly, the two species used, S. epidermis and S. aureus, are oftentimes coisolated from biofilms on indwelling medical devices [133].
3.3. P. mirabilis
P. mirabilis is a major cause of urinary stones and it also forms resilient crystalline biofilms on catheters [63]. The initial formation of large clusters of the bacteria in the bladder lumen may be the etiology of stone formation [62], and urease is considered to be one of the two most important virulence factors in the initiation of cluster development [62,134]. In catheter-associated UTIs, urease-produced ammonium and carbon dioxide bind with Mg2+ and Ca2+, respectively, found in the urine. These minerals precipitate, forming crystalline deposits on catheters or/and aggregates that evolve into macroscopic stones within the urinary system [135]. Adherent bacteria grow and the crystalline biofilm enlarges, so that bacteria and crystals become tightly associated [134]. Later, bacteria can become dissociated and begin the crystallization process elsewhere. P. mirabilis mutant strains that lack urease are unable to form crystalline biofilms [134]. Note that the urea level in human urine is reportedly about 400 mM [119], the average nickel level is approximately 1.7 μg/L (ranging from 0.1 to 20 μg/L) [136] and that the urease activity of P. mirabilis is especially robust.
Not much is known about P. mirabilis’ ability to import and sequester nickel. Based on genome sequence analysis, P. mirabilis has two predicted Ni-transporters (nikAB and yntABCD); both transcriptional units are induced in experimentally-infected mice compared with laboratory-grown cells [63]. While it does not contain Ni-storage proteins, such as the ones found in H. pylori (Hpn, Hpn-like and HspA); nevertheless, P. mirabilis contains two His-rich accessory proteins: UreE, a putative urease accessory protein, contains a His-rich C-terminus domain (eight His residues out of nine residues); HypB, a putative hydrogenase accessory protein, is unusually His-rich, as it contains 17% His; almost all His residues are located in the N-terminus part of PmHypB, bringing the percentage of His residues to an astonishing 39% (Figure 2). It is possible HypB plays a dual role in both hydrogenase and urease maturation in P. mirabilis, as previously demonstrated for both H. pylori [137] and H. hepaticus [138]. In addition, the maturation accessory factor could serve as nickel storage or as a sensor of Ni homeostasis status for the urinary tract pathogen but this remains to be shown.
The extracellular cluster of urease-containing P. mirabilis in the bladder lumen leads to a robust immune response [62]. Adjacent to the P. mirabilis clusters at the bacteria–bladder interface in the mouse model of UTI, neutrophil marker characteristics consistent with antimicrobial peptides in neutrophil extracellular traps/webs and phagocytosed bacteria were all observed. Therefore, the bacterium is likely to be subjected to significant oxidative stress in vivo, and it seems thus conceivable that alternative roles of urease (such as the one recently described for H. pylori urease [99]), could occur at these sites.
3.4. Ureaplasma spp.
Ureaplasma spp. include U. urealyticum, U. parvum, and U. diversum. The first two species are responsible for vaginal infections in humans while the later species cause urogenital tract infection in cattle and small ruminants [42,45]. As the name implies, Ureaplasma species are urease positive, to the extent that the enzyme is actually used as genus-specific diagnostic [42]. At least in U. urealyticum, and probably in other members of this Mollicute class, urease fulfils several roles: (i) the ammonia released in the cytosol contributes to PMF-driven ATP synthesis [43]; (ii) the ammonia also increases the urinary pH, leading to Mg precipitation and subsequent struvite stone formation, as shown in rat bladders [44].
3.5. Eukaryotic Pathogens
In contrast to metals such as copper, zinc, and iron, little is known of the roles of nickel in fungal pathogenesis. Still, ureases play important roles in fungal pathogens, for example in C. neoformans and in C. immitis (Table 1) [140]. In C. neoformans (responsible for human meningoencephalitis), Ni-urease is an important factor for brain invasion, as shown in several independent studies [18,19]. The enzyme can be found in extracellular vesicles, apparently used by the fungus to colonize host tissues [20]. Urease maturation components, sometimes referred to as accessory proteins or maturation chaperones, largely resemble their bacterial counterparts [141]. Although the cryptococcal genome lacks ureE and ureG homologs, one accessory protein, named Ure7, combines the nickel incorporation functions normally assigned to both UreE and UreG [141]. In C. posadasii, the causative agent of San Joaqin Valley fever, the extracellular ammonia generated (by urease) at sites of pulmonary infection contributes to severity of the respiratory disease [23], and urease mutants are less virulent in a mouse intranasal challenge [22]. A nickel permease homolog is present in the Aspergillus fumigatus genome, but the role it plays is not known [140]. It is worth noting, however, that many fungal pathogens apparently have no need for nickel, relying instead on a non-Ni, biotin-requiring urease to metabolize urea.
4. Hydrogenases
Hydrogenases are found in bacteria, archaea, and in some eukarya. They catalyze the conversion of molecular hydrogen (H2) into protons and electrons and the reverse reaction, the generation of H2 [142]. Three classes of hydrogenases have been defined, based on the metallic content of their active site: [NiFe], [FeFe], and [Fe] hydrogenases. Several [NiFe] hydrogenases, (especially of the H2-uptake type) have been shown to be key to colonization and virulence in various organisms such as H. pylori [80] or S. Typhimurium [143]. We review here the major findings on [NiFe]-mediated H2 use by pathogenic bacteria.
4.1. H. pylori
H2-uptake hydrogenase activity was first measured in whole cells of microaerobically grown H. pylori, using an amperometric assay and various artificial and natural electron acceptors, including oxygen [144]. The activity was subsequently shown to be specifically associated with membrane fractions and within these membranes, the hydrogenase enzyme was shown to be poised at a redox potential to oxidize H2 rather than to evolve the gas [144]. Based on genome sequence analysis, H. pylori contains only one hydrogenase, of the H2-uptake type (hydABCDE operon). Transcription of the hyd operon is controlled by various regulatory proteins in response to distinct stimuli (iron, nickel, pH, H2); for instance, hyd genes are transcriptionally repressed by the apo (iron-free) form of Fur, the ferric uptake regulator [145]. Furthermore, the transcription of each of the hydABC structural genes is repressed in wild-type cells grown in nickel-supplemented medium; however, this repression is not observed in a ΔnikR mutant [146,147]. Finally, H2 supplementation increases both hydA transcription and H2-uptake hydrogenase activity in H. pylori [80]; however, neither the H2-sensing mechanism, the H2-responding regulatory mechanism, nor the global H2-responsive proteome, has been characterized in H. pylori.
A unique particularity of nickel trafficking in H. pylori is the interplay between the urease and hydrogenase maturation pathways. Indeed, Olson et al. found that two of the hydrogenase accessory enzymes, HypA and HypB, are required not only for hydrogenase maturation, but also for urease maturation [137]. Additional studies from various groups (including ours) provided further evidence of the interconnectivity between both maturation pathways. Indeed, HypA was shown to physically interact with the urease accessory protein UreE [148,149], and a HypA-(UreE)2 heterotrimeric complex able to bind nickel has been characterized [150]. Furthermore, nickel transfer between both proteins (from HypA to UreE) was demonstrated [151]. Finally, HypB was also found to be physically associated with another urease maturation protein, UreG [152].
Since the H. pylori hydrogenase Km for H2 is approximately 1.8 μM and the concentration of dissolved H2 in animal and human stomachs is in the high micromolar-low millimolar range, the enzyme is predicted to be chronically saturated with H2 [80,153,154]. The [Ni–Fe] hydrogenase is important for virulence: the hydB mutant colonized only 24% of mouse stomachs, while 100% of stomachs inoculated with the parent strain were colonized [80]. Recent studies have showed that the energy (proton motive force, PMF) derived from H2 respiration can drive various important cellular mechanisms in H. pylori. Firstly, a link between H2 utilization and CO2 fixation (in the form of HCO3−) was established in H. pylori [81]. It is interesting to note that this H2-stimulated CO2 fixation (also referred to as “H2-stimulated mixotrophy”) is a growth mode that has never been described for a human pathogen [81]. Secondly, the hydrogenase-mediated H2 respiration can fuel CagA (cytotoxin-associated gene A) translocation into host cells. CagA-positive strains have increased adenocarcinoma incidence [155]. A carcinogenic derivative strain that had greater ability to translocate CagA was found to have higher hydrogenase activity than its noncarcinogenic parent strain [82]. In agreement with this result, a H. pylori Δhyd hydrogenase deletion mutant was unable to translocate CagA into human gastric epithelial AGS cells and the strain did not induce gastric cancer in gerbils [82]. By contrast, 50% of gerbils infected with the wild-type strain (hydrogenase positive, CagA translocating) developed gastric cancers [82]. Finally, albeit a limited strain set was studied, a significantly higher hydrogenase activity was measured in H. pylori strains isolated from cancer patients, compared to those measured in strains isolated from gastritis patients [82]. Taken together, these results suggest a correlation between the H. pylori Ni-hydrogenase and (CagA-mediated) cancer.
4.2. H. hepaticus
H. hepaticus has been shown to induce liver disease in mice, as well as colitis, colorectal cancer, inflammatory bowel disease (IBD), and prostate cancer [156,157]. Based on genome sequence, H. hepaticus possesses only one [NiFe] H2-uptake membrane-bound hydrogenase [158]. Similar to what was reported in H. pylori, hyp hydrogenase accessory genes are present, and mutations in either hypA or hypB abolish both the hydrogenase and the urease activities [138]. Whole cells of H. hepaticus are able to couple H2 oxidation to O2 uptake [159]. H2 concentrations measured in the livers of live adult mice are above 50 μM, which means that H. hepaticus hydrogenase, with an apparent Km of approximately 2.5 μM, is saturated with H2 [159]. Mehta and colleagues showed that the energy derived from H2-oxidation can be used for amino acid uptake, eventually enhancing cell growth; this dual phenotype was observed with the WT strain but not in a ΔhyaB mutant strain [77]. While there was no significant difference in bacterial count numbers between WT and ΔhyaB mutant strains in the liver or cecum of mice, various liver lesions were observed with the WT but not with the mutant [77]. To summarize, the H. hepaticus [NiFe] hydrogenase provides energy (in the form of PMF) to the cell, aiding amino acid transport, bolstering growth and eventually contributing to liver pathogenesis, at least in the established murine model.
4.3. S. Typhimurium
Similar to E. coli, the enteric pathogen S. Typhimurium contains four different [Ni–Fe] hydrogenases: Hya (Hyd-1), Hyb (Hyd-2), Hyc (Hyd-3), and Hyd (Hyd-5) [67]. However, in contrast to E. coli that contains two H2-uptake and two H2-evolving hydrogenases, S. Typhimurium possesses three respiratory (H2-uptake) enzymes (Hya, Hyb, and Hyd) and only one H2-synthesizing enzyme (Hyc); the latter forms the formate-hydrogen-lyase (FHL) system together with the formate dehydrogenase-H (FDH-H), coupling H2 production to formate oxidation, similar to what has been described in E.coli [160]. Each of the three respiratory hydrogenases is coupled to a respiratory pathway that can use O2 as the terminal electron acceptor [143,161]. However, S. Typhimurium can use many terminal acceptors and it can be expected that they could all be coupled to H2 oxidation. The role of each respiratory enzyme as well as their specific expression in various environments (murine macrophages, human polymorphonuclear leukocyte (PMN)-like cells, and mice) was studied using a mutagenesis approach, combined with RIVET (Resolvase In Vivo Expression Technology) [64]. The hya mutant was expressed at low levels in all (mouse) locations tested (e.g., the ileum, the liver and the spleen) and its survival in macrophages was decreased (compared to the WT), a phenotype attributed to the higher acid sensitivity observed for this mutant [64]. The hyd (Hyd-5) gene was found to be highly expressed in the liver and spleen, and weakly expressed in the ileum, at early stages of infection. In the late stages of infection, hyd was expressed at high levels in all organs tested [64]. Expression of the hyb (Hyd-2) gene could not be studied, due to a lack of stability of the hyb RIVET construct.
The role of each enzyme in physiology and virulence was assessed by constructing a series of markerless mutants and testing them using the typhoid fever-mouse model [143]. Double-mutant strains expressing only Hya (Δhyb Δhyd) or only Hyd (Δhya Δhyb) had lower virulence compared to the WT. In contrast, the Δhya Δhyd double mutant strain retaining Hyb activity was almost as virulent as the WT strain, suggesting Hyb is the most important hydrogenase for S. Typhimurium virulence [66,162]. Interestingly, the triple mutant (Δhya Δhyb Δhyd) was found to be avirulent (100% survival in the typhoid fever mouse model) [143]. This was confirmed by an independent study [163].
Based on the analysis of the S. enterica Typhi genome sequence, it appears the causative agent of typhoid fever in humans has the same set of hydrogenases as S. Typhimurium. Given the results of mouse studies with S. Typhimurium, it is expected that one or several of the [Ni–Fe] respiratory hydrogenases of S. Typhi could play an important role in the pathogenicity of typhoid fever in humans.
4.4. C. jejuni
C. jejuni, a leading cause of human diarrheal disease, is a microaerophilic bacterium that possesses a unique, energy-conserving, membrane-bound [Ni–Fe] uptake-type hydrogenase [74,164]. The enzyme is important for both C. jejuni’s growth and virulence. Indeed, in addition to carbon sources formate and fumarate, the respiratory reductant H2 has been found to enhance growth of C. jejuni [74,165]. Disruption of the hydB gene led to abolition of hydrogenase activity, as expected, and the ΔhydB mutant showed severe colonization deficiency of the chicken cecum (compared to the WT) but only in the context of a ΔfdhA (formate dehydrogenase) mutant background [74]. Both ΔfdhA and ΔhydB single mutants showed only modest reduced colonization compared to WT. Finally, the C. jejuni ΔhydB is impaired in cell division (scanning electron microscopy revealed a filamentous phenotype) and is unable to interact with either human intestinal cell lines (INT-407) or with primary chicken intestinal epithelial cells [75]. Thus, similar to what has been observed in H. pylori (which belongs to the same phylogenetic group, the ε-proteobacteria), the [NiFe] H2-uptake hydrogenase plays an important role in C. jejuni metabolism and pathogenesis.
4.5. C. concisus
C. concisus has been found throughout the entire human oral-gastrointestinal tract. The bacterium is associated with various ailments and diseases, such as gingivitis, periodontitis, inflammatory bowel disease, including Crohn’s disease [166]. C. concisus contains genes encoding for two distinct Ni-containing hydrogenase complexes: a H2-uptake type hydrogenase (“Hyd”) similar to those found in other pathogenic ε-proteobacteriae (such as H. pylori or C. jejuni) and a H2-evolving type hydrogenase similar to Hyd-3 (Hyc) and Hyd-4 (Hyf) complexes found in E. coli [167]. The former appears essential, as it is possible to disrupt components of the Hyf complex (hyfB), whereas attempts to generate hyd mutants were unsuccessful [167]. Furthermore, C. concisus has the highest H2-uptake hydrogenase activity reported so far among pathogenic bacteria [167]. In agreement with these observations, H2 was found to be needed for optimal growth under anaerobic conditions, and required for growth under microaerobic conditions, highlighting the importance of the H2-uptake hydrogenase in the pathogen’s metabolism [76,168].
4.6. S. flexneri
Shigella spp. including S. flexneri, S. boydii, S. sonnei, and S. dysenteriae, cause shigellosis (also called bacillary dysentery). Shigella spp. are responsible for approximately 165 million illness episodes worldwide, leading to an estimated 164,000 diarrhoeal deaths annually [169]. Based on genome sequences, Shigella spp. have four predicted unidirectional hydrogenases: two H2-uptake enzymes, Hya and Hyb, and two H2-evolving enzymes, Hyc and Hyf, although the role of the latter remains elusive. McNorton and Maier used a targeted mutagenesis approach to address each enzyme’s respective role in S. flexneri [70]. Both H2-uptake hydrogenases in S. flexneri, and more specifically Hya, can combat severe acid stress through generation of abundant periplasmic proton pools that are hypothesized to act as a barrier against proton influx from the outside [70]. Based on mutant strain analysis, much of the H2 oxidation was attributed to the Hya hydrogenase: its activity was three-fold activated within minutes of acid exposure. This acid activation phenomena has clear pathogen survival consequences, as the Hya enzyme is the hydrogenase shown (in S. Typhimurium) to combat or to resist phagolysosome killing, and a primary method of such killing by immune cells is acidification [170].
5. Other Ni-Dependent Enzymes
Besides urease and hydrogenase, three other Ni-dependent enzymes can be found in a few pathogens: these are the Ni-activated forms of acireductone dioxygenase (ARD) [6,13], glyoxalase I (GloI) [16], and superoxide dismutase (SOD) [15]. Although it could be argued that neither of these three enzymes directly contributes to pathogenesis, the first two (ARD and GloI) play important roles in metabolism of their respective host, while the third (SOD) is a key contributor to oxidative stress resistance in bacteria. Thus, all three Ni-enzymes are expected to play (to a certain degree) a role in metabolism, growth, and virulence of their bacterial hosts. In support of this, heterozygous glo-I mutants of L. donovani (causative agent of visceral leishmaniasis) were found to exhibit reduced methylglyoxal detoxification, and glo-I null mutants were not viable, illustrating the importance of Ni-GloI for this parasite [28].
5.1. Acireductone Dioxygenase (ARD)
The ARD enzyme has two different activities depending on whether it uses Fe2+ or Ni2+ as cofactor [6,13]. The enzyme, which is part of the methionine salvage pathway, uses the same substrates (1,2-dihydroxy-3-keto-5-methylthiopent-1-ene (acireductone) and O2) regardless of the bound metal (Fe2+ or Ni2+); however formate and the ketoacid precursor of methionine, 2-keto-4-methylthiobutyrate are produced in presence of Fe2+, whereas methylthiopropionate, carbon monoxide and formate are produced in presence of Ni2+ [13]. Based on genome sequence analysis, the Ni-containing form of ARD is expected to be found in all pathogenic γ-proteobacteriaceae, as well as in A. baumannii, P. aeruginosa, and S. pneumoniae (Table 1). There is no known Ni-ARD in eukaryotes. The structure of the K. pneumoniae Ni-ARD was revealed by Pochapsky and coworkers, using NMR and X-ray absorption spectroscopy [171].
5.2. Ni-Glyoxalase I
The glyoxalase I (Glo1) enzyme, also called lactoylglutathione lyase, is part of a three-component system aimed at detoxifying methylglyoxal, a chemical that forms adducts with DNA; besides GloI, the system involves the thioesterase glyoxalase II (GloII) and reduced glutathione (GSH), the final product of the detoxification pathway being d-lactate [6,16]. There are two distinct classes of GloI: a Zn2+-dependent class and a Co2+/Ni2+-dependent class, both of which can be found in a variety of eukaryotic and prokaryotic organisms [16]. The former (Zn-GloI) includes Homo sapiens, Saccharomyces cerevisiae, and Pseudomonas putida, while the latter (Ni-GloI) was originally described in E. coli [172]. Since then, nickel has been shown to be the preferred cofactor of GloI in various prokaryotic pathogens such as P. aeruginosa, N. meningitidis, and Y. pestis [173]. In addition, Clostridium acetobutylicum GloI co-crystallized with nickel [36]. Based on sequence analysis, the authors of the study hypothesize other clostridial GloI to be also Ni-activated, including those of pathogenic C. botulinum, C. perfringens, and C. tetani [36]. Since S-lactoylglutathione, a product of GloI, has been shown to play an important role in potassium efflux in E. coli [174], a similar role can be expected for pathogenic E. coli species, as well as for the (Ni) GloI-containing bacteria cited above. In fact, based on genome sequence analysis, the Ni-containing isoform of GloI is widespread among bacterial species. For instance, all Enterobacteriaceae (including E. coli, Enterobacter spp., Klebsiella spp., Morganella spp., Proteus spp., Providencia spp., Serratia spp., Salmonella spp) are expected to have the Ni-GloI type. Finally, Ni-GloI can also be found in protozoan parasites [175], including L. major [27] and T. cruzi [29]. Likewise, the GloI homolog from L. donovani is also expected to be Ni-dependent, based on genome sequence analysis. As stated above, the gloI gene is essential in L. donovani, leading the authors to identify GloI as a potential drug target [28].
5.3. Ni-Superoxide Dismutase (Ni-SOD)
Superoxide dismutases (SOD), which catalyze the dismutation of superoxide radicals (O2•−) into molecular oxygen (O2) or hydrogen peroxide (H2O2), can be found in all domains of life. Three distinct groups of SODs have been defined, based on amino acid sequence homology and preferred metallic cofactors: Cu-Zn-SOD, Fe-SOD and Mn-SOD, and Ni-SOD [15]. The Ni-SOD are seldom encountered; they were first described in a few species of the genus Streptomyces [176], including phytopathogenic species such as S. scabies, S. acidiscabies, and other related species. More recently, the Ni-SOD gene (sodN) has been found in cyanobacteria, marine γ-proteobacteria species, and in a marine eukaryote [177].
6. Nickel Transport and Nickel Metallophores
Pathogens must provide soluble Ni(II) [15] to mobilize the metal into the key nickel enzymes amongst an environment where this metal is in low availability (~0.5 nM) in the host [178]. A number of Ni-binding strategies are used by the pathogens, and the transporters vary in subunit composition, in Ni-binding affinity, and in chelating mechanism and chemistry. Several recent reviews have extensively covered nickel import by bacteria, including in human pathogens [11,179,180]. Therefore, we will only present the latest findings on nickel transport, and limit this to pathogens.
Like other transition metals, nickel needs to be first scavenged and imported from the extracellular environment. These so-called “nickelophores” (by analogy to iron siderophores) are small molecules which can chelate nickel ions before delivering it to specific transporters. Several recent studies have deciphered the structure and the specificity of these metallophores. In the case of nickel, L-His and its derivatives could play such roles [11,179]: for instance, in E. coli, a Ni–(L-His)2 complex with NikA has been revealed by X-ray crystallography [181]. Likewise, S. aureus produces a nicotianamine-like metallophore called staphylopine (StP) to acquire metals under metal-limited conditions [182]. At first, StP was thought to be mostly zinc specific, but recent studies have demonstrated that it can also bind nickel [183,184]. In P. aeruginosa, an organism best known for its high affinity siderophores pyochelin and pyoverdin (the latter being a virulence factor), a recent study from Lhospice et al. has shown that a staphylopine-like metallophore named pseudopaline is able to import nickel in metal scarce environment [185].
In Gram-negative bacteria, the TonB/ExbB/ExbD machinery is needed to energize TonB-dependent transporters, allowing them to transport metals, including nickel, across the outer membrane (OM). For instance, the TonB-dependent FrpB4 protein has been shown to transport nickel through the OM in H. pylori [186].
Two main types of high affinity transporters are used by bacteria to transport Ni(II) across the cytoplasmic membrane. These are the ATP-binding cassette (ABC)-type transporters and the “secondary” nickel/cobalt NiCoT transporters [180]. In addition, a subclass of ABC transporters has been identified: the energy-coupling factor (ECF) transporter, which also requires ATP but uses a membrane-embedded solute binding protein; instead, ABC transporters rely on a soluble periplasmic binding protein (reviewed in [187]). So far, nine nickel ABC importers have been experimentally shown to import nickel in vivo, and only three nickel-binding proteins from human pathogens have been characterized: these are the C. jejuni NikZ, the B. suis NikA and the Y. pestis YntA [179].
Most prokaryotic pathogens mentioned in this review use both types of Ni-transporters. For instance, H. pylori possess both the NiUBDE transporter (ABC-type) and the NixA (NiCoT-type) [188,189]. Both NiuBDE and NixA function as nickel transporters independently of each other, and they are the sole nickel transporters [189]. Although both NiuBDE and NixA participate in nickel acquisition for urease activation, NiuBDE is the only transporter that can operate at both acidic and neutral pH. Furthermore, NiuBDE is also able to transport cobalt or bismuth (this is important, as bismuth is currently used in H. pylori eradication therapy), whereas NixA only transports nickel [189]. Finally, H. pylori nixA mutants retained some colonization ability in two different murine models, in contrast to niuBDE mutants. The latter strains are unable to colonize mouse stomachs, indicating that NiuBDE is required in vivo, but NixA is not [189,190].
In S. aureus, there are three distinct nickel transporters; however, they also fall into the two classes discussed above. Indeed, S. aureus possesses two canonical ABC-importers, the NikABCDE and the (recently discovered) CntABCDF systems, as well as the NixA system that belongs to the NiCoT family [191,192,193]. The Nik system functions in metal-replete medium and is required for urease activity as well as for urinary tract colonization [192]. In contrast to Nik, the multi-cation transporter Cnt is expressed under zinc-depleted conditions. However Cnt also plays an essential role in S. aureus virulence, as it contributes to colonization of the bladder and kidneys in an ascending urinary tract infection model, as well as in systemic infections in mice [193]. Extracytoplasmic nickel-binding components for the Nik and the Cnt ABC-type systems are SaNikA and SaCntA, respectively. A recent study, combining crystallography and mass spectrometry approaches, defined each protein’s substrate specificity: SaNikA is able to bind either a Ni–(L-His)2 complex or a Ni–(L-His) (2-methyl-thiazolidine dicarboxylate) complex (depending on their availability), while SaCntA binds Ni(II) via a different histidine-dependent chelator; however, it cannot bind Ni–(L-His)2 [194].
In E. coli and other enterobacteria, only the ABC-type transporter Nik system is present. As stated above, a Ni–(L-His)2:NikA complex was identified a few year ago [181]. It is worth noting however that transport of Ni–(L-His)2 in E. coli is not a TonB-dependent process, since E. coli ΔtonB mutants are still able to transport it inside the cell [195]. Interestingly, the nikABCDE gene cluster of uropathogenic E. coli (UPEC) is up-expressed in urine samples isolated from UTI patients, as compared to the same UPEC strain cultured in urine from healthy volunteers or grown in lysogeny broth, suggesting that nickel transport is a key fitness factor for the bacteria during human UTI [196]. In agreement with this hypothesis, Δnik mutants were shown to be compromised in fitness in the mouse model of UTI [196]. Even though enterobacteria do not possess the second Ni-transport system (NiCoT-type), it seems they have developed alternate strategies to import metals, including nickel. Indeed, enterobacteria that encode the Yersinia high pathogenicity island (HPI), including strains of E. coli, Klebsiella, and Y. pestis, secrete the metallophore Yersiniabactin (Ybt). Originally shown to chelate iron ions during infection, Ybt can also bind extracellular nickel in UPEC [197]. Ni-Ybt complexes are internalized, then metal-free Ybt is recycled outside the cell while the captured nickel is liberated for use by Ni-requiring enzymes (i.e., hydrogenases and/or urease depending upon the bacterial species). The authors hypothesize the Ybt system can chelate nickel ions that appear to be otherwise inaccessible to the NikABCDE permease [197]. In Mycobacterium avium subsp. paratuberculosis, expression of the dppA gene encoding for a Nickel/dipeptide transporter (ABC type) increased during early infection in an epithelium-macrophage co-culture system [198].
Finally, while nickel importers play a major role in providing Ni-requiring enzymes with the metallic cofactor, nickel exporters are equally important, as they ensure that intracellular nickel levels do not reach toxic concentration. Several nickel export systems have been characterized. For instance, the CznABC (cobalt zinc nickel) export pump of H. pylori was shown to play a critical role in both nickel homeostasis and in vivo stomach colonization: czn mutants had higher urease activities, yet they were unable to colonize in a Mongolian gerbil stomach animal model [199]. Likewise, a P. mirabilis putative nickel export transporter (PMI1518) was found to be essential for CAUTI, in single-species kidney colonization as well as in bladder and kidney colonization coinfection with P. stuartii [200].
7. Nickel Storage, Toxicity, and Metabolism
Among bacterial pathogens, mechanisms used to sequester and store nickel, as well as to remove it due to its toxic properties on macromolecules, are best known for the gastric pathogen H. pylori. This is not surprising as this bacterium contains two nickel enzymes (see above) that are key to the pathogen’s in vivo survival. As such, its demand for nickel is great, but along with the high demand necessarily goes risk for potential toxicity. Much of the bacterium’s regulatory mechanisms are based on sensing nickel levels, thus on nickel-binding proteins. Nickel overload inside the cell is countered by efflux mechanisms and by repression of nickel transport factors, while intracellular levels as well as storage of the metal for later use in Ni-enzyme manufacture is mediated by proteins known within the field as nickel storage proteins. The latter system involves two histidine-rich proteins (termed Hpn and Hpn-like) as well as at least one chaperone (HspA) that plays multiple roles. Most likely, the multiple Ni-binding proteins needed for eventual Ni-enzyme maturation have caused this bacterium to develop complex and unique mechanisms for dealing with nickel. The physiological challenge for the bacterium must then include requirements that minimally must encompass a critical need to discriminately deliver nickel to at least two Ni-enzymes and probably to other proteins, dealing with a very high nickel demand, recognizing nickel amongst a variety of (sometimes competing) metal cations, and balancing metal need with toxicity due to the fluctuating reservoirs of the metal both intracellularly and extracellularly.
7.1. Hpn and Hpn-Like Proteins
H. pylori possess two proteins with remarkably high histidine content. These small His-rich proteins, named Hpn and Hpn-like (referred to herein as Hpnl), contain 47% and 25% His, respectively. Interestingly, both are confined to the gastric colonizing types of Helicobacters [201]. However, Hpn is present in every gastric Helicobacter species, and Hpnl is restricted to H. pylori and its closely related species H. acinonychis, originally isolated from cheetah [202].
These small proteins have apparently redundant functions to one another regarding roles in nickel storage for urease manufacture. Recombinant Hpn exists primarily as a 20-mer with each monomer binding five Ni (II) with a Kd of 7.1 μM [203]. Hpnl monomers bind two Ni (II) with a Kd of 3.8 μM, but it also forms multimeric structures of more than 20 subunits [204]. While the His residues are directly involved in nickel binding, as expected, the additional multiple Gln residues (in Hpnl) are thought to improve stability of the metal complexes [205]. Strains carrying mutations in hpn and hpnl are more sensitive to nickel toxicity, and they influence active urease maturation in a nickel-dependent manner [206]. Nickel release from Hpn and Hpnl is observed under acidic conditions, suggesting that these proteins may supply nickel when urease is also needed (e.g., to combat acidity, see [207]). Expression of the storage proteins confers nickel resistance to E. coli, confirming their ability to sequester excess nickel [201]. Based on studies with pure proteins or on H. pylori mutant strain in-lab phenotypes, the initial suggestions that these proteins might play nickel storage roles in vivo was supported by use of mice maintained under strict nickel-limited conditions [208] or NMRI-specific pathogen-free mice [202]. H. pylori mutant strains lacking either Hpn, or Hpnl, or both storage proteins, were poorer colonizers than their wild type counterparts, when the hosts (C57/Bl6 mice) were subjected to nickel-deficient diets [208].
Purified Hpn was shown to interact with the UreA subunit of urease, while Hpnl interacted most strongly uniquely with the HypA and HypB hydrogenase maturation proteins [202]. Also, Hpn and Hpnl together impact intracellular nickel trafficking, and influence urease activity. However, the authors of the study concluded that Hpn is the primary nickel sequestering reservoir, and that the two storage proteins compete for nickel under low nickel conditions [202]. The result in low nickel is “restricted activation” or basal levels of urease. In high nickel, where both storage proteins are saturated, their nickel delivery roles would ensure Ni-activated levels of urease. In the suggested model, Hpnl would thus not play a nickel detoxification role, due to its more limited capacity to bind nickel and its lower abundance [202].
In contrast to the rather limited roles assigned to the two storage proteins by Vinella et al. [202], the most recent work on the two nickel storage proteins [207] indicates they both play a much broader role than previously reported. Considering their importance in virulence, it seemed reasonable they may play roles in nickel-sensing, regulation, or delivery of nickel; this would likely require many protein–protein interactions. An affinity pulldown approach was used, whereby cross-linking to each storage protein was followed by a Ni-based purification/enrichment; the results indicate the storage proteins interact with a wide array of proteins [207]. The storage proteins interacted with known nickel delivery systems involved in urease and hydrogenase maturation, and hydrogenase activity was severely diminished in a Δhpn Δhpnl double mutant strain when nickel was limited. Interestingly, both storage proteins play roles in ammonia production independent of urease activity, i.e., via amide hydrolysis. Indeed, Hpn and Hpnl were shown to synergistically suppress aliphatic amidase (AmiE) activity [207]. This role makes sense from a physiological view, as the complementary ammonia-producing enzyme (urease) is known to be made in large amounts when cells are provided with nickel [130,131], and the aliphatic amidase AmiE plays a role in acid resistance especially in the absence of urease [209]. In addition, interactions between Hpn and the aminopeptidase PepA were observed [207]. PepA can accept a variety of divalent cations for its activation, and the study implicates the storage proteins role in peptide salvage processes. The interactions between Hpn and Hpnl and AmiE or PepA, respectively, were further confirmed by using purified proteins and a tryptophan (Trp) fluorescence-based method, taking advantage of the concomitant presence of Trp residues in both AmiE and PepA and the lack of Trp in both storage proteins [210] (Figure 3). As expected, neither Hpn nor Hpnl had measurable fluorescence, while both AmiE and PepA, as pure proteins, had observable fluorescence profiles (Figure 3). The maximum fluorescence for both AmiE and PepA fell within the previously established range for Trp fluorescence maxima [211]. Upon addition of either purified Hpn or purified Hpnl, the fluorescence profile of AmiE and PepA shifted markedly (Figure 3). The observed shift suggests that the storage proteins have either altered the microenvironment of the Trp residues within the peptidase and amidase, or that they have caused conformational changes in the target proteins that altered their fluorescence profiles [212]. Bovine serum albumin (BSA), a protein that contains three Trp residues, was used as a negative control. Incubation of either Hpn or Hpnl had no effect on the fluorescence profile of BSA (data not shown), confirming the specificity of the (AmiE and PepA) interactions described above [210].
Ni-acquisition and peptide salvage, maturation of Ni-enzymes, and oxidative stress-combating enzymes are some of the enzymes that may be impacted by interaction with the storage proteins (199). Whether or not nickel delivery to or from these enzymes is involved is not known, and only a few interactions were characterized [207]. Still, the cross-linking results supported that each protein transiently but intimately interacts with perhaps 100 or more proteins. This may at first seems to be a gross overestimate, but considering that (i) nickel metabolism plays a central role in H. pylori; (ii) both storage proteins can bind other metals; (iii) both proteins apparently represent the major nickel reservoirs in the cell; thus, a variety of sensing and delivery roles for Hpn and Hpnl seems more possible. We must also remember that metal binding proteins may comprise a much larger fraction of the total bacterial proteomes than previously appreciated [213].
7.2. HspA
HspA is a nickel-binding protein that has homology to the highly-conserved and essential heat shock protein GroES. However, the H. pylori version has a unique His-rich C-terminus that binds nickel. Although a strain lacking HspA is not recoverable (i.e., lethal), a strain lacking only the C-terminal extension of HspA is viable, and it has been tested for virulence, i.e., mouse colonization capacity [214]. Although the strain had diminished hydrogenase activity and low tolerance to exogenous nickel, the strain was normal in colonization capacity. Of course, this strain still had other nickel-binding proteins. It would be interesting to perform the colonization assays in mice that have reduced nickel levels, to assess HspA’s role in nickel storage when the metal is limiting. Also, comparing a strain lacking both the C-terminus His-rich extension of HspA as well as Hpn and Hpnl, to a strain lacking only the two storage proteins could be an approach to evaluate the (additive) role of HspA in the overall Ni-storage budget. HspA has long been considered to be a candidate for use as anti-H. pylori vaccine. Partial protection against H. pylori in mice was observed after intranasal administration of HspA [215]), while the interesting goal of expressing HpHspA in a probiotic bacterium (Lactococcus lactis) was unfortunately not successful [216].
8. Conclusions
As a required cofactor for some key enzymes, most notably hydrogenase and urease, nickel clearly plays roles in microbial pathogenesis. Still, new information on the role of the metal in pathogens that have other Ni-enzymes is needed, and it is hypothesized that more proteins that use nickel, or respond to fluctuating nickel levels, will be identified. In addition to uncovering the molecular mechanisms of nickel trafficking and homeostasis in the Ni-requiring pathogens, large gaps in our knowledge on nickel in vivo availability exist. These include understanding the dietary nickel sources, host metabolic factors that modulate accessibility of nickel to the pathogens and how nickel availability in the host is impacted by the (Ni-utilizing) host intestinal microbiota composition. These represent just some research areas that are in need of development. Availability of the metal within the host certainly varies, and it is important that we know how generally accessible the metal is within specific host organs, specific tissue types, and within specific host cell (e.g., epithelial cell, immune cell, blood cell) types. Finally, one promising avenue for future nickel-related research is the fact that numerous mammalian pathogens (at least 39 prokaryotes and nine eukaryotes) require the metal (for various enzymes, e.g., Ni-ARD, Ni-GloI, [Fe-Ni] hydrogenase, urease, and Ni-SOD), while their host do not; this presents an opportunity to specifically target pathogens via nickel sequestration. It seems such sequestration naturally occurs in higher plants, since plants use nickel (in the form of Ni-urease), while at the same time, the number of nickel-utilizing plant pathogens is very limited: indeed, only S. scabies and a few other related Streptomyces species are known plant pathogens that contain a Ni-enzyme (Ni-SOD). This interesting two-kingdom conundrum (plants use nickel/few Ni-requiring pathogens; mammals do not require nickel/many Ni-utilizing pathogens) not only gives us insights on the evolution of host/pathogens competition for nickel, but can perhaps provide us with a roadmap for future projects aimed at inhibiting or eradicating the nickel-requiring human or mammal pathogens.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zambelli, B.; Uversky, V.N.; Ciurli, S. Nickel impact on human health: An intrinsic disorder perspective. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2016, 1864, 1714–1731. [Google Scholar] [CrossRef]
- Rutherford, J.C. The emerging role of urease as a general microbial virulence factor. PLoS Pathog. 2014, 10, e1004062. [Google Scholar] [CrossRef]
- Benoit, S.L.; Maier, R.J. Hydrogen and nickel metabolism in Helicobacter species. Ann. N. Y. Acad. Sci. 2008, 1125, 242–251. [Google Scholar] [CrossRef]
- Maier, R.J. Use of molecular hydrogen as an energy substrate by human pathogenic bacteria. Biochem. Soc. Trans. 2005, 33, 83–85. [Google Scholar] [CrossRef] [Green Version]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef]
- Maroney, M.J.; Ciurli, S. Nonredox nickel enzymes. Chem. Rev. 2013, 114, 4206–4228. [Google Scholar] [CrossRef]
- Mazzei, L.; Musiani, F.; Ciurli, S. Urease. In The Biological Chemistry of Nickel; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 60–97. [Google Scholar]
- Tai, H.; Higuchi, Y.; Hirota, S. Comprehensive reaction mechanisms at and near the Ni–Fe active sites of [NiFe] hydrogenases. Dalton Trans. 2018, 47, 4408–4423. [Google Scholar] [CrossRef]
- Ogata, H.; Lubitz, W.; Higuchi, Y. Structure and function of [NiFe] hydrogenases. J. Biochem. 2016, 160, 251–258. [Google Scholar] [CrossRef] [Green Version]
- De Reuse, H.; Vinella, D.; Cavazza, C. Common themes and unique proteins for the uptake and trafficking of nickel, a metal essential for the virulence of Helicobacter pylori. Front. Cell. Infect. Microbiol. 2013, 3, 94. [Google Scholar] [CrossRef]
- Zeer-Wanklyn, C.J.; Zamble, D.B. Microbial nickel: Cellular uptake and delivery to enzyme centers. Curr. Opin. Chem. Biol. 2017, 37, 80–88. [Google Scholar] [CrossRef]
- Gaddy, J.A.; Haley, K.P. Metalloregulation of Helicobacter pylori physiology and pathogenesis. Front. Microbiol. 2015, 6, 911. [Google Scholar]
- Deshpande, A.R.; Pochapsky, T.C.; Ringe, D. The Metal Drives the Chemistry: Dual Functions of Acireductone Dioxygenase. Chem. Rev. 2017, 117, 10474–10501. [Google Scholar] [CrossRef]
- Miller, A.-F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef]
- Ryan, K.C.; Guce, A.I.; Johnson, O.E.; Brunold, T.C.; Cabelli, D.E.; Garman, S.C.; Maroney, M.J. Nickel superoxide dismutase: Structural and functional roles of His1 and its H-bonding network. Biochemistry 2015, 54, 1016–1027. [Google Scholar] [CrossRef]
- Honek, J.F. Nickel Glyoxalase I. In The Biological Chemistry of Nickel; Zamble, D., Rowinska-Zyrek, M., Kozlowski, H., Eds.; Royal Society of Chemistry: Cambridge, UK, 2017. [Google Scholar]
- Cox, G.M.; Mukherjee, J.; Cole, G.T.; Casadevall, A.; Perfect, J.R. Urease as a virulence factor in experimental cryptococcosis. Infect. Immun. 2000, 68, 443–448. [Google Scholar] [CrossRef]
- Olszewski, M.A.; Noverr, M.C.; Chen, G.-H.; Toews, G.B.; Cox, G.M.; Perfect, J.R.; Huffnagle, G.B. Urease expression by Cryptococcus neoformans promotes microvascular sequestration, thereby enhancing central nervous system invasion. Am. J. Pathol. 2004, 164, 1761–1771. [Google Scholar] [CrossRef]
- Fu, M.S.; Coelho, C.; De Leon-Rodriguez, C.M.; Rossi, D.C.P.; Camacho, E.; Jung, E.H.; Kulkarni, M.; Casadevall, A. Cryptococcus neoformans urease affects the outcome of intracellular pathogenesis by modulating phagolysosomal pH. PLoS Pathog. 2018, 14, e1007144. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nakayasu, E.S.; Oliveira, D.L.; Nimrichter, L.; Nosanchuk, J.D.; Almeida, I.C.; Casadevall, A. Extracellular vesicles produced by Cryptococcus neoformans contain protein components associated with virulence. Eukaryot. Cell 2008, 7, 58–67. [Google Scholar] [CrossRef]
- Feder, V.; Kmetzsch, L.; Staats, C.C.; Vidal-Figueiredo, N.; Ligabue-Braun, R.; Carlini, C.R.; Vainstein, M.H. Cryptococcus gattii urease as a virulence factor and the relevance of enzymatic activity in cryptococcosis pathogenesis. FEBS J. 2015, 282, 1406–1418. [Google Scholar] [CrossRef]
- Mirbod-Donovan, F.; Schaller, R.; Hung, C.Y.; Xue, J.; Reichard, U.; Cole, G.T. Urease produced by Coccidioides posadasii contributes to the virulence of this respiratory pathogen. Infect. Immun. 2006, 74, 504–515. [Google Scholar] [CrossRef]
- Wise, H.Z.; Hung, C.-Y.; Whiston, E.; Taylor, J.W.; Cole, G.T. Extracellular ammonia at sites of pulmonary infection with Coccidioides posadasii contributes to severity of the respiratory disease. Microb. Pathog. 2013, 59, 19–28. [Google Scholar] [CrossRef]
- Baltazar, L.M.; Zamith-Miranda, D.; Burnet, M.C.; Choi, H.; Nimrichter, L.; Nakayasu, E.S.; Nosanchuk, J.D. Concentration-dependent protein loading of extracellular vesicles released by Histoplasma capsulatum after antibody treatment and its modulatory action upon macrophages. Sci. Rep. 2018, 8, 8065. [Google Scholar] [CrossRef]
- Costa, M.; Borges, C.L.; Bailao, A.M.; Meirelles, G.V.; Mendonça, Y.A.; Dantas, S.F.; de Faria, F.P.; Felipe, M.S.; Molinari-Madlum, E.N.E.; Mendes-Giannini, M.J. Transcriptome profiling of Paracoccidioides brasiliensis yeast-phase cells recovered from infected mice brings new insights into fungal response upon host interaction. Microbiology 2007, 153, 4194–4207. [Google Scholar] [CrossRef]
- Rujirawat, T.; Patumcharoenpol, P.; Lohnoo, T.; Yingyong, W.; Kumsang, Y.; Payattikul, P.; Tangphatsornruang, S.; Suriyaphol, P.; Reamtong, O.; Garg, G.; et al. Probing the Phylogenomics and Putative Pathogenicity Genes of Pythium insidiosum by Oomycete Genome Analyses. Sci. Rep. 2018, 8, 4135. [Google Scholar] [CrossRef]
- Ariza, A.; Vickers, T.J.; Greig, N.; Armour, K.A.; Dixon, M.J.; Eggleston, I.M.; Fairlamb, A.H.; Bond, C.S. Specificity of the trypanothione-dependent Leishmania major glyoxalase I: Structure and biochemical comparison with the human enzyme. Mol. Microbiol. 2006, 59, 1239–1248. [Google Scholar] [CrossRef]
- Chauhan, S.C.; Madhubala, R. Glyoxalase I gene deletion mutants of Leishmania donovani exhibit reduced methylglyoxal detoxification. PLoS ONE 2009, 4, e6805. [Google Scholar] [CrossRef]
- Greig, N.; Wyllie, S.; Vickers, T.J.; Fairlamb, A.H. Trypanothione-dependent glyoxalase I in Trypanosoma cruzi. Biochem. J. 2006, 400, 217–223. [Google Scholar] [CrossRef]
- Morou-Bermudez, E.; Burne, R.A. Genetic and physiologic characterization of urease of Actinomyces naeslundii. Infect. Immun. 1999, 67, 504–512. [Google Scholar]
- Salem, N.; Salem, L.; Saber, S.; Ismail, G.; Bluth, M.H. Corynebacterium urealyticum: A comprehensive review of an understated organism. Infect. Drug Resist. 2015, 8, 129–145. [Google Scholar]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989–12994. [Google Scholar] [CrossRef] [Green Version]
- Fontán, P.; Aris, V.; Ghanny, S.; Soteropoulos, P.; Smith, I. Global transcriptional profile of Mycobacterium tuberculosis during THP-1 human macrophage infection. Infect. Immun. 2008, 76, 717–725. [Google Scholar] [CrossRef]
- Schnappinger, D.; Ehrt, S.; Voskuil, M.I.; Liu, Y.; Mangan, J.A.; Monahan, I.M.; Dolganov, G.; Efron, B.; Butcher, P.D.; Nathan, C. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J. Exp. Med. 2003, 198, 693–704. [Google Scholar] [CrossRef]
- Lin, W.; Mathys, V.; Ang, E.L.; Koh, V.H.; Martinez Gomez, J.M.; Ang, M.L.; Zainul Rahim, S.Z.; Tan, M.P.; Pethe, K.; Alonso, S. Urease activity represents an alternative pathway for Mycobacterium tuberculosis nitrogen metabolism. Infect. Immun. 2012, 80, 2771–2779. [Google Scholar] [CrossRef]
- Suttisansanee, U.; Lau, K.; Lagishetty, S.; Rao, K.N.; Swaminathan, S.; Sauder, J.M.; Burley, S.K.; Honek, J.F. Structural Variation in Bacterial Glyoxalase I Enzymes Investigation of the Metalloenzyme Glyoxalase I from Clostridium acetobutylicum. J. Biol. Chem. 2011, 286, 38367–38374. [Google Scholar] [CrossRef]
- Resch, A.; Rosenstein, R.; Nerz, C.; Gotz, F. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl. Environ. Microbiol. 2005, 71, 2663–2676. [Google Scholar] [CrossRef]
- Zhou, C.; Bhinderwala, F.; Lehman, M.K.; Thomas, V.C.; Chaudhari, S.S.; Yamada, K.J.; Foster, K.W.; Powers, R.; Kielian, T.; Fey, P.D. Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 2019, 15, e1007538. [Google Scholar] [CrossRef]
- Vandecandelaere, I.; Van Nieuwerburgh, F.; Deforce, D.; Coenye, T. Metabolic activity, urease production, antibiotic resistance and virulence in dual species biofilms of Staphylococcus epidermidis and Staphylococcus aureus. PLoS ONE 2017, 12, e0172700. [Google Scholar] [CrossRef]
- Gatermann, S.; John, J.; Marre, R. Staphylococcus saprophyticus urease: Characterization and contribution to uropathogenicity in unobstructed urinary tract infection of rats. Infect. Immun. 1989, 57, 110–116. [Google Scholar]
- Chen, Y.Y.; Weaver, C.A.; Burne, R.A. Dual functions of Streptococcus salivarius urease. J. Bacteriol. 2000, 182, 4667–4669. [Google Scholar] [CrossRef]
- Kokkayil, P.; Dhawan, B. Ureaplasma: Current perspectives. Indian J. Med. Microbiol. 2015, 33, 205–214. [Google Scholar]
- Smith, D.; Russell, W.; Ingledew, W.; Thirkell, D. Hydrolysis of urea by Ureaplasma urealyticum generates a transmembrane potential with resultant ATP synthesis. J. Bacteriol. 1993, 175, 3253–3258. [Google Scholar] [CrossRef]
- Grenabo, L.; Hedelin, H.; Pettersson, S. Urinary infection stones caused by Ureaplasma urealyticum: A review. Scand. J. Infect. Dis. Suppl. 1988, 53, 46–49. [Google Scholar]
- Silva, J.; Marques, L.; Timenetsky, J.; de Farias, S.T. Ureaplasma diversum protein interaction networks: Evidence of horizontal gene transfer and evolution of reduced genomes among the Mollicutes. Can. J. Microbiol. 2019. [Google Scholar] [CrossRef]
- Sangari, F.J.; Seoane, A.; Rodriguez, M.C.; Aguero, J.; Garcia Lobo, J.M. Characterization of the urease operon of Brucella abortus and assessment of its role in virulence of the bacterium. Infect. Immun. 2007, 75, 774–780. [Google Scholar] [CrossRef]
- Abkar, M.; Amani, J.; Sahebghadam Lotfi, A.; Nikbakht Brujeni, G.; Alamian, S.; Kamali, M. Subcutaneous immunization with a novel immunogenic candidate (urease) confers protection against Brucella abortus and Brucella melitensis infections. APMIS 2015, 123, 667–675. [Google Scholar] [CrossRef]
- Bandara, A.B.; Contreras, A.; Contreras-Rodriguez, A.; Martins, A.M.; Dobrean, V.; Poff-Reichow, S.; Rajasekaran, P.; Sriranganathan, N.; Schurig, G.G.; Boyle, S.M. Brucella suis urease encoded by ure 1 but not ure 2 is necessary for intestinal infection of BALB/c mice. BMC Microbiol. 2007, 7, 57. [Google Scholar] [CrossRef]
- Smith, M.G.; Gianoulis, T.A.; Pukatzki, S.; Mekalanos, J.J.; Ornston, L.N.; Gerstein, M.; Snyder, M. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 2007, 21, 601–614. [Google Scholar] [CrossRef]
- Rathinavelu, S.; Zavros, Y.; Merchant, J.L. Acinetobacter lwoffii infection and gastritis. Microbes Infect. 2003, 5, 651–657. [Google Scholar] [CrossRef]
- Bossé, J.T.; MacInnes, J.I. Urease activity may contribute to the ability of Actinobacillus pleuropneumoniae to establish infection. Can. J. Vet. Res. 2000, 64, 145. [Google Scholar]
- Klitgaard, K.; Friis, C.; Jensen, T.K.; Angen, O.; Boye, M. Transcriptional portrait of Actinobacillus pleuropneumoniae during acute disease—Potential strategies for survival and persistence in the host. PLoS ONE 2012, 7, e35549. [Google Scholar] [CrossRef]
- Pinske, C.; Sawers, R.G. Anaerobic Formate and Hydrogen Metabolism. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef]
- Steyert, S.R.; Kaper, J.B. Contribution of urease to colonization by Shiga toxin-producing Escherichia coli. Infect. Immun. 2012, 80, 2589–2600. [Google Scholar] [CrossRef]
- Li, M.F.; Sun, L. Edwardsiella tarda Sip2: A Serum-Induced Protein That Is Essential to Serum Survival, Acid Resistance, Intracellular Replication, and Host Infection. Front. Microbiol. 2018, 9, 1084. [Google Scholar] [CrossRef]
- Murphy, T.F.; Brauer, A.L. Expression of urease by Haemophilus influenzae during human respiratory tract infection and role in survival in an acid environment. BMC Microbiol. 2011, 11, 183. [Google Scholar] [CrossRef]
- Maroncle, N.; Rich, C.; Forestier, C. The role of Klebsiella pneumoniae urease in intestinal colonization and resistance to gastrointestinal stress. Res. Microbiol. 2006, 157, 184–193. [Google Scholar] [CrossRef]
- Young, G.M.; Amid, D.; Miller, V.L. A bifunctional urease enhances survival of pathogenic Yersinia enterocolitica and Morganella morganii at low pH. J. Bacteriol. 1996, 178, 6487–6495. [Google Scholar] [CrossRef]
- Armbruster, C.E.; Smith, S.N.; Yep, A.; Mobley, H.L. Increased incidence of urolithiasis and bacteremia during Proteus mirabilis and Providencia stuartii coinfection due to synergistic induction of urease activity. J. Infect. Dis. 2014, 209, 1524–1532. [Google Scholar] [CrossRef]
- Alteri, C.J.; Himpsl, S.D.; Engstrom, M.D.; Mobley, H.L. Anaerobic respiration using a complete oxidative TCA cycle drives multicellular swarming in Proteus mirabilis. MBio 2012, 3, e00365-12. [Google Scholar] [CrossRef]
- Johnson, D.; Russell, R.; Lockatell, C.; Zulty, J.; Warren, J.; Mobley, H. Contribution of Proteus mirabilis urease to persistence, urolithiasis, and acute pyelonephritis in a mouse model of ascending urinary tract infection. Infect. Immun. 1993, 61, 2748–2754. [Google Scholar]
- Schaffer, J.N.; Norsworthy, A.N.; Sun, T.T.; Pearson, M.M. Proteus mirabilis fimbriae- and urease-dependent clusters assemble in an extracellular niche to initiate bladder stone formation. Proc. Natl. Acad. Sci. USA 2016, 113, 4494–4499. [Google Scholar] [CrossRef]
- Armbruster, C.E.; Mobley, H.L.T.; Pearson, M.M. Pathogenesis of Proteus mirabilis Infection. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Zbell, A.L.; Maier, S.E.; Maier, R.J. Salmonella enterica serovar Typhimurium NiFe uptake-type hydrogenases are differentially expressed in vivo. Infect. Immun. 2008, 76, 4445–4454. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Barthel, M.; Stecher, B.; Maier, R.J.; Gunn, J.S.; Hardt, W.-D. Salmonella Typhimurium strain ATCC14028 requires H2-hydrogenases for growth in the gut, but not at systemic sites. PLoS ONE 2014, 9, e110187. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.H.; Monack, D.M. Intraspecies competition for niches in the distal gut dictate transmission during persistent Salmonella infection. PLoS Pathog. 2014, 10, e1004527. [Google Scholar] [CrossRef] [PubMed]
- Zbell, A.L.; Benoit, S.L.; Maier, R.J. Differential expression of NiFe uptake-type hydrogenase genes in Salmonella enterica serovar Typhimurium. Microbiology 2007, 153, 3508–3516. [Google Scholar] [CrossRef] [PubMed]
- Parkin, A.; Bowman, L.; Roessler, M.M.; Davies, R.A.; Palmer, T.; Armstrong, F.A.; Sargent, F. How Salmonella oxidises H2 under aerobic conditions. FEBS Lett. 2012, 586, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane-Khadka, R.; Benoit, S.L.; Miller-Parks, E.F.; Maier, R.J. Host hydrogen rather than that produced by the pathogen is important for Salmonella enterica serovar Typhimurium virulence. Infect. Immun. 2015, 83, 311–316. [Google Scholar] [CrossRef]
- McNorton, M.M.; Maier, R.J. Roles of H2 uptake hydrogenases in Shigella flexneri acid tolerance. Microbiology 2012, 158, 2204–2212. [Google Scholar] [CrossRef]
- Cai, Y.; Ni, Y. Purification, characterization, and pathogenicity of urease produced by Vibrio parahaemolyticus. J. Clin. Lab. Anal. 1996, 10, 70–73. [Google Scholar] [CrossRef]
- De Koning-Ward, T.F.; Robins-Browne, R.M. Contribution of urease to acid tolerance in Yersinia enterocolitica. Infect. Immun. 1995, 63, 3790–3795. [Google Scholar]
- Da Silva, S.M.; Venceslau, S.S.; Fernandes, C.L.; Valente, F.M.; Pereira, I.A. Hydrogen as an energy source for the human pathogen Bilophila wadsworthia. Antonie Leeuwenhoek 2008, 93, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Weerakoon, D.R.; Borden, N.J.; Goodson, C.M.; Grimes, J.; Olson, J.W. The role of respiratory donor enzymes in Campylobacter jejuni host colonization and physiology. Microb. Pathog. 2009, 47, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Kassem, I.; Khatri, M.; Esseili, M.A.; Sanad, Y.M.; Saif, Y.M.; Olson, J.W.; Rajashekara, G. Respiratory proteins contribute differentially to Campylobacter jejuni’s survival and in vitro interaction with hosts’ intestinal cells. BMC Microbiol. 2012, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.L.; Maier, R.J. Site-directed mutagenesis of Campylobacter concisus respiratory genes provides insight into the pathogen’s growth requirements. Sci. Rep. 2018, 8, 14203. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.S.; Benoit, S.; Mysore, J.V.; Sousa, R.S.; Maier, R.J. Helicobacter hepaticus hydrogenase mutants are deficient in hydrogen-supported amino acid uptake and in causing liver lesions in A/J mice. Infect. Immun. 2005, 73, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Lee, A.; Whary, M.T.; Rogers, A.B.; Maurer, K.J.; Taylor, N.S.; Schauer, D.B.; Fox, J.G. Helicobacter hepaticus urease is not required for intestinal colonization but promotes hepatic inflammation in male A/JCr mice. Microb. Pathog. 2008, 45, 18–24. [Google Scholar] [CrossRef]
- Andrutis, K.A.; Fox, J.G.; Schauer, D.B.; Marini, R.P.; Murphy, J.C.; Yan, L.; Solnick, J.V. Inability of an isogenic urease-negative mutant stain of Helicobacter mustelae to colonize the ferret stomach. Infect. Immun. 1995, 63, 3722–3725. [Google Scholar]
- Olson, J.W.; Maier, R.J. Molecular hydrogen as an energy source for Helicobacter pylori. Science 2002, 298, 1788–1790. [Google Scholar] [CrossRef]
- Kuhns, L.G.; Benoit, S.L.; Bayyareddy, K.; Johnson, D.; Orlando, R.; Evans, A.L.; Waldrop, G.L.; Maier, R.J. Carbon fixation driven by Molecular Hydrogen Results in Chemolithoautotrophically Enhanced Growth of Helicobacter pylori. J. Bacteriol. 2016, 198, 1423–1428. [Google Scholar] [CrossRef]
- Wang, G.; Romero-Gallo, J.; Benoit, S.L.; Piazuelo, M.B.; Dominguez, R.L.; Morgan, D.R.; Peek, R.M., Jr.; Maier, R.J. Hydrogen metabolism in Helicobacter pylori plays a role in gastric carcinogenesis through facilitating CagA translocation. MBio 2016, 7, e01022-16. [Google Scholar] [CrossRef]
- Segal, E.D.; Shon, J.; Tompkins, L.S. Characterization of Helicobacter pylori urease mutants. Infect. Immun. 1992, 60, 1883–1889. [Google Scholar] [PubMed]
- Tsuda, M.; Karita, M.; Morshed, M.G.; Okita, K.; Nakazawa, T. A urease-negative mutant of Helicobacter pylori constructed by allelic exchange mutagenesis lacks the ability to colonize the nude mouse stomach. Infect. Immun. 1994, 62, 3586–3589. [Google Scholar] [PubMed]
- Eaton, K.A.; Krakowka, S. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect. Immun. 1994, 62, 3604–3607. [Google Scholar] [PubMed]
- Harris, P.R.; Ernst, P.B.; Kawabata, S.; Kiyono, H.; Graham, M.F.; Smith, P.D. Recombinant Helicobacter pylori urease activates primary mucosal macrophages. J. Infect. Dis. 1998, 178, 1516–1520. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.; Mobley, H.; Perez-Perez, G.; Blaser, M.; Smith, P. Helicobacter pylori urease is a potent stimulus of mononuclear phagocyte activation and inflammatory cytokine production. Gastroenterology 1996, 111, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Gunasena, H.; Cheng, Z.; Espejo, R.; Crowe, S.E.; Ernst, P.B.; Reyes, V.E. Helicobacter pylori urease binds to class II MHC on gastric epithelial cells and induces their apoptosis. J. Immunol. 2000, 165, 1918–1924. [Google Scholar] [CrossRef] [PubMed]
- Kavermann, H.; Burns, B.P.; Angermuller, K.; Odenbreit, S.; Fischer, W.; Melchers, K.; Haas, R. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J. Exp. Med. 2003, 197, 813–822. [Google Scholar] [CrossRef]
- Wirth, H.P.; Beins, M.H.; Yang, M.; Tham, K.T.; Blaser, M.J. Experimental infection of Mongolian gerbils with wild-type and mutant Helicobacter pylori strains. Infect. Immun. 1998, 66, 4856–4866. [Google Scholar]
- Kuwahara, H.; Miyamoto, Y.; Akaike, T.; Kubota, T.; Sawa, T.; Okamoto, S.; Maeda, H. Helicobacter pylori urease suppresses bactericidal activity of peroxynitrite via carbon dioxide production. Infect. Immun. 2000, 68, 4378–4383. [Google Scholar] [CrossRef]
- Lytton, S.D.; Fischer, W.; Nagel, W.; Haas, R.; Beck, F.X. Production of ammonium by Helicobacter pylori mediates occludin processing and disruption of tight junctions in Caco-2 cells. Microbiology 2005, 151, 3267–3276. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Shen, L.; Ogden, S.; Romero-Gallo, J.; Lapierre, L.A.; Israel, D.A.; Turner, J.R.; Peek, R.M., Jr. Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology 2009, 136, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, G.E.; Olivera-Severo, D.; Uberti, A.F.; Carlini, C.R. Helicobacter pylori urease activates blood platelets through a lipoxygenase-mediated pathway. J. Cell. Mol. Med. 2010, 14, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Perrais, M.; Rousseaux, C.; Ducourouble, M.-P.; Courcol, R.; Vincent, P.; Jonckheere, N.; Van Seuningen, I. Helicobacter pylori urease and flagellin alter mucin gene expression in human gastric cancer cells. Gastric Cancer 2014, 17, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Debowski, A.W.; Walton, S.M.; Chua, E.G.; Tay, A.C.; Liao, T.; Lamichhane, B.; Himbeck, R.; Stubbs, K.A.; Marshall, B.J.; Fulurija, A.; et al. Helicobacter pylori gene silencing in vivo demonstrates urease is essential for chronic infection. PLoS Pathog. 2017, 13, e1006464. [Google Scholar] [CrossRef] [PubMed]
- Olivera-Severo, D.; Uberti, A.F.; Marques, M.S.; Pinto, M.T.; Gomez-Lazaro, M.; Figueiredo, C.; Leite, M.; Carlini, C.R. A new role for Helicobacter pylori urease: Contributions to angiogenesis. Front. Microbiol. 2017, 8, 1883. [Google Scholar] [CrossRef] [PubMed]
- Scopel-Guerra, A.; Olivera-Severo, D.; Staniscuaski, F.; Uberti, A.F.; Callai-Silva, N.; Jaeger, N.; Porto, B.N.; Carlini, C.R. The impact of Helicobacter pylori urease upon platelets and consequent contributions to inflammation. Front. Microbiol. 2017, 8, 2447. [Google Scholar] [CrossRef] [PubMed]
- Schmalstig, A.A.; Benoit, S.L.; Misra, S.K.; Sharp, J.S.; Maier, R.J. Noncatalytic antioxidant role for Helicobacter pylori urease. J. Bacteriol. 2018, 200, e00124-18. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, G.V.; Kollmer, W.E.; Bowen, H.J.M. The Elemental Composition of Human Tissues and Body Fluids: A Compilation of Values for Adults; Verlag Chemie: Weinheim, Germany, 1978. [Google Scholar]
- Maret, W. Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics 2010, 2, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ochoa, V.E.; Jellbauer, S.; Klaus, S.; Raffatellu, M. Transition metal ions at the crossroads of mucosal immunity and microbial pathogenesis. Front. Cell. Infect. Microbiol. 2014, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Zackular, J.P.; Chazin, W.J.; Skaar, E.P. Nutritional Immunity: S100 Proteins at the host-pathogen interface. J. Biol. Chem. 2015, 290, 18991–18998. [Google Scholar] [CrossRef]
- Choby, J.E.; Mike, L.A.; Mashruwala, A.A.; Dutter, B.F.; Dunman, P.M.; Sulikowski, G.A.; Boyd, J.M.; Skaar, E.P. A small-molecule inhibitor of iron-sulfur cluster assembly uncovers a link between virulence regulation and metabolism in Staphylococcus aureus. Cell Chem. Biol. 2016, 23, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.D.; Skaar, E.P. Transition Metals and Virulence in Bacteria. Annu. Rev. Genet. 2016, 50, 67–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashige, T.G.; Zygiel, E.M.; Drennan, C.L.; Nolan, E.M. Nickel sequestration by the host-defense protein human calprotectin. J. Am. Chem. Soc. 2017, 139, 8828–8836. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.R.; Roy, K.; Patel, Y.; Zhou, S.F.; Singh, M.R.; Singh, D.; Nasir, M.; Sehgal, R.; Sehgal, A.; Singh, R.S.; et al. Multifunctional iron bound lactoferrin and nanomedicinal approaches to enhance its bioactive functions. Molecules 2015, 20, 9703–9731. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Iron and infection. Int. J. Hematol. 2018, 107, 7–15. [Google Scholar] [CrossRef]
- Kulprachakarn, K.; Chen, Y.L.; Kong, X.; Arno, M.C.; Hider, R.C.; Srichairatanakool, S.; Bansal, S.S. Copper(II) binding properties of hepcidin. J. Biol. Inorg. Chem. 2016, 21, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Stefanova, D.; Raychev, A.; Deville, J.; Humphries, R.; Campeau, S.; Ruchala, P.; Nemeth, E.; Ganz, T.; Bulut, Y. Hepcidin protects against lethal Escherichia coli sepsis in mice inoculated with isolates from septic patients. Infect. Immun. 2018, 86, e00253-18. [Google Scholar] [CrossRef]
- Stefanova, D.; Raychev, A.; Arezes, J.; Ruchala, P.; Gabayan, V.; Skurnik, M.; Dillon, B.J.; Horwitz, M.A.; Ganz, T.; Bulut, Y.; et al. Endogenous hepcidin and its agonist mediate resistance to selected infections by clearing non-transferrin-bound iron. Blood 2017, 130, 245–257. [Google Scholar] [CrossRef]
- Stafford, S.L.; Bokil, N.J.; Achard, M.E.; Kapetanovic, R.; Schembri, M.A.; McEwan, A.G.; Sweet, M.J. Metal ions in macrophage antimicrobial pathways: Emerging roles for zinc and copper. Biosci. Rep. 2013, 33, e00049. [Google Scholar] [CrossRef]
- Cellier, M.F.M. Developmental Control of NRAMP1 (SLC11A1) Expression in Professional Phagocytes. Biology 2017, 6, 28. [Google Scholar] [CrossRef]
- Corbin, B.D.; Seeley, E.H.; Raab, A.; Feldmann, J.; Miller, M.R.; Torres, V.J.; Anderson, K.L.; Dattilo, B.M.; Dunman, P.M.; Gerads, R.; et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 2008, 319, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Rowinska-Zyrek, M.; Zakrzewska-Czerwinska, J.; Zawilak-Pawlik, A.; Kozlowski, H. Ni2+ chemistry in pathogens–a possible target for eradication. Dalton Trans. 2014, 43, 8976–8989. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.J. Availability and use of molecular hydrogen as an energy substrate for Helicobacter species. Microbes Infect. 2003, 5, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.A.; Fraser, J.A. Is the nickel-dependent urease complex of Cryptococcus the pathogen’s Achilles’ heel? MBio 2013, 4, e00408-13. [Google Scholar] [CrossRef] [PubMed]
- Mora, D.; Arioli, S. Microbial urease in health and disease. PLoS Pathog. 2014, 10, e1004472. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-T.; Chen, M.-L.; Huang, L.-L.; Mao, I.-F. Uric acid and urea in human sweat. Chin. J. Physiol. 2002, 45, 109–116. [Google Scholar] [PubMed]
- Lasisi, T.J.; Raji, Y.R.; Salako, B.L. Salivary creatinine and urea analysis in patients with chronic kidney disease: A case control study. BMC Nephrol. 2016, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Bishai, W.; Timmins, G. Potential for breath test diagnosis of urease positive pathogens in lung infections. J. Breath Res. 2019, 13, 032002. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.L.; Roux, C.M.; Olson, M.W.; Luong, T.T.; Lee, C.Y.; Olson, R.; Dunman, P.M. Characterizing the effects of inorganic acid and alkaline shock on the Staphylococcus aureus transcriptome and messenger RNA turnover. FEMS Immunol. Med. Microbiol. 2010, 60, 208–250. [Google Scholar] [CrossRef] [PubMed]
- Pot, R.G.; Stoof, J.; Nuijten, P.J.; De Haan, L.A.; Loeffen, P.; Kuipers, E.J.; Van Vliet, A.H.; Kusters, J.G. UreA2B2: A second urease system in the gastric pathogen Helicobacter felis. FEMS Immunol. Med. Microbiol. 2007, 50, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Stoof, J.; Breijer, S.; Pot, R.G.; van der Neut, D.; Kuipers, E.J.; Kusters, J.G.; van Vliet, A.H. Inverse nickel-responsive regulation of two urease enzymes in the gastric pathogen Helicobacter mustelae. Environ. Microbiol. 2008, 10, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.L.; Tronrud, D.E.; Taber, S.R.; Karplus, P.A.; Hausinger, R.P. Iron-containing urease in a pathogenic bacterium. Proc. Natl. Acad. Sci. USA 2011, 108, 13095–13099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomons, N.W.; Viteri, F.; Shuler, T.R.; Nielsen, F.H. Bioavailability of nickel in man: Effects of foods and chemically-defined dietary constituents on the absorption of inorganic nickel. J. Nutr. 1982, 112, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Bauerfeind, P.; Garner, R.; Dunn, B.; Mobley, H. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut 1997, 40, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Gupta, A.; Chandra, M.; Koowar, S. Role of Helicobacter pylori infection in the pathogenesis of minimal hepatic encephalopathy and effect of its eradication. Indian J. Gastroenterol. 2011, 30, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Stingl, K.; De Reuse, H. Staying alive overdosed: How does Helicobacter pylori control urease activity? Int. J. Med. Microbiol. 2005, 295, 307–315. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, A.H.; Poppelaars, S.W.; Davies, B.J.; Stoof, J.; Bereswill, S.; Kist, M.; Penn, C.W.; Kuipers, E.J.; Kusters, J.G. NikR mediates nickel-responsive transcriptional induction of urease expression in Helicobacter pylori. Infect. Immun. 2002, 70, 2846–2852. [Google Scholar] [CrossRef]
- van Vliet, A.H.; Kuipers, E.J.; Waidner, B.; Davies, B.J.; de Vries, N.; Penn, C.W.; Vandenbroucke-Grauls, C.M.; Kist, M.; Bereswill, S.; Kusters, J.G. Nickel-responsive induction of urease expression in Helicobacter pylori is mediated at the transcriptional level. Infect. Immun. 2001, 69, 4891–4897. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Molina-Manso, D.; del Prado, G.; Ortiz-Perez, A.; Manrubia-Cobo, M.; Gomez-Barrena, E.; Cordero-Ampuero, J.; Esteban, J. In vitro susceptibility to antibiotics of staphylococci in biofilms isolated from orthopaedic infections. Int. J. Antimicrob. Agents 2013, 41, 521–523. [Google Scholar] [CrossRef]
- Norsworthy, A.N.; Pearson, M.M. From Catheter to Kidney Stone: The uropathogenic lifestyle of Proteus mirabilis. Trends Microbiol. 2017, 25, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Bichler, K.H.; Eipper, E.; Naber, K.; Braun, V.; Zimmermann, R.; Lahme, S. Urinary infection stones. Int. J. Antimicrob. Agents 2002, 19, 488–498. [Google Scholar] [CrossRef]
- Heitland, P.; Köster, H.D. Biomonitoring of 30 trace elements in urine of children and adults by ICP-MS. Clin. Chim. Acta 2006, 365, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.W.; Mehta, N.S.; Maier, R.J. Requirement of nickel metabolism proteins HypA and HypB for full activity of both hydrogenase and urease in Helicobacter pylori. Mol. Microbiol. 2001, 39, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.L.; Zbell, A.L.; Maier, R.J. Nickel enzyme maturation in Helicobacter hepaticus: Roles of accessory proteins in hydrogenase and urease activities. Microbiology 2007, 153, 3748–3756. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.M.; Sebaihia, M.; Churcher, C.; Quail, M.A.; Seshasayee, A.S.; Luscombe, N.M.; Abdellah, Z.; Arrosmith, C.; Atkin, B.; Chillingworth, T.; et al. Complete genome sequence of uropathogenic Proteus mirabilis, a master of both adherence and motility. J. Bacteriol. 2008, 190, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Gerwien, F.; Skrahina, V.; Kasper, L.; Hube, B.; Brunke, S. Metals in fungal virulence. FEMS Microbiol. Rev. 2018, 42. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Panting, R.J.; Varma, A.; Saijo, T.; Waldron, K.J.; Jong, A.; Ngamskulrungroj, P.; Chang, Y.C.; Rutherford, J.C.; Kwon-Chung, K.J. Factors required for activation of urease as a virulence determinant in Cryptococcus neoformans. MBio 2013, 4, e00220-13. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Maier, R.J.; Olczak, A.; Maier, S.; Soni, S.; Gunn, J. Respiratory hydrogen use by Salmonella enterica serovar Typhimurium is essential for virulence. Infect. Immun. 2004, 72, 6294–6299. [Google Scholar] [CrossRef]
- Maier, R.J.; Fu, C.; Gilbert, J.; Moshiri, F.; Olson, J.; Plaut, A.G. Hydrogen uptake hydrogenase in Helicobacter pylori. FEMS Microbiol. Lett. 1996, 141, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Ernst, F.D.; Bereswill, S.; Waidner, B.; Stoof, J.; Mader, U.; Kusters, J.G.; Kuipers, E.J.; Kist, M.; van Vliet, A.H.; Homuth, G. Transcriptional profiling of Helicobacter pylori Fur- and iron-regulated gene expression. Microbiology 2005, 151, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Bahlawane, C.; Aubert, S.; Delay, C.M.; Schauer, K.; Michaud-Soret, I.; De Reuse, H. Hierarchical regulation of the NikR-mediated nickel response in Helicobacter pylori. Nucleic Acids Res. 2011, 39, 7564–7575. [Google Scholar] [CrossRef] [PubMed]
- Contreras, M.; Thiberge, J.M.; Mandrand-Berthelot, M.A.; Labigne, A. Characterization of the roles of NikR, a nickel-responsive pleiotropic autoregulator of Helicobacter pylori. Mol. Microbiol. 2003, 49, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.L.; Mehta, N.; Weinberg, M.V.; Maier, C.; Maier, R.J. Interaction between the Helicobacter pylori accessory proteins HypA and UreE is needed for urease maturation. Microbiology 2007, 153, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.L.; McMurry, J.L.; Hill, S.A.; Maier, R.J. Helicobacter pylori hydrogenase accessory protein HypA and urease accessory protein UreG compete with each other for UreE recognition. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Q.; Huang, H.-T.; Maroney, M.J. The Helicobacter pylori HypA· UreE2 Complex Contains a Novel High-Affinity Ni(II)-Binding Site. Biochemistry 2018, 57, 2932–2942. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, H.; Cheng, T.; Xia, W.; Lai, Y.-T.; Sun, H. Nickel translocation between metallochaperones HypA and UreE in Helicobacter pylori. Metallomics 2014, 6, 1731–1736. [Google Scholar] [CrossRef]
- Stingl, K.; Schauer, K.; Ecobichon, C.; Labigne, A.; Lenormand, P.; Rousselle, J.-C.; Namane, A.; de Reuse, H. In vivo interactome of Helicobacter pylori urease revealed by tandem affinity purification. Mol. Cell. Proteom. 2008, 7, 2429–2441. [Google Scholar] [CrossRef]
- Levitt, M.D. Production and excretion of hydrogen gas in man. N. Engl. J. Med. 1969, 281, 122–127. [Google Scholar] [CrossRef]
- Kanazuru, T.; Sato, E.F.; Nagata, K.; Matsui, H.; Watanabe, K.; Kasahara, E.; Jikumaru, M.; Inoue, J.; Inoue, M. Role of hydrogen generation by Klebsiella pneumoniae in the oral cavity. J. Microbiol. 2010, 48, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J.; Perez-Perez, G.I.; Kleanthous, H.; Cover, T.L.; Peek, R.M.; Chyou, P.H.; Stemmermann, G.N.; Nomura, A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55, 2111–2115. [Google Scholar] [PubMed]
- Fox, J.G.; Ge, Z.; Whary, M.T.; Erdman, S.E.; Horwitz, B.H. Helicobacter hepaticus infection in mice: Models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 2011, 4, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Poutahidis, T.; Cappelle, K.; Levkovich, T.; Lee, C.W.; Doulberis, M.; Ge, Z.; Fox, J.G.; Horwitz, B.H.; Erdman, S.E. Pathogenic intestinal bacteria enhance prostate cancer development via systemic activation of immune cells in mice. PLoS ONE 2013, 8, e73933. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Josenhans, C.; Sterzenbach, T.; Drescher, B.; Brandt, P.; Bell, M.; Droge, M.; Fartmann, B.; Fischer, H.P.; Ge, Z.; et al. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl. Acad. Sci. USA 2003, 100, 7901–7906. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.J.; Olson, J.; Olczak, A. Hydrogen-oxidizing capabilities of Helicobacter hepaticus and in vivo availability of the substrate. J. Bacteriol. 2003, 185, 2680–2682. [Google Scholar] [CrossRef] [PubMed]
- McDowall, J.S.; Murphy, B.J.; Haumann, M.; Palmer, T.; Armstrong, F.A.; Sargent, F. Bacterial formate hydrogenlyase complex. Proc. Natl. Acad. Sci. USA 2014, 111, E3948–E3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbell, A.L.; Maier, R.J. Role of the Hya hydrogenase in recycling of anaerobically produced H2 in Salmonella enterica serovar Typhimurium. Appl. Environ. Microbiol. 2009, 75, 1456–1459. [Google Scholar] [CrossRef]
- Maier, L.; Vyas, R.; Cordova, C.D.; Lindsay, H.; Schmidt, T.S.; Brugiroux, S.; Periaswamy, B.; Bauer, R.; Sturm, A.; Schreiber, F.; et al. Microbiota-derived hydrogen fuels Salmonella typhimurium invasion of the gut ecosystem. Cell Host Microbe 2013, 14, 641–651. [Google Scholar] [CrossRef]
- Craig, M.; Sadik, A.Y.; Golubeva, Y.A.; Tidhar, A.; Slauch, J.M. Twin-arginine translocation system (tat) mutants of Salmonella are attenuated due to envelope defects, not respiratory defects. Mol. Microbiol. 2013, 89, 887–902. [Google Scholar] [CrossRef]
- Parkhill, J.; Wren, B.W.; Mungall, K.; Ketley, J.M.; Churcher, C.; Basham, D.; Chillingworth, T.; Davies, R.M.; Feltwell, T.; Holroyd, S.; et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 2000, 403, 665–668. [Google Scholar] [CrossRef]
- Carlone, G.M.; Lascelles, J. Aerobic and anaerobic respiratory systems in Campylobacter fetus subsp. jejuni grown in atmospheres containing hydrogen. J. Bacteriol. 1982, 152, 306–314. [Google Scholar]
- Istivan, T.; Ward, P.; Coloe, P. Current Research, Technology and Education Topics in Applied Microbiology and Microbial Biotechnology; Méndez-Vilas, A., Ed.; FORMATEX: Badajoz, Spain, 2010; pp. 626–634. [Google Scholar]
- Benoit, S.L.; Holland, A.A.; Johnson, M.K.; Maier, R.J. Iron-sulfur protein maturation in Helicobacter pylori: Identifying a Nfu-type cluster carrier protein and its iron-sulfur protein targets. Mol. Microbiol. 2018, 108, 379–396. [Google Scholar] [CrossRef]
- Lee, H.; Ma, R.; Grimm, M.C.; Riordan, S.M.; Lan, R.; Zhong, L.; Raftery, M.; Zhang, L. Examination of the anaerobic growth of Campylobacter concisus strains. Int. J. Microbiol. 2014, 2014, 476047. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Riddle, M.S.; Platts-Mills, J.A.; Pavlinac, P.; Zaidi, A.K. Shigellosis. Lancet 2018, 391, 801–812. [Google Scholar] [CrossRef]
- Kissing, S.; Saftig, P.; Haas, A. Vacuolar ATPase in phago (lyso) some biology. Int. J. Med. Microbiol. 2018, 308, 58–67. [Google Scholar] [CrossRef]
- Pochapsky, T.C.; Pochapsky, S.S.; Ju, T.; Mo, H.; Al-Mjeni, F.; Maroney, M.J. Modeling and experiment yields the structure of acireductone dioxygenase from Klebsiella pneumoniae. Nat. Struct. Mol. Biol. 2002, 9, 966. [Google Scholar] [CrossRef]
- Clugston, S.L.; Barnard, J.F.; Kinach, R.; Miedema, D.; Ruman, R.; Daub, E.; Honek, J.F. Overproduction and characterization of a dimeric non-zinc glyoxalase I from Escherichia coli: Evidence for optimal activation by nickel ions. Biochemistry 1998, 37, 8754–8763. [Google Scholar] [CrossRef]
- Sukdeo, N.; Clugston, S.L.; Daub, E.; Honek, J.F. Distinct classes of glyoxalase I: Metal specificity of the Yersinia pestis, Pseudomonas aeruginosa and Neisseria meningitidis enzymes. Biochem. J. 2004, 384, 111–117. [Google Scholar] [CrossRef]
- Ozyamak, E.; Black, S.S.; Walker, C.A.; Maclean, M.J.; Bartlett, W.; Miller, S.; Booth, I.R. The critical role of S-lactoylglutathione formation during methylglyoxal detoxification in Escherichia coli. Mol. Microbiol. 2010, 78, 1577–1590. [Google Scholar] [CrossRef]
- Deponte, M. Glyoxalase diversity in parasitic protists. Biochem. Soc. Trans. 2014, 42, 473–478. [Google Scholar] [CrossRef]
- Youn, H.D.; Kim, E.J.; Roe, J.H.; Hah, Y.C.; Kang, S.O. A novel nickel-containing superoxide dismutase from Streptomyces spp. Biochem. J. 1996, 318 Pt 3, 889–896. [Google Scholar] [CrossRef]
- Dupont, C.; Neupane, K.; Shearer, J.; Palenik, B. Diversity, function and evolution of genes coding for putative Ni-containing superoxide dismutases. Environ. Microbiol. 2008, 10, 1831–1843. [Google Scholar] [CrossRef]
- Zambelli, B.; Ciurli, S. Nickel and human health. In Interrelations between Essential Metal Ions and Human Diseases; Springer: Dordrecht, The Netherlands, 2013; pp. 321–357. [Google Scholar]
- Lebrette, H.; Brochier-Armanet, C.; Zambelli, B.; de Reuse, H.; Borezee-Durant, E.; Ciurli, S.; Cavazza, C. Promiscuous nickel import in human pathogens: Structure, thermodynamics, and evolution of extracytoplasmic nickel-binding proteins. Structure 2014, 22, 1421–1432. [Google Scholar] [CrossRef]
- Chivers, P.T. Nickel recognition by bacterial importer proteins. Metallomics 2015, 7, 590–595. [Google Scholar] [CrossRef] [Green Version]
- Lebrette, H.; Iannello, M.; Fontecilla-Camps, J.C.; Cavazza, C. The binding mode of Ni-(L-His) 2 in NikA revealed by X-ray crystallography. J. Inorg. Biochem. 2013, 121, 16–18. [Google Scholar] [CrossRef]
- Ghssein, G.; Brutesco, C.; Ouerdane, L.; Fojcik, C.; Izaute, A.; Wang, S.; Hajjar, C.; Lobinski, R.; Lemaire, D.; Richaud, P. Biosynthesis of a broad-spectrum nicotianamine-like metallophore in Staphylococcus aureus. Science 2016, 352, 1105–1109. [Google Scholar] [CrossRef]
- Grim, K.P.; San Francisco, B.; Radin, J.N.; Brazel, E.B.; Kelliher, J.L.; Solórzano, P.K.P.; Kim, P.C.; McDevitt, C.A.; Kehl-Fie, T.E. The metallophore staphylopine enables Staphylococcus aureus to compete with the host for zinc and overcome nutritional immunity. MBio 2017, 8, e01281-17. [Google Scholar] [CrossRef]
- Song, L.; Zhang, Y.; Chen, W.; Gu, T.; Zhang, S.-Y.; Ji, Q. Mechanistic insights into staphylopine-mediated metal acquisition. Proc. Natl. Acad. Sci. USA 2018, 115, 3942–3947. [Google Scholar] [CrossRef] [Green Version]
- Lhospice, S.; Gomez, N.O.; Ouerdane, L.; Brutesco, C.; Ghssein, G.; Hajjar, C.; Liratni, A.; Wang, S.; Richaud, P.; Bleves, S. Pseudomonas aeruginosa zinc uptake in chelating environment is primarily mediated by the metallophore pseudopaline. Sci. Rep. 2017, 7, 17132. [Google Scholar] [CrossRef]
- Schauer, K.; Gouget, B.; Carrière, M.; Labigne, A.; De Reuse, H. Novel nickel transport mechanism across the bacterial outer membrane energized by the TonB/ExbB/ExbD machinery. Mol. Microbiol. 2007, 63, 1054–1068. [Google Scholar] [CrossRef]
- Zhang, P. Structure and mechanism of energy-coupling factor transporters. Trends Microbiol. 2013, 21, 652–659. [Google Scholar] [CrossRef]
- Bauerfeind, P.; Garner, R.M.; Mobley, L. Allelic exchange mutagenesis of nixA in Helicobacter pylori results in reduced nickel transport and urease activity. Infect. Immun. 1996, 64, 2877–2880. [Google Scholar]
- Fischer, F.; Robbe-Saule, M.; Turlin, E.; Mancuso, F.; Michel, V.; Richaud, P.; Veyrier, F.J.; De Reuse, H.; Vinella, D. Characterization in Helicobacter pylori of a nickel transporter essential for colonization that was acquired during evolution by gastric Helicobacter species. PLoS Pathog. 2016, 12, e1006018. [Google Scholar] [CrossRef]
- Nolan, K.J.; McGee, D.J.; Mitchell, H.M.; Kolesnikow, T.; Harro, J.M.; O’Rourke, J.; Wilson, J.E.; Danon, S.J.; Moss, N.D.; Mobley, H.L. In vivo behavior of a Helicobacter pylori SS1 nixA mutant with reduced urease activity. Infect. Immun. 2002, 70, 685–691. [Google Scholar] [CrossRef]
- Eitinger, T.; Suhr, J.; Moore, L.; Smith, J.A.C. Secondary transporters for nickel and cobalt ions: Theme and variations. Biometals 2005, 18, 399–405. [Google Scholar] [CrossRef]
- Hiron, A.; Posteraro, B.; Carriere, M.; Remy, L.; Delporte, C.; La Sorda, M.; Sanguinetti, M.; Juillard, V.; Borezee-Durant, E. A nickel ABC-transporter of Staphylococcus aureus is involved in urinary tract infection. Mol. Microbiol. 2010, 77, 1246–1260. [Google Scholar] [CrossRef]
- Remy, L.; Carrière, M.; Derré-Bobillot, A.; Martini, C.; Sanguinetti, M.; Borezée-Durant, E. The Staphylococcus aureus Opp1 ABC transporter imports nickel and cobalt in zinc-depleted conditions and contributes to virulence. Mol. Microbiol. 2013, 87, 730–743. [Google Scholar] [CrossRef]
- Lebrette, H.; Borezée-Durant, E.; Martin, L.; Richaud, P.; Erba, E.B.; Cavazza, C. Novel insights into nickel import in Staphylococcus aureus: The positive role of free histidine and structural characterization of a new thiazolidine-type nickel chelator. Metallomics 2015, 7, 613–621. [Google Scholar] [CrossRef]
- Chivers, P.T.; Benanti, E.L.; Heil-Chapdelaine, V.; Iwig, J.S.; Rowe, J.L. Identification of Ni-(L-His)2 as a substrate for NikABCDE-dependent nickel uptake in Escherichia coli. Metallomics 2012, 4, 1043–1050. [Google Scholar] [CrossRef]
- Subashchandrabose, S.; Hazen, T.H.; Brumbaugh, A.R.; Himpsl, S.D.; Smith, S.N.; Ernst, R.D.; Rasko, D.A.; Mobley, H.L. Host-specific induction of Escherichia coli fitness genes during human urinary tract infection. Proc. Natl. Acad. Sci. USA 2014, 111, 18327–18332. [Google Scholar] [CrossRef]
- Robinson, A.E.; Lowe, J.E.; Koh, E.I.; Henderson, J.P. Uropathogenic enterobacteria use the yersiniabactin metallophore system to acquire nickel. J. Biol. Chem. 2018, 293, 14953–14961. [Google Scholar] [CrossRef] [Green Version]
- Lamont, E.A.; Xu, W.W.; Sreevatsan, S. Host-Mycobacterium avium subsp. paratuberculosis interactome reveals a novel iron assimilation mechanism linked to nitric oxide stress during early infection. BMC Genom. 2013, 14, 694. [Google Scholar] [CrossRef]
- Stahler, F.N.; Odenbreit, S.; Haas, R.; Wilrich, J.; Van Vliet, A.H.; Kusters, J.G.; Kist, M.; Bereswill, S. The novel Helicobacter pylori CznABC metal efflux pump is required for cadmium, zinc, and nickel resistance, urease modulation, and gastric colonization. Infect. Immun. 2006, 74, 3845–3852. [Google Scholar] [CrossRef]
- Armbruster, C.E.; Forsyth-DeOrnellas, V.; Johnson, A.O.; Smith, S.N.; Zhao, L.; Wu, W.; Mobley, H.L.T. Genome-wide transposon mutagenesis of Proteus mirabilis: Essential genes, fitness factors for catheter-associated urinary tract infection, and the impact of polymicrobial infection on fitness requirements. PLoS Pathog. 2017, 13, e1006434. [Google Scholar] [CrossRef]
- De Reuse, H. Nickel and Virulence in Bacterial Pathogens. In The Biological Chemistry of Nickel; Zamble, D., Rowińska-Żyrek, M., Kozlowski, H., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 339–356. [Google Scholar]
- Vinella, D.; Fischer, F.; Vorontsov, E.; Gallaud, J.; Malosse, C.; Michel, V.; Cavazza, C.; Robbe-Saule, M.; Richaud, P.; Chamot-Rooke, J.; et al. Evolution of Helicobacter: Acquisition by gastric species of two histidine-rich proteins essential for colonization. PLoS Pathog. 2015, 11, e1005312. [Google Scholar] [CrossRef]
- Ge, R.; Watt, R.M.; Sun, X.; Tanner, J.A.; He, Q.Y.; Huang, J.D.; Sun, H. Expression and characterization of a histidine-rich protein, Hpn: Potential for Ni2+ storage in Helicobacter pylori. Biochem. J. 2006, 393, 285–293. [Google Scholar] [CrossRef]
- Zeng, Y.-B.; Zhang, D.-M.; Li, H.; Sun, H. Binding of Ni2+ to a histidine-and glutamine-rich protein, Hpn-like. JBIC J. Biol. Inorg. Chem. 2008, 13, 1121. [Google Scholar] [CrossRef]
- Chiera, N.M.; Rowinska-Zyrek, M.; Wieczorek, R.; Guerrini, R.; Witkowska, D.; Remelli, M.; Kozlowski, H. Unexpected impact of the number of glutamine residues on metal complex stability. Metallomics 2013, 5, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Seshadri, S.; Benoit, S.L.; Maier, R.J. Roles of His-rich hpn and hpn-like proteins in Helicobacter pylori nickel physiology. J. Bacteriol. 2007, 189, 4120–4126. [Google Scholar] [CrossRef]
- Saylor, Z.; Maier, R. Helicobacter pylori nickel storage proteins: Recognition and modulation of diverse metabolic targets. Microbiology 2018, 164, 1059–1068. [Google Scholar] [CrossRef]
- Benoit, S.L.; Miller, E.F.; Maier, R.J. Helicobacter pylori stores nickel to aid its host colonization. Infect. Immun. 2013, 81, 580–584. [Google Scholar] [CrossRef]
- Skouloubris, S.; Labigne, A.; De Reuse, H. The AmiE aliphatic amidase and AmiF formamidase of Helicobacter pylori: Natural evolution of two enzyme paralogues. Mol. Microbiol. 2001, 40, 596–609. [Google Scholar] [CrossRef]
- Saylor, Z.J. Helicobacter pylori nickel storage proteins: Recognition and modulation of diverse metabolic targets. Ph.D. Thesis, The University of Georgia, Athens, GA, USA, 2018. [Google Scholar]
- Lopez, A.J.; Martinez, L. Parametric models to compute tryptophan fluorescence wavelengths from classical protein simulations. J. Comput. Chem. 2018, 39, 1249–1258. [Google Scholar] [CrossRef]
- Deshayes, S.; Divita, G. Fluorescence technologies for monitoring interactions between biological molecules in vitro. Prog. Mol. Biol. Transl. Sci. 2013, 113, 109–143. [Google Scholar]
- Cvetkovic, A.; Menon, A.L.; Thorgersen, M.P.; Scott, J.W.; Poole, F.L., II; Jenney, F.E., Jr.; Lancaster, W.A.; Praissman, J.L.; Shanmukh, S.; Vaccaro, B.J. Microbial metalloproteomes are largely uncharacterized. Nature 2010, 466, 779. [Google Scholar] [CrossRef]
- Schauer, K.; Muller, C.; Carrière, M.; Labigne, A.; Cavazza, C.; De Reuse, H. The Helicobacter pylori GroES cochaperonin HspA functions as a specialized nickel chaperone and sequestration protein through its unique C-terminal extension. J. Bacteriol. 2010, 192, 1231–1237. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Yang, F.; Wu, W.; Sun, H.; Xie, Q.; Si, W.; Zou, Q.; Yang, Z. Immunization with heat shock protein A and gamma-glutamyl transpeptidase induces reduction on the Helicobacter pylori colonization in mice. PLoS ONE 2015, 10, e0130391. [Google Scholar]
- Zhang, X.J.; Feng, S.Y.; Li, Z.T.; Feng, Y.M. Expression of Helicobacter pylori hspA Gene in Lactococcus lactis NICE System and Experimental Study on Its Immunoreactivity. Gastroenterol. Res. Pract. 2015, 2015, 750932. [Google Scholar] [CrossRef]
Figure 1.
Dual role for H. pylori urease. (A) Holo (Ni-bound) urease can convert urea into ammonia and carbon dioxide (catalytic activity, #1) and can protect against oxidative stress (nonspecific quenching through Msr-repairable Met, activity #2). (B) Apo-urease (Ni-free) is non ureolytic yet it retains the nonspecific oxidative stress combatting activity (#2).
Figure 1.
Dual role for H. pylori urease. (A) Holo (Ni-bound) urease can convert urea into ammonia and carbon dioxide (catalytic activity, #1) and can protect against oxidative stress (nonspecific quenching through Msr-repairable Met, activity #2). (B) Apo-urease (Ni-free) is non ureolytic yet it retains the nonspecific oxidative stress combatting activity (#2).
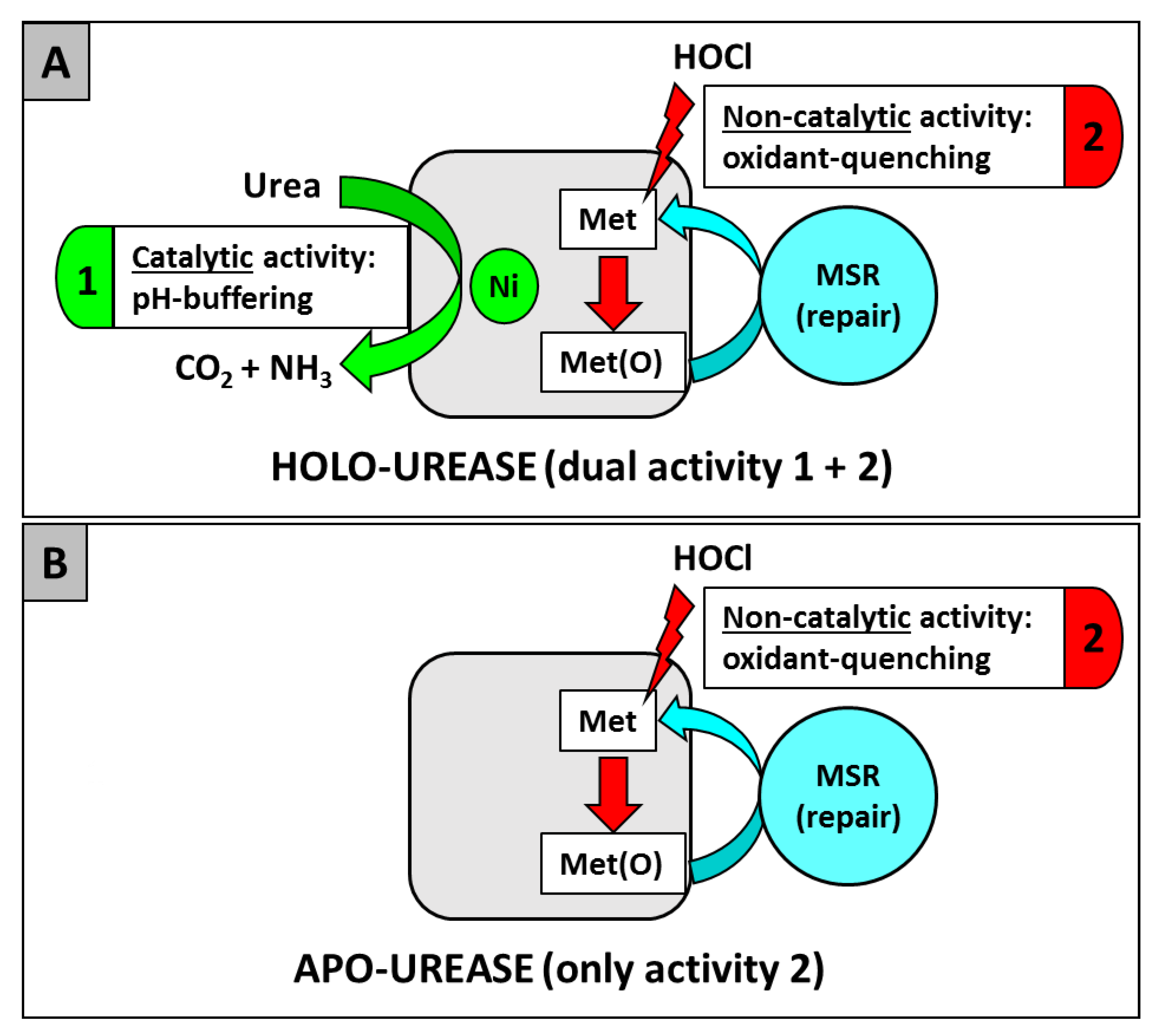
Figure 2.
The P. mirabilis HypB hydrogenase accessory protein is exceptionally His-rich. The sequence shown was translated from the hypB nucleotide sequence, as found in genome sequence of P. mirabilis strain HI4320 [139]. His residues are shown in red. The His-rich sequence suggests PmHypB might play additional roles in Ni-enzyme maturation and/or Ni-storage besides its expected role as hydrogenase accessory protein.
Figure 2.
The P. mirabilis HypB hydrogenase accessory protein is exceptionally His-rich. The sequence shown was translated from the hypB nucleotide sequence, as found in genome sequence of P. mirabilis strain HI4320 [139]. His residues are shown in red. The His-rich sequence suggests PmHypB might play additional roles in Ni-enzyme maturation and/or Ni-storage besides its expected role as hydrogenase accessory protein.

Figure 3.
Tryptophan fluorescence as a way to detect Hpn and Hpnl protein interactions with (A) AmiE or (B) PepA. Purified proteins were incubated overnight at 4 °C with 1 mM DMS (crosslinker). Tryptophan fluorescence was recorded on a BioTek SynergyMx spectrophotometer (excitation at 295 nm and emission at 315–415 nm). Samples were normalized against their respective (buffer-only) controls and plotted as relative fluorescence against wavelength. See [210].
Figure 3.
Tryptophan fluorescence as a way to detect Hpn and Hpnl protein interactions with (A) AmiE or (B) PepA. Purified proteins were incubated overnight at 4 °C with 1 mM DMS (crosslinker). Tryptophan fluorescence was recorded on a BioTek SynergyMx spectrophotometer (excitation at 295 nm and emission at 315–415 nm). Samples were normalized against their respective (buffer-only) controls and plotted as relative fluorescence against wavelength. See [210].
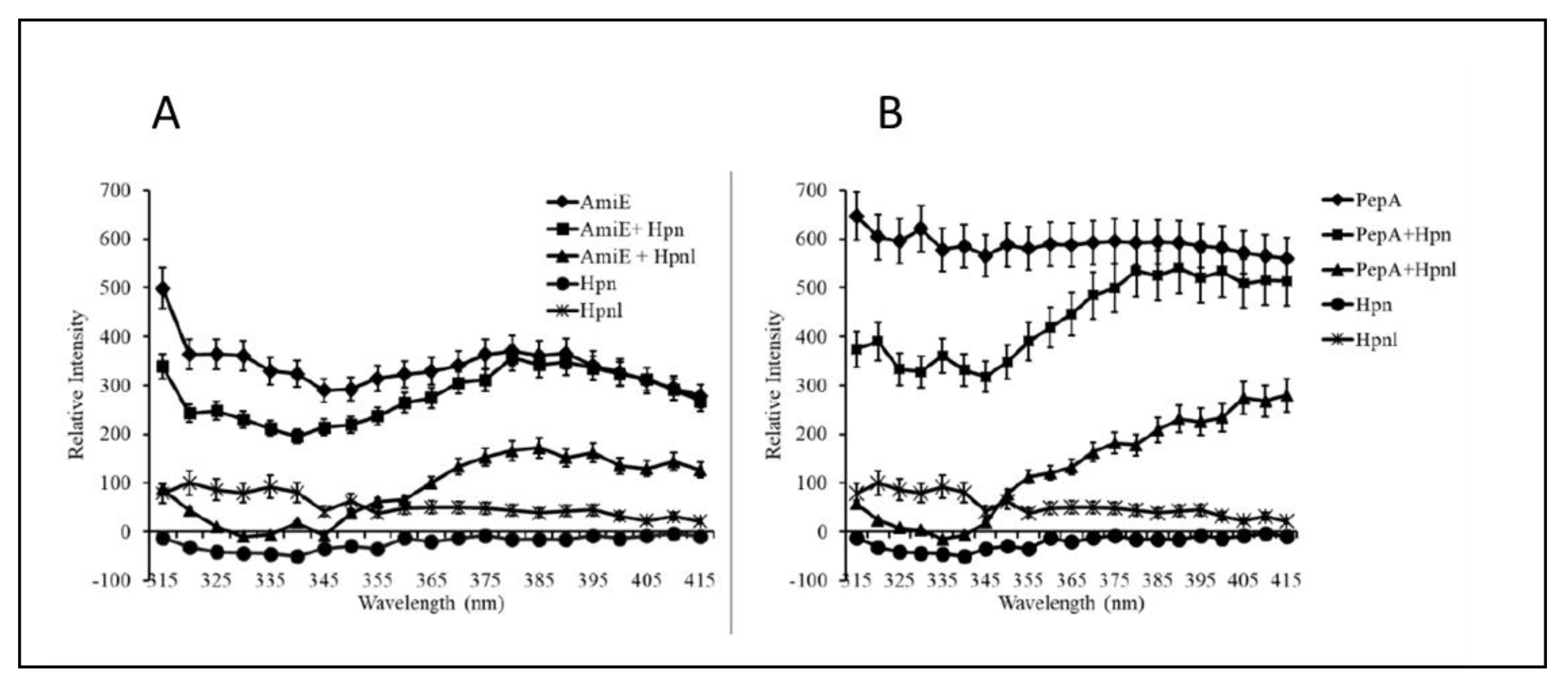

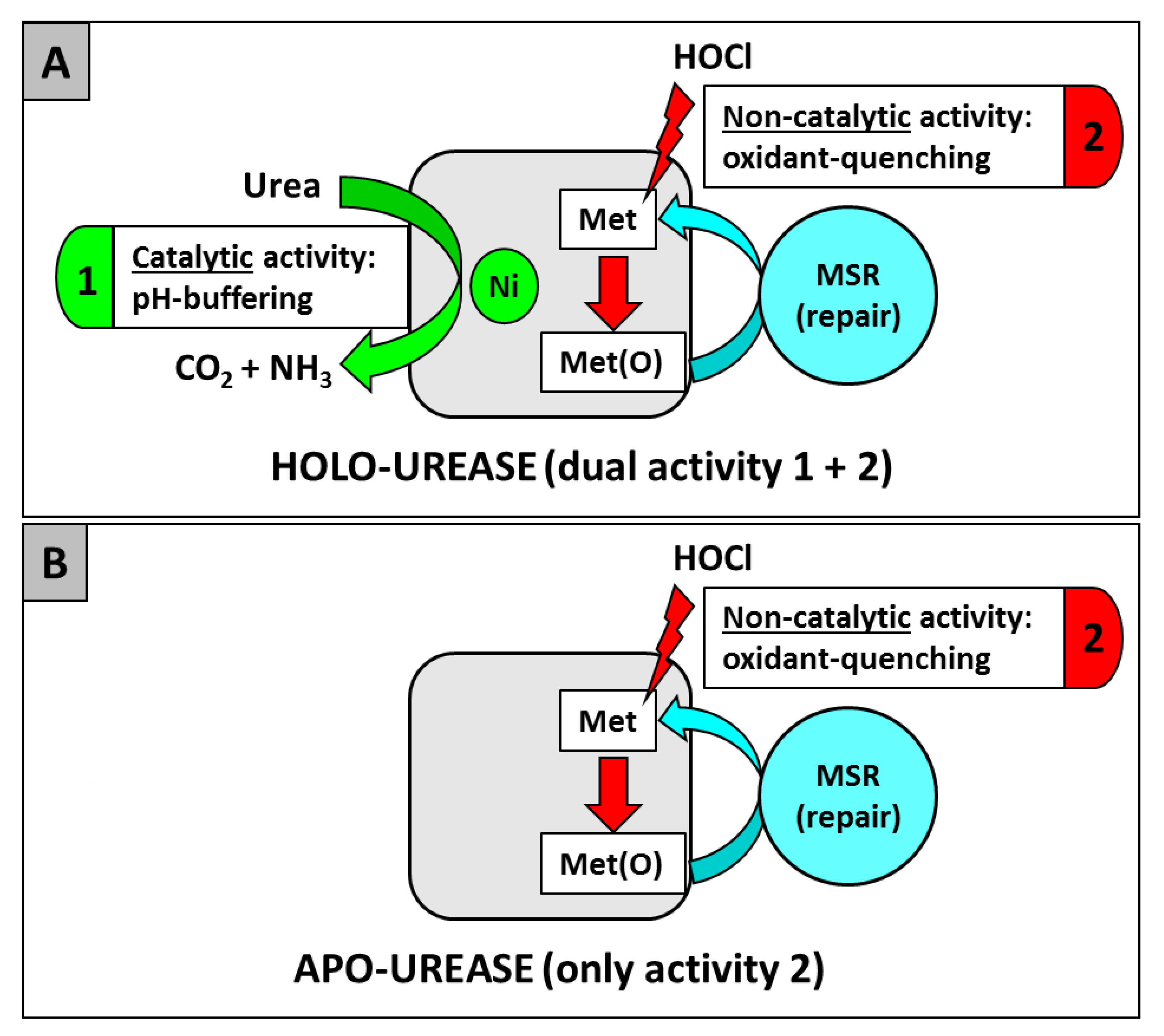{kind=link}
{kind=link}
{kind=link}
Table 1.
List of eukaryotic and prokaryotic pathogens with Ni-enzymes and their putative or demonstrated role in pathogenesis.
Table 1.
List of eukaryotic and prokaryotic pathogens with Ni-enzymes and their putative or demonstrated role in pathogenesis.
| Pathogen | Ni-enzyme * | Role in Pathogenesis (Reference) |
|---|---|---|
| EUKARYOTES | ||
| Human fungi | ||
| Cryptococcus neoformans | Ure | Virulence factor in experimental cryptococcosis [17] |
| Required for microvascular sequestration and mouse brain invasion [18] | ||
| Modulates phagolysosomal pH; important for mouse brain infection [19] | ||
| Released via extracellular vesicles [20] | ||
| Cryptococcus gattii | Ure | Virulence factor in mice [21] |
| Coccidioides posadasii | Ure | Coccidioidomycosis in mice [22,23] |
| Histoplasma capsulatum | Ure | Released via extracellular vesicles [24] |
| Paracoccidioides brasiliensis | Ure | Up-expressed in mouse infection model [25] |
| Oomycetes | ||
| Pythium insidiosum | Ure | Putative virulence factor for pythiosis [26] |
| Protists | ||
| Leishmania major | Glo-I | Important for parasite metabolism: methylglyoxal detoxification [27] |
| Leishmania donovani | Glo-I | Essential for growth; suggested as drug target [28] |
| Trypanosoma cruzi | Ard | Important for parasite metabolism: methionine salvage pathway |
| Glo-I | Important for parasite metabolism: methylglyoxal detoxification [29] | |
| PROKARYOTES | ||
| Actinobacteria | ||
| Actinomyces naeslundii | Ure | Needed in acidic environment; promotes plaque formation [30] |
| Corynebacterium urealyticum | Ure | Plays an important role in urinary tract infection [31] |
| Mycobacterium tuberculosis | Hyc | Essential for optimal growth [32] |
| Up-regulated during infection of human macrophage-like cells [33] | ||
| Up-expressed in resting and active murine bone marrow macrophages [34] | ||
| Ure | Important for survival under nitrogen-limited environment [35] | |
| Streptomyces scabies | Sod | Important against oxidative stress encountered in host (plant) |
| Firmicutes | ||
| Clostridia | Glo-I | Important for metabolism: methylglyoxal detoxification [36] |
| Staphylococcus aureus | Ure | Increased expression of structural and accessory genes in biofilms [37] |
| Required for acid response and persistent murine kidney infection [38] | ||
| Decreased activity in mixed source (S. epidermidis) biofilms [39] | ||
| Staphylococcus epidermidis | Ure | Decreased activity in mixed source (S. aureus) biofilms [39] |
| Staphylococcus saprophyticus | Ure | Important for bladder infection and bladder stones in rats [40] |
| Streptococcus salivarius | Ure | Important as source of nitrogen and to combat acid stress [41] |
| Mollicutes | ||
| Ureaplasma urealyticum | Ure | Role in human vaginal infection; used for diagnostic [42] |
| Ammonia contributes to PMF-driven ATP synthesis [43] | ||
| Ammonia generates struvite stone formation in rat bladders [44] | ||
| Ureaplasma parvum | Ure | Role in human vaginal infection; used for diagnostic [42] |
| Ureaplasma diversum | Ure | Role in vaginal infection of cattle and small ruminants [45] |
| Proteobacteria | ||
| Alphaproteobacteria | ||
| Brucella abortus | Ure | Needed for intestinal colonization in a murine model [46] |
| Immunization with B. a. urease protects against B. abortus infection in mice [47] | ||
| Brucella melitensis | Ure | Immunization with B. a. urease protects against B. melitensis in mice [47] |
| Brucella suis | Ure | Required for intestinal colonization in a murine model [48] |
| Immunization with B. a. urease protects against B. suis infection in mice [47] | ||
| Betaproteobacteria | ||
| Neisseria meningitides | Glo-I | Important for methylglyoxal detoxification and potassium efflux (hypothesized) |
| Neisseria gonorrhoeae | Glo-I | Important for methylglyoxal detoxification and potassium efflux (hypothesized) |
| Gammaproteobacteria | ||
| All γ-proteobacteria | Ard | Important for metabolism: methionine salvage pathway |
| All γ-proteobacteria | Glo-I | Important for methylglyoxal detoxification, potassium efflux |
| Acinetobacter baumannii | Ure | Virulence factor in worm and amoeba hosts [49] |
| Acinetobacter lwoffii | Ure | Needed to survive in the stomach [50] |
| Actinobacillus | Ure | Important for swine respiratory tract infection [51,52] |
| pleuropneumoniae | Hyd-1 | Important for (PMF-driven) metabolism and motility |
| Escherichia coli | Hyd-2 | Important for (PMF-driven) metabolism and motility |
| Hyc | Needed (as part of FHL) to dissipate formic acid-induced acidity [53] | |
| E. coli (Shiga-toxin producing) | Ure | Needed for colonization in the murine model [54] |
| Edwardsiella tarda | Hyd | Hyd. accessory protein Sip2 essential for acid resistance and host infection [55] |
| Haemophilus influenzae | Ure | Important for acid resistance, expressed during human pulmonary infection [56] |
| Klebsiella pneumoniae | Ure | Required for colonization in murine intestinal model [57] |
| Morganella morganii | Ure | Needed for survival at low pH [58,59] |
| Proteus mirabilis | Hyd | Important for swarming motility [60] |
| Ure | Role in persistence, urolithiasis, and acute pyelonephritis in a mouse model [61] | |
| Role in extracellular crystal stone cluster formation in the bladder [62] | ||
| Induced in polymicrobial biofilms [63] | ||
| Providencia stuartii | Ure | Involved in crystal stones formation; induced in polymicrobial biofilms [59] |
| Pseudomonas aeruginosa | Glo-I | Important for methylglyoxal detoxification and potassium efflux (hypothesized) |
| Salmonella Typhimurium | Hyd-1 | Important for acid resistance and macrophage colonization [64] |
| Hyd-2 | Most important hydrogenase for gut invasion [65,66] | |
| Hyd-5 | Expressed under aerobic conditions and in macrophages [64,67,68] | |
| Hyc | Important for anaerobic acid resistance [69] | |
| Shigella flexneri | Hyd | Important for acid resistance [70] |
| Vibrio parahaemolyticus | Ure | Important for pathogenicity [71] |
| Yersinia enterocolitica | Ure | Important for survival at low pH [58,72] |
| Yersinia pestis | Glo-I | Important for methylglyoxal detoxification and potassium efflux (hypothesized) |
| Deltaproteobacteria | ||
| Bilophila wadsworthia | Hyd | H2 used as energy source, optimal growth in presence of H2 and taurine [73] |
| Epsilonproteobacteria | ||
| Campylobacter jejuni | Hyd | Important for chicken cecum colonization [74] |
| Essential for chicken colonization in absence of formate dehydrogenase [74] | ||
| Required for in vitro interaction with human intestinal cells [75] | ||
| Campylobacter concisus | Hyd | Essential for growth under microaerobic conditions [76] |
| Helicobacter hepaticus | Hyd | Role in amino-acid transport and causing liver lesions in mice [77] |
| Ure | Promotes hepatic inflammation in mice [78] | |
| Helicobacter mustelae | Ure | Essential for ferret stomach colonization [79] |
| Helicobacter pylori | Hyd | Needed for mouse stomach colonization [80] |
| Role in CO2 fixation [81] | ||
| Role in CagA translocation [82] | ||
| Ure | Cytotoxic effect on Caco-2 cells [83] | |
| Needed for nude mouse stomach colonization [84] | ||
| Essential for gnotobiotic piglet stomach colonization [85] | ||
| Activates human phagocytes and macrophages [86,87] | ||
| Binds to class II MHC on gastric epithelial cells and induces their apoptosis [88] | ||
| Essential for Mongolian gerbil colonization [89,90] | ||
| Urease-produced CO2 protects against host peroxynitrite [91] | ||
| Urease-produced ammonia disrupts tight cell junction integrity [92] | ||
| Dysregulates epithelial tight junctions through myosin activation [93] | ||
| Activates blood platelets through a lipoxygenase-mediated pathway [94] | ||
| Alters mucin gene expression in human gastric cells [95] | ||
| Essential for chronic mouse infection [96] | ||
| Role in angiogenesis, endothelial cells and chicken embryo models [97] | ||
| Induces blood platelets inflammatory pathways [98] | ||
| Non catalytic, oxidative stress-combatting role [99] |
* Abreviations: Ard: acireductone dioxygenase; Glo-I: Glyoxalase I; Hyc: H2-evolving hydrogenase; Hyd: H2-uptake hydrogenase; Sod: superoxide dismutase; Ure: urease.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Maier, R.J.; Benoit, S.L. Role of Nickel in Microbial Pathogenesis. Inorganics 2019, 7, 80. https://doi.org/10.3390/inorganics7070080
AMA Style
Maier RJ, Benoit SL. Role of Nickel in Microbial Pathogenesis. Inorganics. 2019; 7(7):80. https://doi.org/10.3390/inorganics7070080
Chicago/Turabian StyleMaier, Robert J., and Stéphane L. Benoit. 2019. "Role of Nickel in Microbial Pathogenesis" Inorganics 7, no. 7: 80. https://doi.org/10.3390/inorganics7070080
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.