The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms



{kind=link}
Abstract
:1. Introduction
1.1. Nickel Overview
1.2. Nickel-Induced Carcinogenesis
1.3. Non-Coding RNA
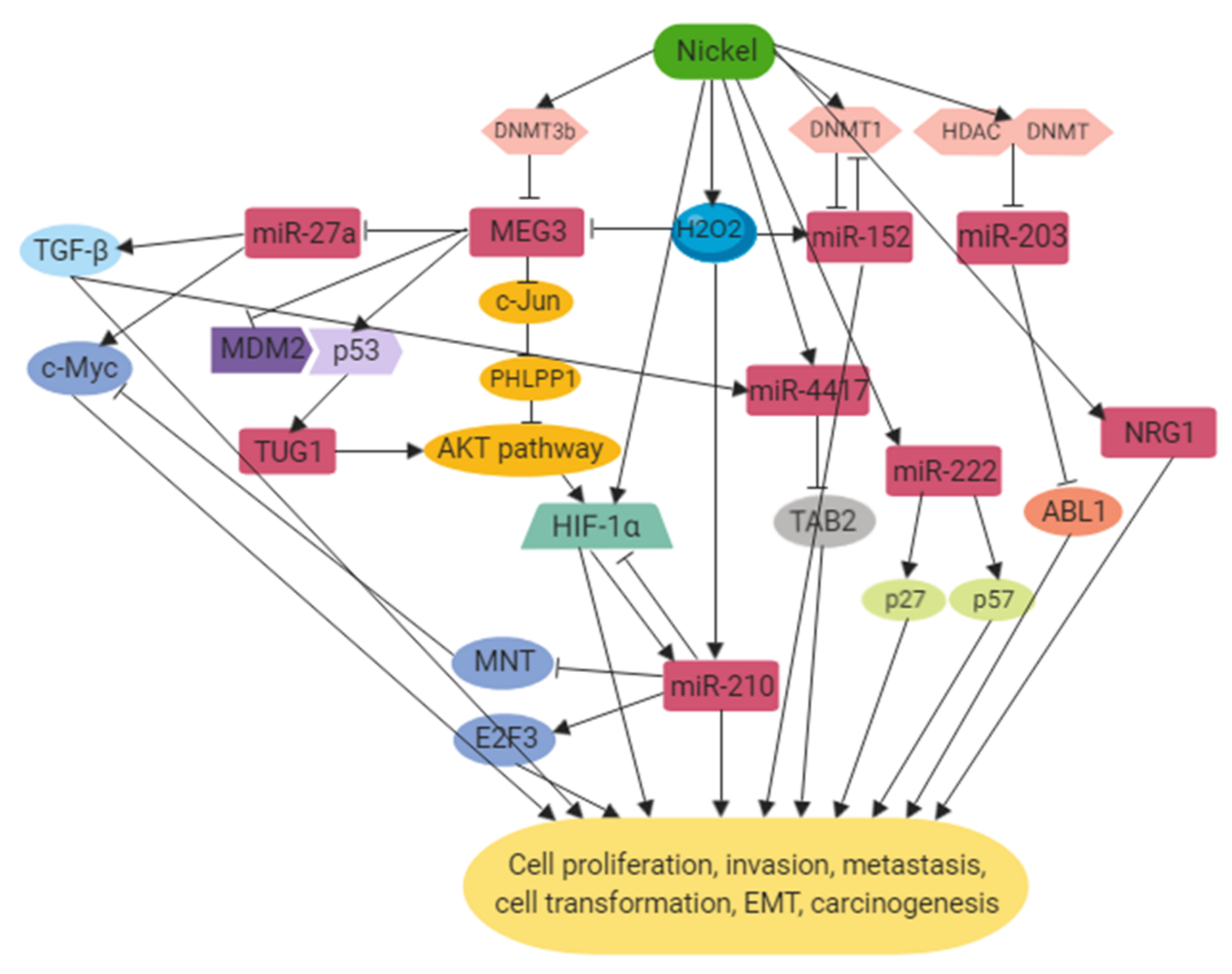
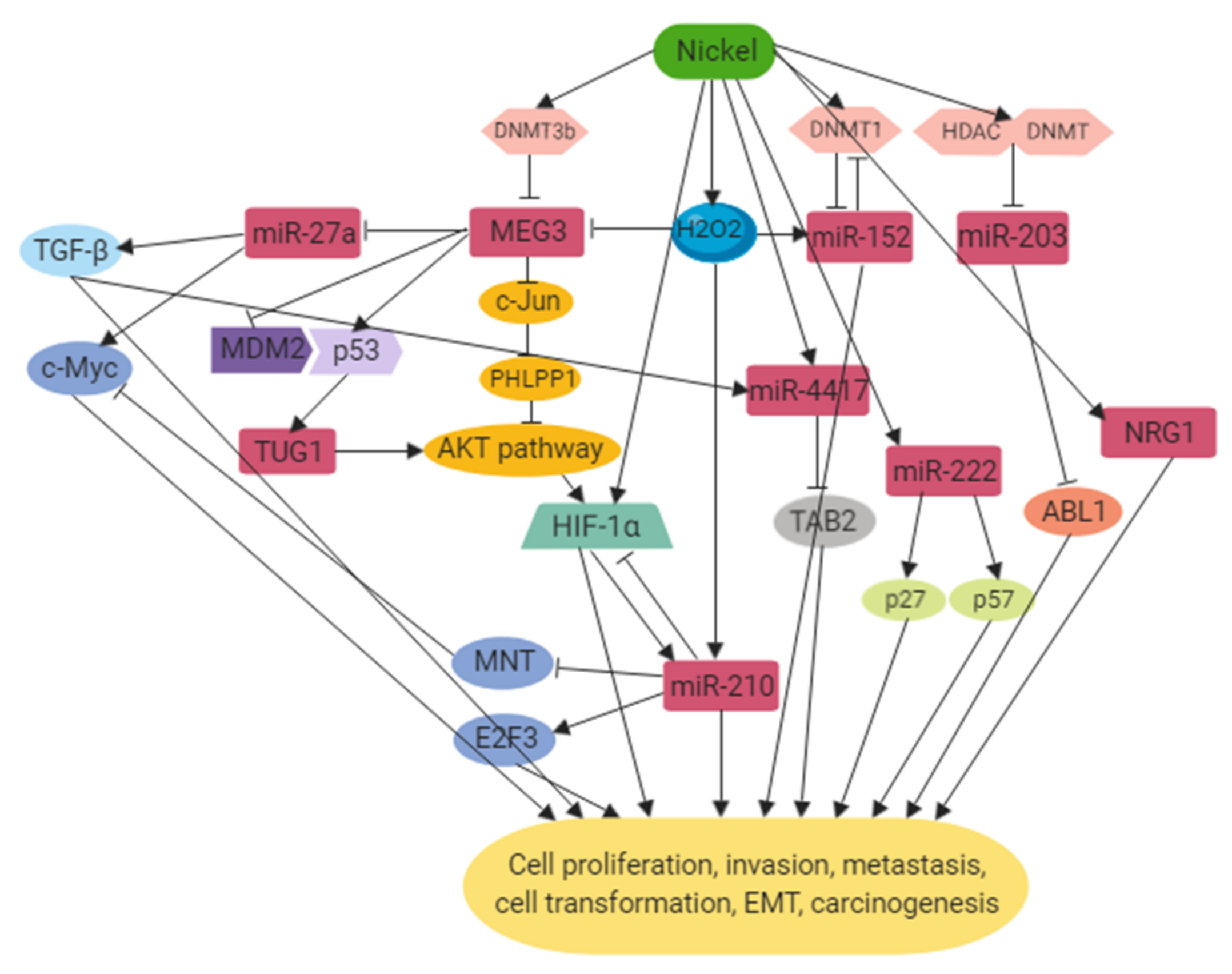
2. Mechanisms
2.1. DNA Hypermethylation
2.2. Hypoxia-Inducible Pathway
2.3. Oxidative Stress
2.4. Tumor Suppressor Gene and Proto-Oncogene Alterations
3. Conclusions
Funding
Conflicts of Interest
References
- Schmidt, M.; Goebeler, M. Nickel allergies: Paying the Toll for innate immunity. J. Mol. Med. 2011, 89, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Cempel, M.; Nikel, G. Nickel: A Review of Its Sources and Environmental Toxicology. Pol. J. Environ. Stud. 2006, 15, 375–382. [Google Scholar]
- Zambelli, B.; Uversky, V.N.; Ciurli, S. Nickel impact on human health: An intrinsic disorder perspective. Biochim. Biophys. Acta 2016, 1864, 1714–1731. [Google Scholar] [CrossRef] [PubMed]
- Coogan, T.P.; Latta, D.M.; Snow, E.T.; Costa, M. Toxicity and carcinogenicity of nickel compounds. Crit. Rev. Toxicol. 1989, 19, 341–384. [Google Scholar] [CrossRef] [PubMed]
- Barceloux, D.G. Nickel. J. Toxicol. Clin. Toxicol. 1999, 37, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Rezuke, W.N.; Knight, J.A.; Sunderman, F.W., Jr. Reference values for nickel concentrations in human tissues and bile. Am. J. Ind. Med. 1987, 11, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Duda-Chodak, A.D.A.; Błaszczyk, U. The impact of nickel on human health. J. Elementol. 2008, 13, 685–696. [Google Scholar]
- Morgan, L.G.; Usher, V. Health problems associated with nickel refining and use. Ann. Occup. Hyg. 1994, 38, 189–198. [Google Scholar]
- Berge, S.R.; Skyberg, K. Radiographic evidence of pulmonary fibrosis and possible etiologic factors at a nickel refinery in Norway. J. Environ. Monit. 2003, 5, 681–688. [Google Scholar] [CrossRef]
- Oller, A.R.; Costa, M.; Oberdorster, G. Carcinogenicity assessment of selected nickel compounds. Toxicol. Appl. Pharmacol. 1997, 143, 152–166. [Google Scholar] [CrossRef]
- Doll, R. Report of the International Committee on Nickel Carcinogenesis in Man; Final Report (No. PB-91-109801/XAB); Program Resources, Inc.: Research Triangle Park, NC, USA, 1990; p. 84. [Google Scholar]
- Huvinen, M.; Pukkala, E. Cancer incidence among Finnish ferrochromium and stainless steel production workers in 1967–2011: A cohort study. BMJ Open 2013, 3, e003819. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, T.K.; Andersen, A. Evidence of carcinogenicity in humans of water-soluble nickel salts. J. Occup. Med. Toxicol. 2010, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.; Berge, S.R.; Engeland, A.; Norseth, T. Exposure to nickel compounds and smoking in relation to incidence of lung and nasal cancer among nickel refinery workers. Occup. Environ. Med. 1996, 53, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Seilkop, S.K.; Oller, A.R. Respiratory cancer risks associated with low-level nickel exposure: An integrated assessment based on animal, epidemiological, and mechanistic data. Regul. Toxicol. Pharmacol. 2003, 37, 173–190. [Google Scholar] [CrossRef]
- Moulin, J.J.; Clavel, T.; Roy, D.; Dananche, B.; Marquis, N.; Fevotte, J.; Fontana, J.M. Risk of lung cancer in workers producing stainless steel and metallic alloys. Int. Arch. Occup. Environ. Health 2000, 73, 171–180. [Google Scholar] [CrossRef]
- Anttila, A.; Pukkala, E.; Aitio, A.; Rantanen, T.; Karjalainen, S. Update of cancer incidence among workers at a copper/nickel smelter and nickel refinery. Int. Arch. Occup. Environ. Health 1998, 71, 245–250. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Zhao, Y.; Srivastava, D. A developmental view of microRNA function. Trends Biochem. Sci. 2007, 32, 189–197. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef]
- Uddin, A.; Chakraborty, S. Role of miRNAs in lung cancer. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Ofir, M.; Hacohen, D.; Ginsberg, D. MiR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Mol. Cancer Res. 2011, 9, 440–447. [Google Scholar] [CrossRef]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sempere, L.F.; Ouyang, H.; Memoli, V.A.; Andrew, A.S.; Luo, Y.; Demidenko, E.; Korc, M.; Shi, W.; Preis, M.; et al. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J. Clin. Investig. 2010, 120, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, T.; Liu, H.; Lv, T.; Yuan, D.; Yao, Y.; Lv, Y.; Song, Y. MiR-101 and Mcl-1 in non-small-cell lung cancer: Expression profile and clinical significance. Med. Oncol. 2012, 29, 1681–1686. [Google Scholar] [CrossRef]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Sullenger, B.A.; Nair, S. From the RNA world to the clinic. Science 2016, 352, 1417–1420. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.M.; Zhou, G.B. Long Non-coding RNAs and Their Roles in Non-small-cell Lung Cancer. Genom. Proteom. Bioinform. 2016, 14, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Chen, L.; Wang, Y.; Jiang, X.; Xia, H.; Zhuang, Z. Long noncoding RNA MALAT1 promotes brain metastasis by inducing epithelial-mesenchymal transition in lung cancer. J. Neurooncol. 2015, 121, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Loewen, G.; Jayawickramarajah, J.; Zhuo, Y.; Shan, B. Functions of lncRNA HOTAIR in lung cancer. J. Hematol. Oncol. 2014, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, C.; Wang, J.; Huang, H.; Li, J.; Xie, Q.; Liu, Y.; Zhu, J.; Li, Y.; Zhang, D.; et al. LncRNA MEG3 downregulation mediated by DNMT3b contributes to nickel malignant transformation of human bronchial epithelial cells via modulating PHLPP1 transcription and HIF-1α translation. Oncogene 2017, 36, 3878–3889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, Y.; Wu, Y.; Ma, L.; Fan, Y.; Kang, X.; Shi, H.; Zhang, J. Isolation and characterization of a novel noncoding RNA from nickel-induced lung cancer. Biol. Trace. Elem. Res. 2012, 150, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Cameron, K.S.; Buchner, V.; Tchounwou, P.B. Exploring the molecular mechanisms of nickel-induced genotoxicity and carcinogenicity: A literature review. Rev. Environ. Health 2011, 26, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar] [PubMed]
- Lee, Y.W.; Klein, C.B.; Kargacin, B.; Salnikow, K.; Kitahara, J.; Dowjat, K.; Zhitkovich, A.; Christie, N.T.; Costa, M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: A new model for epigenetic carcinogens. Mol. Cell. Biol. 1995, 15, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Ohtani-Fujita, N.; Fujita, T.; Aoike, A.; Osifchin, N.E.; Robbins, P.D.; Sakai, T. CpG methylation inactivates the promoter activity of the human retinoblastoma tumor-suppressor gene. Oncogene 1993, 8, 1063–1067. [Google Scholar] [PubMed]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Greger, V.; Debus, N.; Lohmann, D.; Hopping, W.; Passarge, E.; Horsthemke, B. Frequency and parental origin of hypermethylated RB1 alleles in retinoblastoma. Hum. Genet. 1994, 94, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Gartler, S.M.; Scott, C.R.; Chen, S.H.; Laird, C.D. Methylation analysis of CGG sites in the CpG island of the human FMR1 gene. Hum. Mol. Genet. 1992, 1, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Yasaei, H.; Gilham, E.; Pickles, J.C.; Roberts, T.P.; O’Donovan, M.; Newbold, R.F. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene 2013, 32, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Broday, L.; Costa, M. Effects of nickel on DNA methyltransferase activity and genomic DNA methylation levels. Mutagen. Res. 1998, 415, 213–218. [Google Scholar] [CrossRef]
- Zingg, J.M.; Jones, P.A. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis 1997, 18, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Tang, S.C.; Wang, P.H.; Lee, H.; Ko, J.L. Nickel-induced epithelial-mesenchymal transition by reactive oxygen species generation and E-cadherin promoter hypermethylation. J. Biol. Chem. 2012, 287, 25292–25302. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Ono, T.; Kawahara, A.; Kawaguchi, T.; Tanaka, H.; Shimamatsu, K.; Kakuma, T.; Akiba, J.; Umeno, H.; Yano, H. Prognostic impact of p16 and PD-L1 expression in patients with oropharyngeal squamous cell carcinoma receiving a definitive treatment. J. Clin. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; DesMarais, T.; Costa, M. Metals and Mechanisms of Carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 537–554. [Google Scholar] [CrossRef]
- Lee, Y.W.; Pons, C.; Tummolo, D.M.; Klein, C.B.; Rossman, T.G.; Christie, N.T. Mutagenicity of soluble and insoluble nickel compounds at the gpt locus in G12 Chinese hamster cells. Environ. Mol. Mutagen. 1993, 21, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Ellen, T.P.; Kluz, T.; Harder, M.E.; Xiong, J.; Costa, M. Heterochromatinization as a potential mechanism of nickel-induced carcinogenesis. Biochemistry 2009, 48, 4626–4632. [Google Scholar] [CrossRef] [PubMed]
- Carone, D.M.; Lawrence, J.B. Heterochromatin instability in cancer: From the Barr body to satellites and the nuclear periphery. Semin. Cancer Biol. 2013, 23, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.A.; Shilatifard, A. Chromatin signatures of cancer. Genes Dev. 2015, 29, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, N.; Wagatsuma, H.; Wakana, S.; Shiroishi, T.; Nomura, M.; Aisaka, K.; Kohda, T.; Surani, M.A.; Kaneko-Ishino, T.; Ishino, F. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 2000, 5, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Klibanski, A. MEG3 noncoding RNA: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, R45–R53. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Vartholomatos, G.; Hatzimichael, E. MEG3 imprinted gene contribution in tumorigenesis. Int. J. Cancer 2011, 129, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yang, L.; Yu, L.; Yuan, J.; Hu, D.; Zhang, W.; Yang, J.; Pang, Y.; Li, W.; Lu, J.; et al. Epigenetic silencing of O6-methylguanine DNA methyltransferase gene in NiS-transformed cells. Carcinogenesis 2008, 29, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yang, L.; Yuan, J.; Yang, L.; Zhang, M.; Qi, D.; Duan, X.; Xuan, A.; Zhang, W.; Lu, J.; et al. MicroRNA-152 targets DNA methyltransferase 1 in NiS-transformed cells via a feedback mechanism. Carcinogenesis 2013, 34, 446–453. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Wu, Y.J.; Li, M.J.; Wang, R.J.; Huang, S.Q.; Gao, R.R.; Ma, L.; Shi, H.J.; Zhang, J. Hyper-methylated miR-203 dysregulates ABL1 and contributes to the nickel-induced tumorigenesis. Toxicol. Lett. 2013, 223, 42–51. [Google Scholar] [CrossRef]
- Salnikow, K.; Davidson, T.; Zhang, Q.; Chen, L.C.; Su, W.; Costa, M. The involvement of hypoxia-inducible transcription factor-1-dependent pathway in nickel carcinogenesis. Cancer Res 2003, 63, 3524–3530. [Google Scholar]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Costa, M.; Davidson, T.L.; Chen, H.; Ke, Q.; Zhang, P.; Yan, Y.; Huang, C.; Kluz, T. Nickel carcinogenesis: Epigenetics and hypoxia signaling. Mutagen. Res. 2005, 592, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, H.; Huang, X.; Costa, M. Effects of 12 metal ions on iron regulatory protein 1 (IRP-1) and hypoxia-inducible factor-1 alpha (HIF-1α) and HIF-regulated genes. Toxicol. Appl. Pharmacol. 2006, 213, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.L.; Chen, H.; Di Toro, D.M.; D’Angelo, G.; Costa, M. Soluble nickel inhibits HIF-prolyl-hydroxylases creating persistent hypoxic signaling in A549 cells. Mol. Carcinog. 2006, 45, 479–489. [Google Scholar] [CrossRef]
- Davidson, T.; Chen, H.; Garrick, M.D.; D’Angelo, G.; Costa, M. Soluble nickel interferes with cellular iron homeostasis. Mol. Cell. Biochem. 2005, 279, 157–162. [Google Scholar] [CrossRef]
- Salnikow, K.; Blagosklonny, M.V.; Ryan, H.; Johnson, R.; Costa, M. Carcinogenic Nickel Induces Genes Involved with Hypoxic Stress. Cancer Res. 2000, 60, 38–41. [Google Scholar]
- Rezvani, H.R.; Mahfouf, W.; Ali, N.; Chemin, C.; Ged, C.; Kim, A.L.; de Verneuil, H.; Taieb, A.; Bickers, D.R.; Mazurier, F. Hypoxia-inducible factor-1alpha regulates the expression of nucleotide excision repair proteins in keratinocytes. Nucleic Acids Res. 2010, 38, 797–809. [Google Scholar] [CrossRef]
- Chan, N.; Ali, M.; McCallum, G.P.; Kumareswaran, R.; Koritzinsky, M.; Wouters, B.G.; Wells, P.G.; Gallinger, S.; Bristow, R.G. Hypoxia provokes base excision repair changes and a repair-deficient, mutator phenotype in colorectal cancer cells. Mol. Cancer Res. 2014, 12, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, R.; Ferracin, M.; Negrini, M.; Calin, G.A.; Davuluri, R.V.; Ivan, M. Regulation of microRNA expression: The hypoxic component. Cell Cycle 2007, 6, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Lu, Y.; Xu, S.; Mao, L.; Zhang, L.; Duan, W.; Liu, C.; Pi, H.; Zhang, Y.; Zhong, M.; et al. MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron-sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells. Cell Death Dis. 2014, 5, e1090. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, R.; Ferracin, M.; Wojcik, S.E.; Garzon, R.; Alder, H.; Agosto-Perez, F.J.; Davuluri, R.; Liu, C.G.; Croce, C.M.; Negrini, M.; et al. A microRNA signature of hypoxia. Mol. Cell. Biol. 2007, 27, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Flach, H.; Onizawa, M.; Wei, L.; McManus, M.T.; Weiss, A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat. Immunol. 2014, 15, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Pero, R.W.; Roush, G.C.; Markowitz, M.M.; Miller, D.G. Oxidative stress, DNA repair, and cancer susceptibility. Cancer Detect. Prev. 1990, 14, 555–561. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, 14s–22s. [Google Scholar] [CrossRef]
- Lee, J.D.; Cai, Q.; Shu, X.O.; Nechuta, S.J. The Role of Biomarkers of Oxidative Stress in Breast Cancer Risk and Prognosis: A Systematic Review of the Epidemiologic Literature. J. Womens Health 2017, 26, 467–482. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Dalal, N.S.; Kasprzak, K.S. Generation of free radicals from lipid hydroperoxides by Ni2+ in the presence of oligopeptides. Arch. Biochem. Biophys. 1992, 299, 154–162. [Google Scholar] [CrossRef]
- Tsao, Y.C.; Gu, P.W.; Liu, S.H.; Tzeng, I.S.; Chen, J.Y.; Luo, J.J. Nickel exposure and plasma levels of biomarkers for assessing oxidative stress in nickel electroplating workers. Biomarkers 2017, 22, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Di Bucchianico, S.; Gliga, A.R.; Åkerlund, E.; Skoglund, S.; Wallinder, I.O.; Fadeel, B.; Karlsson, H.L. Calcium-dependent cyto- and genotoxicity of nickel metal and nickel oxide nanoparticles in human lung cells. Part. Fibre Toxicol. 2018, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Klein, C.B.; Costa, M. Crystalline Ni3S2 specifically enhances the formation of oxidants in the nuclei of CHO cells as detected by dichlorofluorescein. Carcinogenesis 1994, 15, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Lynn, S.; Yew, F.H.; Chen, K.S.; Jan, K.Y. Reactive oxygen species are involved in nickel inhibition of DNA repair. Environ. Mol. Mutagen. 1997, 29, 208–216. [Google Scholar] [CrossRef]
- Scanlon, S.E.; Scanlon, C.D.; Hegan, D.C.; Sulkowski, P.L.; Glazer, P.M. Nickel induces transcriptional down-regulation of DNA repair pathways in tumorigenic and non-tumorigenic lung cells. Carcinogenesis 2017, 38, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Cui, R.; Gu, J.; Zhang, X.; Xiang, X.; Liu, C.; Qu, K.; Lin, T. Identification of Four Oxidative Stress-Responsive MicroRNAs, miR-34a-5p, miR-1915-3p, miR-638, and miR-150-3p, in Hepatocellular Carcinoma. Oxid. Med. Cell. Longev. 2017, 2017, 5189138. [Google Scholar] [CrossRef]
- Sun, W.; Zhao, L.; Song, X.; Zhang, J.; Xing, Y.; Liu, N.; Yan, Y.; Li, Z.; Lu, Y.; Wu, J.; et al. MicroRNA-210 Modulates the Cellular Energy Metabolism Shift During H2O2-Induced Oxidative Stress by Repressing ISCU in H9c2 Cardiomyocytes. Cell. Physiol. Biochem. 2017, 43, 383–394. [Google Scholar] [CrossRef]
- Ali, T.; Mushtaq, I.; Maryam, S.; Farhan, A.; Saba, K.; Jan, M.I.; Sultan, A.; Anees, M.; Duygu, B.; Hamera, S.; et al. Interplay of N acetyl cysteine and melatonin in regulating oxidative stress-induced cardiac hypertrophic factors and microRNAs. Arch. Biochem. Biophys. 2019, 661, 56–65. [Google Scholar] [CrossRef]
- Clemens, F.; Verma, R.; Ramnath, J.; Landolph, J.R. Amplification of the Ect2 proto-oncogene and over-expression of Ect2 mRNA and protein in nickel compound and methylcholanthrene-transformed 10T1/2 mouse fibroblast cell lines. Toxicol. Appl. Pharmacol. 2005, 206, 138–149. [Google Scholar] [CrossRef]
- Chiocca, S.M.; Sterner, D.A.; Biggart, N.W.; Murphy, E.C., Jr. Nickel mutagenesis: Alteration of the MuSVts110 thermosensitive splicing phenotype by a nickel-induced duplication of the 3’ splice site. Mol. Carcinog. 1991, 4, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Maehle, L.; Metcalf, R.A.; Ryberg, D.; Bennett, W.P.; Harris, C.C.; Haugen, A. Altered p53 gene structure and expression in human epithelial cells after exposure to nickel. Cancer Res. 1992, 52, 218–221. [Google Scholar] [PubMed]
- Shi, Y.; Lv, C.; Shi, L.; Tu, G. MEG3 inhibits proliferation and invasion and promotes apoptosis of human osteosarcoma cells. Oncol. Lett. 2018, 15, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Li, W.; Liu, X.H.; Sun, M.; Zhang, M.L.; Wu, W.Q.; Xie, W.P.; Hou, Y.Y. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer 2013, 13, 461. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Su, C.; Zhao, F.; Tao, J.; Hu, D.; Shi, A.; Pan, J.; Zhang, Y. Paclitaxel promotes lung cancer cell apoptosis via MEG3-P53 pathway activation. Biochem. Biophys. Res. Commun. 2018, 504, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 2003, 88, 5119–5126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Deng, W.; Zhou, X.T.; Chen, R.P.; Xiang, M.Q.; Guo, Y.T.; Pu, Z.J.; Li, R.; Wang, G.F.; Wu, L.F. The mechanism of adenosine-mediated activation of lncRNA MEG3 and its antitumor effects in human hepatoma cells. Int. J. Oncol. 2016, 48, 421–429. [Google Scholar] [CrossRef]
- Lv, D.; Sun, R.; Yu, Q.; Zhang, X. The long non-coding RNA maternally expressed gene 3 activates p53 and is downregulated in esophageal squamous cell cancer. Tumour. Biol. 2016, 37, 16259–16267. [Google Scholar] [CrossRef]
- Liu, J.; Wan, L.; Lu, K.; Sun, M.; Pan, X.; Zhang, P.; Lu, B.; Liu, G.; Wang, Z. The Long Noncoding RNA MEG3 Contributes to Cisplatin Resistance of Human Lung Adenocarcinoma. PLoS ONE 2015, 10, e0114586. [Google Scholar] [CrossRef]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Liao, X.; Jin, H.; Xie, F.; Zheng, F.; Li, J.; Zhou, C.; Jiang, G.; Wu, X.R.; Huang, C. MEG3, as a Competing Endogenous RNA, Binds with miR-27a to Promote PHLPP2 Protein Translation and Impairs Bladder Cancer Invasion. Mol. Ther. Nucleic Acids 2019, 16, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Zhang, S.; Xu, M.; Zhang, R.; Sui, P.; Yang, Q. MicroRNA-27a contributes to the malignant behavior of gastric cancer cells by directly targeting PH domain and leucine-rich repeat protein phosphatase 2. J. Exp. Clin. Cancer Res. 2017, 36, 45. [Google Scholar] [CrossRef] [PubMed]
- Chae, D.K.; Ban, E.; Yoo, Y.S.; Kim, E.E.; Baik, J.H.; Song, E.J. MIR-27a regulates the TGF-β signaling pathway by targeting SMAD2 and SMAD4 in lung cancer. Mol. Carcinog. 2017, 56, 1992–1998. [Google Scholar] [CrossRef]
- Khandelwal, A.; Bacolla, A.; Vasquez, K.M.; Jain, A. Long non-coding RNA: A new paradigm for lung cancer. Mol. Carcinog. 2015, 54, 1235–1251. [Google Scholar] [CrossRef]
- Gao, C.; He, Z.; Li, J.; Li, X.; Bai, Q.; Zhang, Z.; Zhang, X.; Wang, S.; Xiao, X.; Wang, F.; et al. Specific long non-coding RNAs response to occupational PAHs exposure in coke oven workers. Toxicol. Rep. 2016, 3, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.B.; Yin, D.D.; Sun, M.; Kong, R.; Liu, X.H.; You, L.H.; Han, L.; Xia, R.; Wang, K.M.; Yang, J.S.; et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis. 2014, 5, e1243. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, H.; Dai, H.; Walsh, R.M.; Imakura, M.; Schelter, J.; Burchard, J.; Dai, X.; Chang, A.N.; Diaz, R.L.; et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 2009, 8, 2756–2768. [Google Scholar] [CrossRef] [Green Version]
- Giannakakis, A.; Sandaltzopoulos, R.; Greshock, J.; Liang, S.; Huang, J.; Hasegawa, K.; Li, C.; O’Brien-Jenkins, A.; Katsaros, D.; Weber, B.L.; et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther. 2008, 7, 255–264. [Google Scholar] [CrossRef] [Green Version]
- He, R.Q.; Cen, W.L.; Cen, J.M.; Cen, W.N.; Li, J.Y.; Li, M.W.; Gan, T.Q.; Hu, X.H.; Chen, G. Clinical Significance of miR-210 and its Prospective Signaling Pathways in Non-Small Cell Lung Cancer: Evidence from Gene Expression Omnibus and the Cancer Genome Atlas Data Mining with 2763 Samples and Validation via Real-Time Quantitative PCR. Cell. Physiol. Biochem. 2018, 46, 925–952. [Google Scholar] [CrossRef]
- Yang, J.S.; Li, B.J.; Lu, H.W.; Chen, Y.; Lu, C.; Zhu, R.X.; Liu, S.H.; Yi, Q.T.; Li, J.; Song, C.H. Serum miR-152, miR-148a, miR-148b, and miR-21 as novel biomarkers in non-small cell lung cancer screening. Tumour. Biol. 2015, 36, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.J.; Perez de Castro, I.; Gomez de Cedron, M.; Santos, J.; Calin, G.A.; Cigudosa, J.C.; Croce, C.M.; Fernandez-Piqueras, J.; Malumbres, M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell 2008, 13, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Craig, V.J.; Cogliatti, S.B.; Rehrauer, H.; Wundisch, T.; Muller, A. Epigenetic silencing of microRNA-203 dysregulates ABL1 expression and drives Helicobacter-associated gastric lymphomagenesis. Cancer Res. 2011, 71, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Kozaki, K.I.; Tanaka, S.; Arii, S.; Imoto, I.; Inazawa, J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 2010, 31, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Mori, T.; Otsuka, Y.; Yamada, N.; Yasui, Y.; Iwasaki, J.; Kumazaki, M.; Maruo, K.; Akao, Y. Anti-oncogenic microRNA-203 induces senescence by targeting E2F3 protein in human melanoma cells. J. Biol. Chem. 2012, 287, 11769–11777. [Google Scholar] [CrossRef]
- Yi, R.; Poy, M.N.; Stoffel, M.; Fuchs, E. A skin microRNA promotes differentiation by repressing ‘stemness’. Nature 2008, 452, 225–229. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Ma, L.; Huang, S.; Wang, R.; Gao, R.; Wu, Y.; Shi, H.; Zhang, J. The alteration of miR-222 and its target genes in nickel-induced tumor. Biol. Trace Elem. Res. 2013, 152, 267–274. [Google Scholar] [CrossRef]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafre, S.A.; et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef]
- Fornari, F.; Gramantieri, L.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Giovannini, C.; Croce, C.M.; Bolondi, L.; et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene 2008, 27, 5651–5661. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Hsiao, Y.M.; Yeh, K.T.; Tsou, T.C.; Chen, C.Y.; Wu, M.F.; Ko, J.L. Upregulation of microRNA-4417 and Its Target Genes Contribute to Nickel Chloride-promoted Lung Epithelial Cell Fibrogenesis and Tumorigenesis. Sci. Rep. 2017, 7, 15320. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Chen, Q.Y.; Li, A.H.; Costa, M. The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms. Inorganics 2019, 7, 81. https://doi.org/10.3390/inorganics7070081
Zhu Y, Chen QY, Li AH, Costa M. The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms. Inorganics. 2019; 7(7):81. https://doi.org/10.3390/inorganics7070081
Chicago/Turabian StyleZhu, Yusha, Qiao Yi Chen, Alex Heng Li, and Max Costa. 2019. "The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms" Inorganics 7, no. 7: 81. https://doi.org/10.3390/inorganics7070081
APA StyleZhu, Y., Chen, Q. Y., Li, A. H., & Costa, M. (2019). The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms. Inorganics, 7(7), 81. https://doi.org/10.3390/inorganics7070081