Hexaborate(2−) and Dodecaborate(6−) Anions as Ligands to Zinc(II) Centres: Self-Assembly and Single-Crystal XRD Characterization of [Zn{κ3O-B6O7(OH)6}(κ3N-dien)]·0.5H2O (dien = NH(CH2–CH2NH2)2), (NH4)2[Zn{κ2O-B6O7(OH)6}2 (H2O)2]·2H2O and (1,3-pnH2)3[(κ1N-H3N{CH2}3NH2) Zn{κ3O-B12O18(OH)6}]2·14H2O (1,3-pn = 1,3-diaminopropane)



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
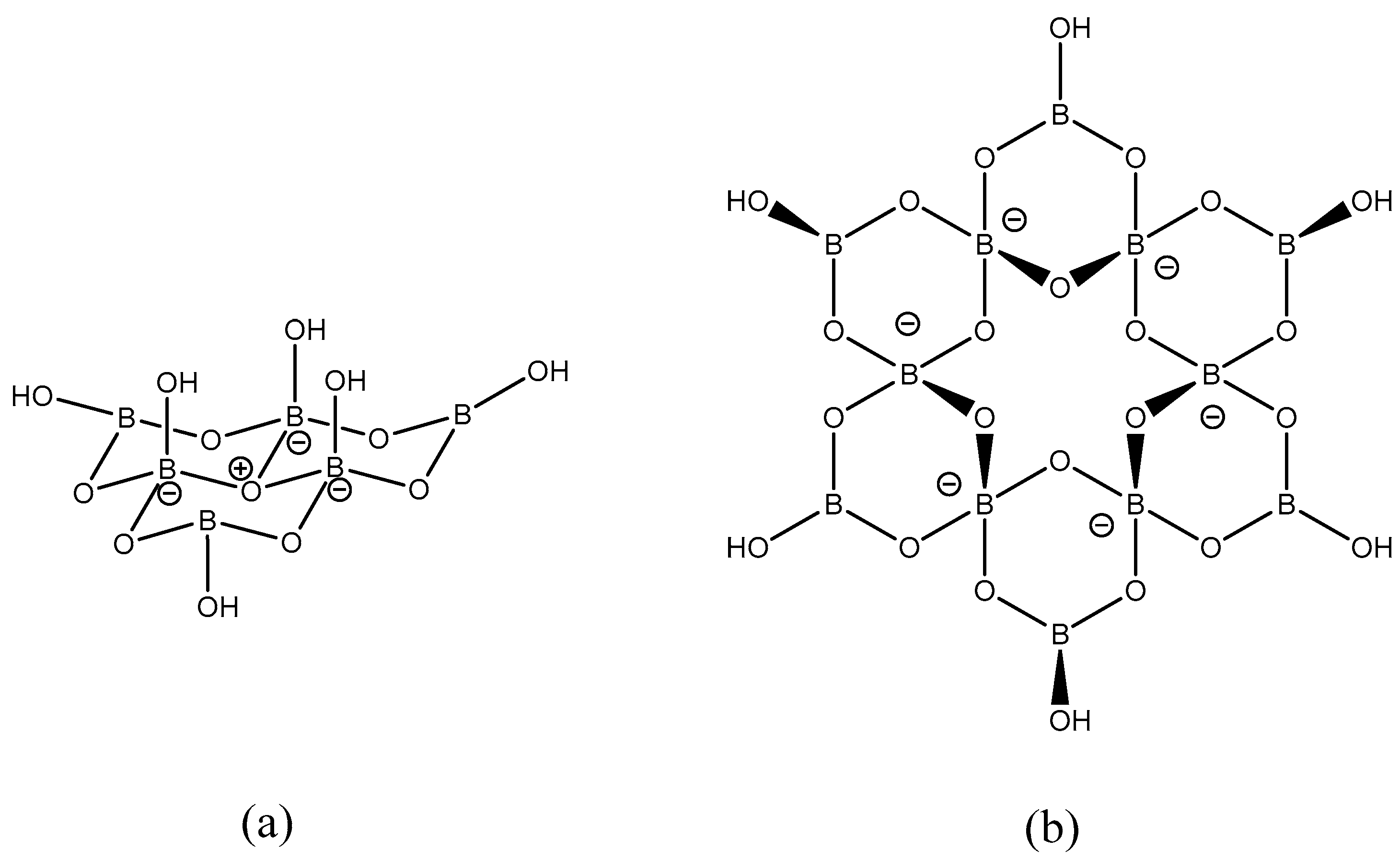
2. Results and Discussion
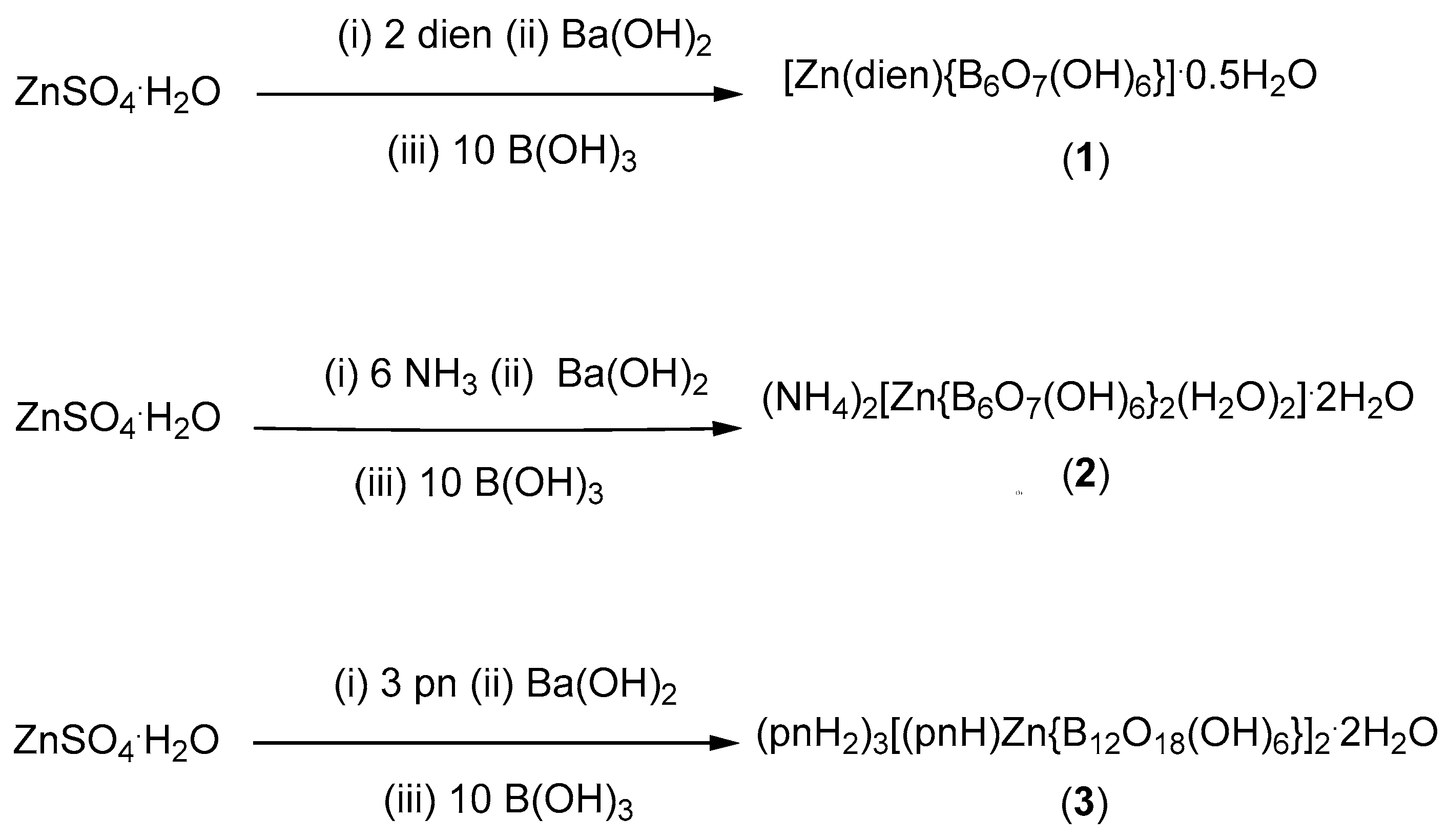
2.1. Synthesis and Characterization
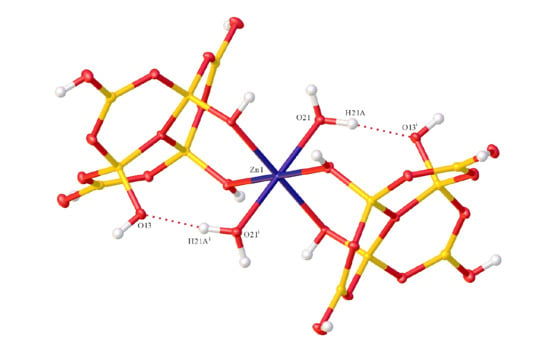
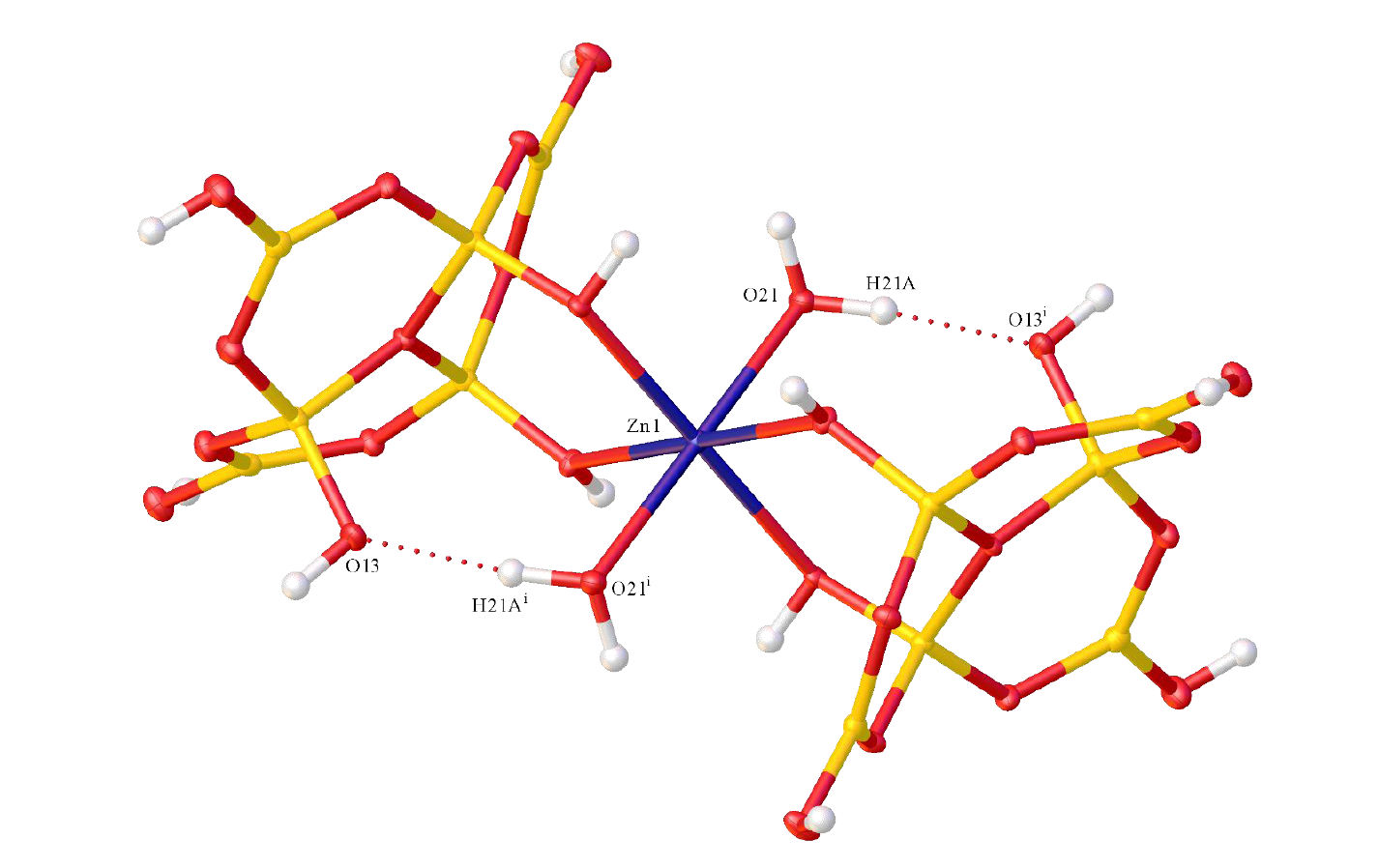
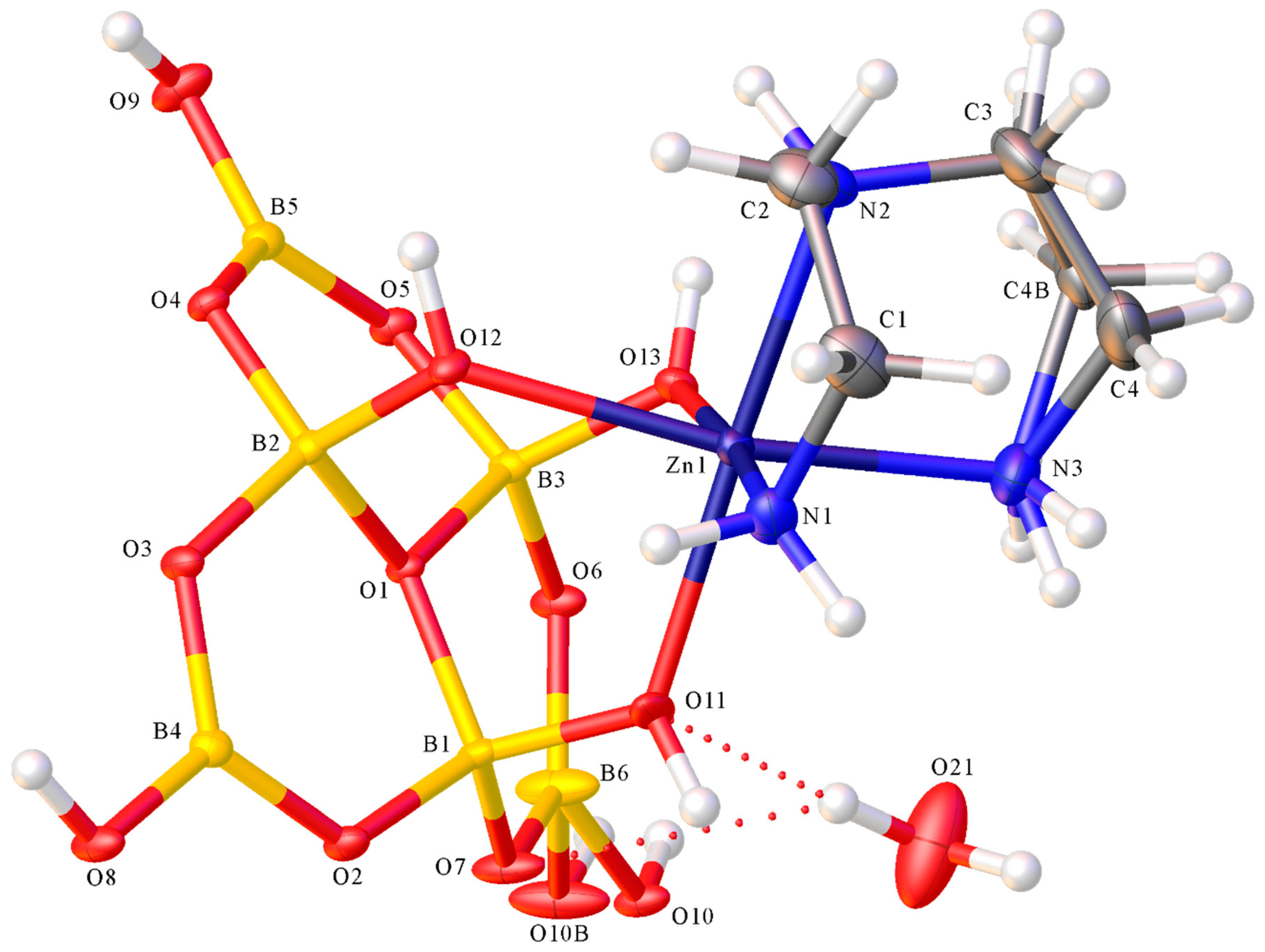
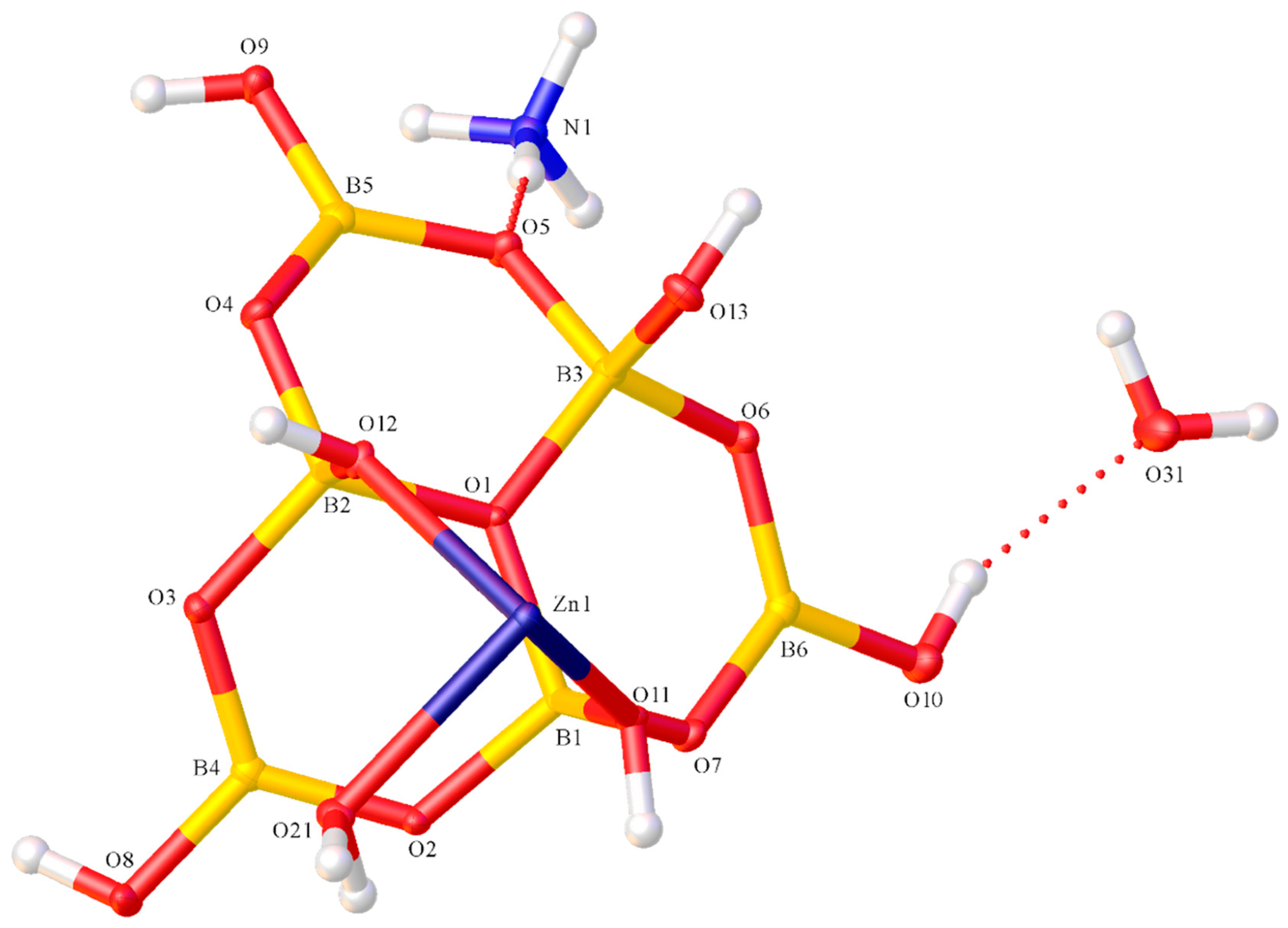
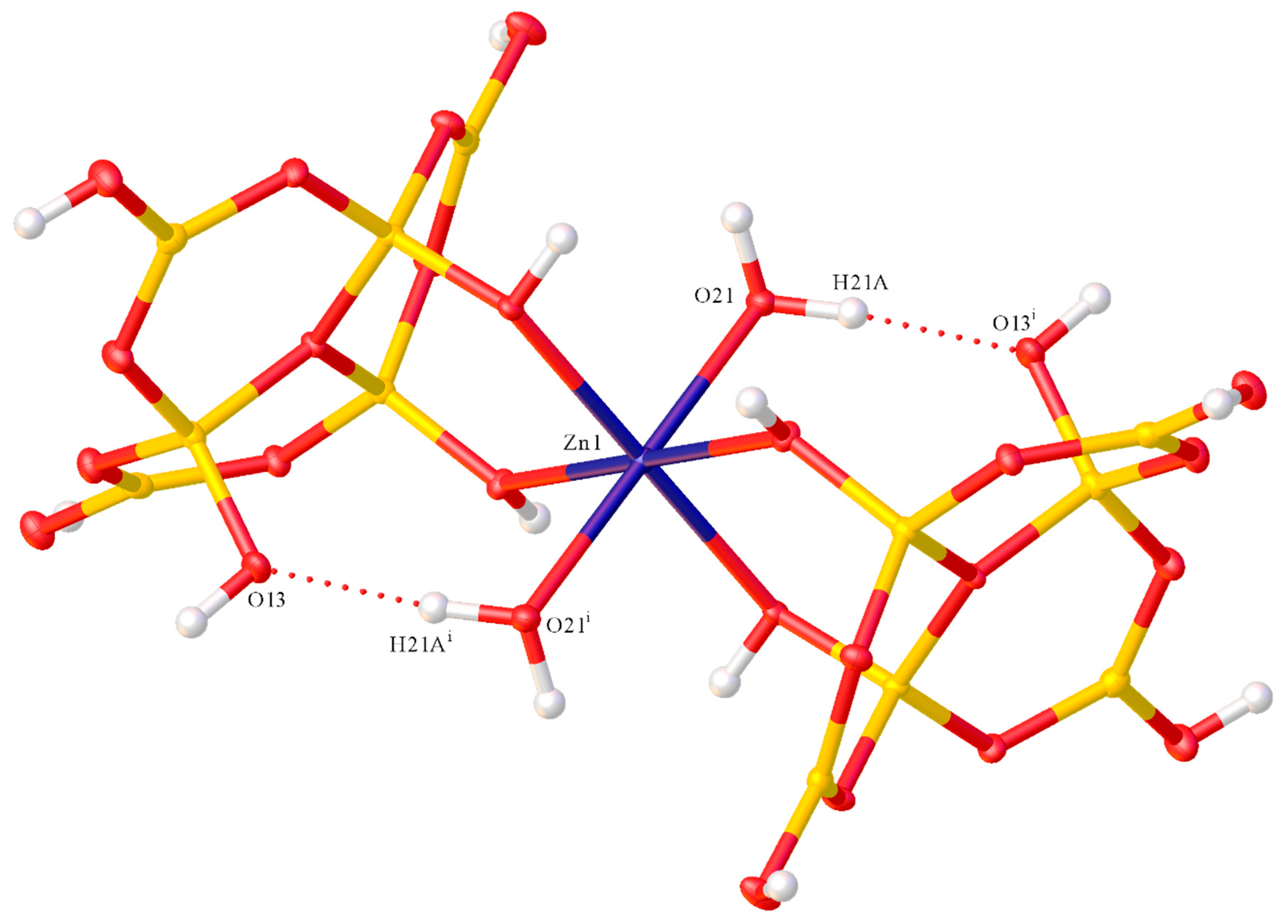
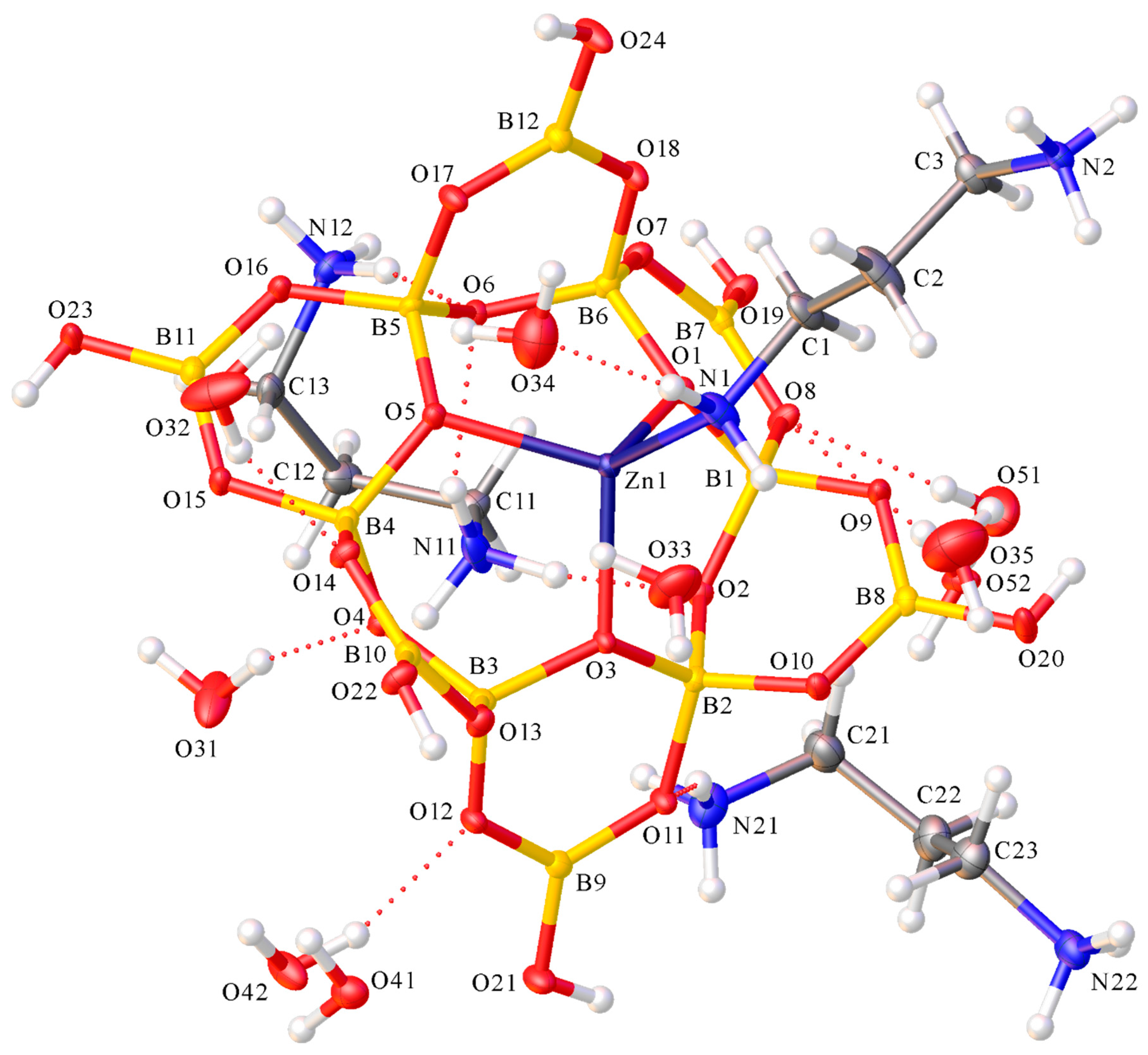
2.2. X-ray Diffraction Studies
3. Experimental
3.1. General
3.2. Synthesis, Spectroscopic, Analytical and Crystallographic data for 1
3.3. Synthesis, Spectroscopic, Analytical and Crystallographic Data for 2
3.4. Synthesis, Spectroscopic, Analytical and Crystallographic Data for 3
3.5. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schubert, D.M.; Knobler, C.B. Recent studies of polyborate anions. Phys. Chem. Glasses Eur. J. Glass Sci. Technol. B 2009, 50, 71–78. [Google Scholar]
- Grice, J.D.; Burns, P.C.; Hawthorne, F.C. Borate minerals II. A hierarchy of structures based upon the borate fundamental building block. Can. Mineral. 1999, 37, 731–762. [Google Scholar]
- Becker, P. A contribution to borate crystal chemistry: Rules for the occurrence of polyborate anion types. Z. Kristallogr. 2001, 216, 523–533. [Google Scholar] [CrossRef]
- Heller, G. A survey of structural types of borates and polyborates. Top. Curr. Chem. 1986, 131, 39–98. [Google Scholar]
- Belokoneva, E.L. Borate crystal chemistry in terms of the extended OD theory: Topology and symmetry analysis. Crystallogr. Rev. 2005, 11, 151–198. [Google Scholar] [CrossRef]
- Christ, C.L.; Clark, J.R. A crystal-chemical classification of borate structures with emphasis on hydrated borates. Phys. Chem. Miner. 1977, 2, 59–87. [Google Scholar] [CrossRef]
- Beckett, M.A. Recent Advances in crystalline hydrated borates with non-metal or transition-metal complex cations. Coord. Chem. Rev. 2016, 323, 2–14. [Google Scholar] [CrossRef]
- Wiebcke, M.; Freyhardt, C.C.; Felsche, J.; Engelhardt, G. Clathrates with three-dimensional host structures of hydrogen bonded pentaborate [B5O6(OH)4]− ions: Pentaborates with the cations NMe4+, NEt4+, NPhMe3+ and pipH+ (pipH+ = piperidinium). Z. Naturforsch. 1993, 48b, 978–985. [Google Scholar] [CrossRef]
- Visi, M.Z.; Knobler, C.B.; Owen, J.J.; Khan, M.I.; Schubert, D.M. Structures of self-assembled nonmetal borates derived from α,ω-diaminoalkanes. Cryst. Growth Des. 2006, 6, 538–545. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Davies, R.A.; Horton, P.N.; Jones, C.L. Pentaborate(1−) salts templated by substituted pyrrolidinium cations: Synthesis, structural characterization, and modelling of solid-state H-bond interactions by DFT calculations. Dalton Trans. 2015, 44, 7032–7040. [Google Scholar] [CrossRef] [PubMed]
- Beckett, M.A.; Bland, C.C.; Horton, P.N.; Hursthouse, M.B.; Varma, K.S. Supramolecular structures containing “isolated” pentaborate anions and non-metal cations: Crystal structures of [Me3NCH2CH2OH][B5O6(OH)4] and [4-Mepy,4-MepyH][B5O6(OH)4]. J. Organomet. Chem. 2007, 692, 2832–2838. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, C.J.; Horton, P.N. A new polyborate anion [B7O9(OH)6]3−: Self-assembly, XRD and thermal properties of s-fac-[Co(en)3][B7O9(OH)6]·9H2O. Inorg. Chem. Commun. 2015, 59, 95–98. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, C.J.; Horton, P.N. A new decaoxidooctaborate(2−) anion, [B8O10(OH)6]2−: Synthesis and characterization of [Co(en)3][B5O6(OH)4][B8O10(OH)6]·5H2O (en = 1,2-diaminoethane). Inorg. Chem. 2015, 54, 412–414. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, C.J.; Horton, P.N. Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [H3NCH2CH2NH2)Zn{κ3O,O’,O’’-B12O18(OH)6-κ1O’’’} Zn(en)(NH2CH2CH2NH3)]·8H2O: A neutral bimetallic zwiterionic polyborate system containing the “isolated” dodecaborate(6−) anion. Pure Appl. Chem. 2018, 90, 625–632. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, C.J.; Horton, P.N. Two 1-D Coordination Polymers containing Zinc(II) Hexaborates: [Zn(en){B6O7(OH)6}]·2H2O (en = 1,2-diaminoethane) and [Zn(pn){B6O7(OH)6}]·1.5H2O (pn = (+/−) 1,2-diaminopropane). Crystals 2018, 8, 470. [Google Scholar] [CrossRef]
- Wang, G.-M.; Sun, Y.-Q.; Yang, G.-Y. Synthesis and crystal structures of two new pentaborates. J. Solid State Chem. 2005, 178, 729–735. [Google Scholar] [CrossRef]
- He, Y.; Yang, J.; Xi, C.-Y.; Chen, J.-S. Solvothermal synthesis and crystal structure of Zn(en)3B5O7(OH)3. Chem. Res. Chin. Univ. 2006, 22, 271–273. [Google Scholar] [CrossRef]
- Jiang, H.; Yang, B.-F.; Wang, G.-M. [Zn(dap)3][Zn(dap)B5O8(OH)2]2: A novel organic-inorganic hybrid chain-like zincoborate made up of [B5O8(OH)2]3− and [Zn(dap)]2+ linkers. J. Clust. Sci. 2017, 28, 1421–1429. [Google Scholar] [CrossRef]
- Wei, L.; Sun, A.-H.; Xue, Z.-Z.; Pan, J.; Wang, G.-M.; Wang, Y.-X.; Wang, Z.-H. Hydrothermal synthesis and structural characterization of a new hybrid zinc borate, [Zn(dap)2][B4O6(OH)2]. J. Clust. Sci. 2017, 28, 1453–1462. [Google Scholar] [CrossRef]
- Zhao, P.; Cheng, L.; Yang, G.Y. Synthesis and characterization of a new organic-inorganic hybrid borate [Zn(dab)0.5(dab’)0.5(B4O6(OH)2]·H2O. Inorg. Chem. Commun. 2012, 20, 138–141. [Google Scholar] [CrossRef]
- Paul, A.V.; Sachidananda, K.; Natarajan, S. [B4O9H2] cyclic borate units as the building unit in a family of zinc borate structures. Cryst. Growth Des. 2010, 10, 456–464. [Google Scholar] [CrossRef]
- Pan, R.; Chen, C.-A.; Yang, B.-F. Two new octaborates constructed of two different sub-clusters and supported by metal complexes. J. Clust. Sci. 2017, 28, 1237–1248. [Google Scholar] [CrossRef]
- Zhao, P.; Lin, Z.-E.; Wei, Q.; Cheng, L.; Yang, G.-Y. A pillared-layered zincoborate with an anionic network containing unprecedented zinc oxide chains. Chem. Commun. 2014, 50, 3592–3594. [Google Scholar] [CrossRef]
- Schubert, D.M.; Alam, F.; Visi, M.Z.; Knobler, C.B. Structural characterization and chemistry of an industrially important zinc borate, Zn[B3O4(OH)3]. Chem Mater. 2003, 15, 866–871. [Google Scholar] [CrossRef]
- Sola, J.; Lafuente, M.; Atcher, J.; Alfonso, I. Constitutional self-selection from dynamic combinatorial libraries in aqueous solution through supramolecular interactions. Chem. Commun. 2014, 50, 4564–4566. [Google Scholar] [CrossRef] [PubMed]
- Corbett, P.T.; Leclaire, J.; Vial, L.; West, K.R.; Wietor, J.-L.; Sanders, J.K.M.; Otto, S. Dynamic combinatorial chemistry. Chem. Rev. 2006, 106, 3652–3711. [Google Scholar] [CrossRef] [PubMed]
- Salentine, G. High-field 11B NMR of alkali borate. Aqueous polyborate equilibria. Inorg. Chem. 1983, 22, 3920–3924. [Google Scholar] [CrossRef]
- Anderson, J.L.; Eyring, E.M.; Whittaker, M.P. Temperature jump rate studies of polyborate formation in aqueous boric acid. J. Phys. Chem. 1964, 68, 1128–1132. [Google Scholar] [CrossRef]
- Taube, H. Rates and mechanisms of substitutions in inorganic complexes in aqueous solution. Chem. Rev. 1952, 50, 69–126. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Gavezzotti, A. Supramolecular synthons: Validation and ranking of intermolecular interaction energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Synthesis and characterization of polyborates template by cationic copper(II) complexes: Structural (XRD), spectroscopic, thermal (TGA/DSC) and magnetic properties. Polyhedron 2017, 135, 247–257. [Google Scholar] [CrossRef]
- Wang, G.-M.; Sun, Y.-Q.; Yang, G.-Y. Synthesis and crystal structures of three new borates templated by transition-metal complexes in situ. J. Solid State Chem. 2006, 179, 1545–1553. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Zhu, J.; Liu, R.-B.; Xu, J.; Meng, C.-C. A new mixed ligand copper pentaborate with square-like, rectangular-like and ellipse-like channels formed via hydrogen bonds. Inorg. Chim. Acta 2011, 376, 401–407. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Zhang, J.-J.; Zhang, W.-J. Synthesis, crystal structure and vibrational spectroscopy of a novel mixed ligands Ni(II) pentaborate [Ni(C4H10N2)(C2H8N2)2][B5O6(OH)4]2. Inorg. Chim. Acta 2006, 359, 519–524. [Google Scholar] [CrossRef]
- Li, J.; Xia, S.; Gao, S. FT-IR and Raman spectroscopic study of hydrated borates. Spectrochim. Acta 1995, 51A, 519–532. [Google Scholar] [CrossRef]
- Archibald, S.J. Zinc. In Comprehensive Coordination Chemistry II, 2nd ed.; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 6, pp. 1147–1251. [Google Scholar]
- Natarajan, S.; Klein, W.; Panthoefer, M.; Wuellen, L.V.; Jansen, M. Solution mediated synthesis and structure of the first anionic bis(hexaborato)-zincate prepared in the presence of organic base. Z. Anorg. Allg. Chem. 2003, 629, 959–962. [Google Scholar] [CrossRef]
- Jemai, N.; Rzaigui, S.; Akriche, S. Piperazine-1,4-diium bis(hexahydroxidoheptaoxidohexaborato-κ3O,O’,O’’) cobaltate(II) hexahydrate. Acta Cryst. 2014, E70, m167–m169. [Google Scholar] [CrossRef] [PubMed]
- Beckett, M.A.; Hibbs, D.E.; Hursthouse, M.B.; Malik, K.M.A.; Owen, P.; Varma, K.S. cyclo-Boratrisiloxane and cyclo-diboratetrasiloxane derivatives and their reactions with amines: Crystal and molecular structure of (p-BrC6H4BO)2(Ph2SiO)2. J. Organomet. Chem. 2000, 595, 241–247. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic chemistry. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Skakibaie-Moghadam, M.; Heller, G.; Timper, U. Die kristallstruktur von Ag6[B12O18(OH)6].3H2O einen neuen dokekaborat. Z. Kristallogr. 1990, 190, 85. [Google Scholar] [CrossRef]
- Menchetti, M.; Sabelli, C. A new borate polyanion in the structure of Na8[B12O20(OH)4]. Acta Cryst. 1979, B35, 2488–2493. [Google Scholar] [CrossRef]
- Choudhury, A.; Neeraj, S.; Natarajan, S.; Rao, C.N.R. An open-framework zincoborate formed by Zn6B12O24 clusters. J. Chem. Soc. Dalton Trans. 2002, 1535–1538. [Google Scholar] [CrossRef]
- Zhang, T.-J.; Pan, R.; He, H.; Yang, B.-F.; Yang, G.-Y. Solvothermal synthesis and structure of two new boranes containing [B7O9(OH)5]2− and [B12O18(OH)6]6− clusters. J. Clust. Sci. 2016, 27, 625. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Zhang, J.; Zheng, S.T.; Yang, G.-Y. K7{(BO3)Mn[B12O18(OH)6]}·H2O: First manganese borate based on covalently linked B12O18(OH)6 clusters and BO3 units via Mn2+ cations. Inorg. Chem. Commun. 2004, 7, 781–783. [Google Scholar] [CrossRef]
- Rong, C.; Jiang, J.; Li, Q.-L. Synthesis and transitional metal borates K7{(BO3)Zn[B12O18(OH)6]}·H2O and quantum chemistry study. Chinese J. Inorg. Chem. 2012, 28, 2217–2222. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. ShelXT-intergrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Hexaborate(2−) and Dodecaborate(6−) Anions as Ligands to Zinc(II) Centres: Self-Assembly and Single-Crystal XRD Characterization of [Zn{κ3O-B6O7(OH)6}(κ3N-dien)]·0.5H2O (dien = NH(CH2–CH2NH2)2), (NH4)2[Zn{κ2O-B6O7(OH)6}2 (H2O)2]·2H2O and (1,3-pnH2)3[(κ1N-H3N{CH2}3NH2) Zn{κ3O-B12O18(OH)6}]2·14H2O (1,3-pn = 1,3-diaminopropane). Inorganics 2019, 7, 44. https://doi.org/10.3390/inorganics7040044
Altahan MA, Beckett MA, Coles SJ, Horton PN. Hexaborate(2−) and Dodecaborate(6−) Anions as Ligands to Zinc(II) Centres: Self-Assembly and Single-Crystal XRD Characterization of [Zn{κ3O-B6O7(OH)6}(κ3N-dien)]·0.5H2O (dien = NH(CH2–CH2NH2)2), (NH4)2[Zn{κ2O-B6O7(OH)6}2 (H2O)2]·2H2O and (1,3-pnH2)3[(κ1N-H3N{CH2}3NH2) Zn{κ3O-B12O18(OH)6}]2·14H2O (1,3-pn = 1,3-diaminopropane). Inorganics. 2019; 7(4):44. https://doi.org/10.3390/inorganics7040044
Chicago/Turabian StyleAltahan, Mohammed A., Michael A. Beckett, Simon J. Coles, and Peter N. Horton. 2019. "Hexaborate(2−) and Dodecaborate(6−) Anions as Ligands to Zinc(II) Centres: Self-Assembly and Single-Crystal XRD Characterization of [Zn{κ3O-B6O7(OH)6}(κ3N-dien)]·0.5H2O (dien = NH(CH2–CH2NH2)2), (NH4)2[Zn{κ2O-B6O7(OH)6}2 (H2O)2]·2H2O and (1,3-pnH2)3[(κ1N-H3N{CH2}3NH2) Zn{κ3O-B12O18(OH)6}]2·14H2O (1,3-pn = 1,3-diaminopropane)" Inorganics 7, no. 4: 44. https://doi.org/10.3390/inorganics7040044
APA StyleAltahan, M. A., Beckett, M. A., Coles, S. J., & Horton, P. N. (2019). Hexaborate(2−) and Dodecaborate(6−) Anions as Ligands to Zinc(II) Centres: Self-Assembly and Single-Crystal XRD Characterization of [Zn{κ3O-B6O7(OH)6}(κ3N-dien)]·0.5H2O (dien = NH(CH2–CH2NH2)2), (NH4)2[Zn{κ2O-B6O7(OH)6}2 (H2O)2]·2H2O and (1,3-pnH2)3[(κ1N-H3N{CH2}3NH2) Zn{κ3O-B12O18(OH)6}]2·14H2O (1,3-pn = 1,3-diaminopropane). Inorganics, 7(4), 44. https://doi.org/10.3390/inorganics7040044