Computational Characterization of Single-Electron Transfer Steps in Water Oxidation


Abstract
1. Introduction
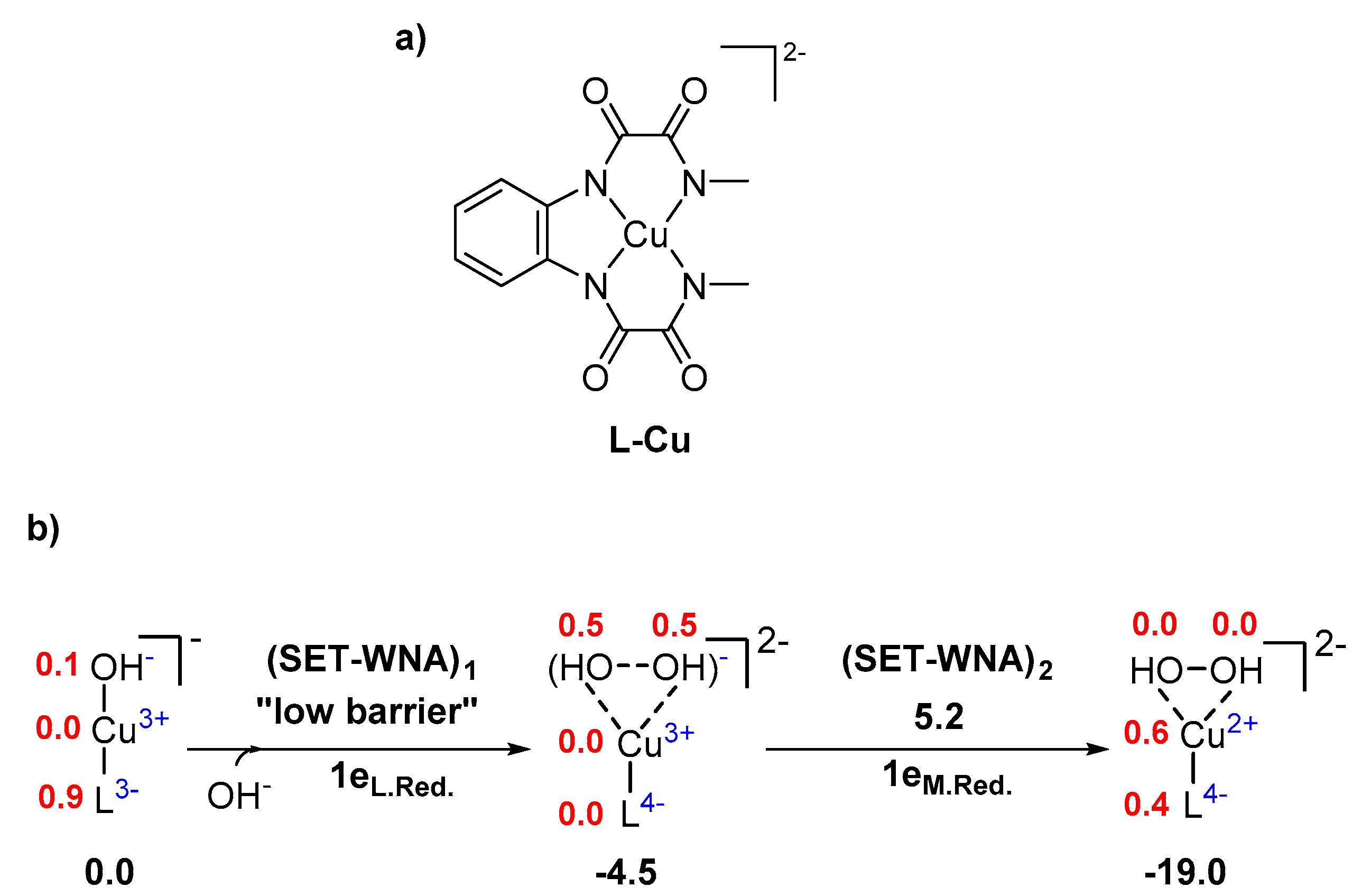
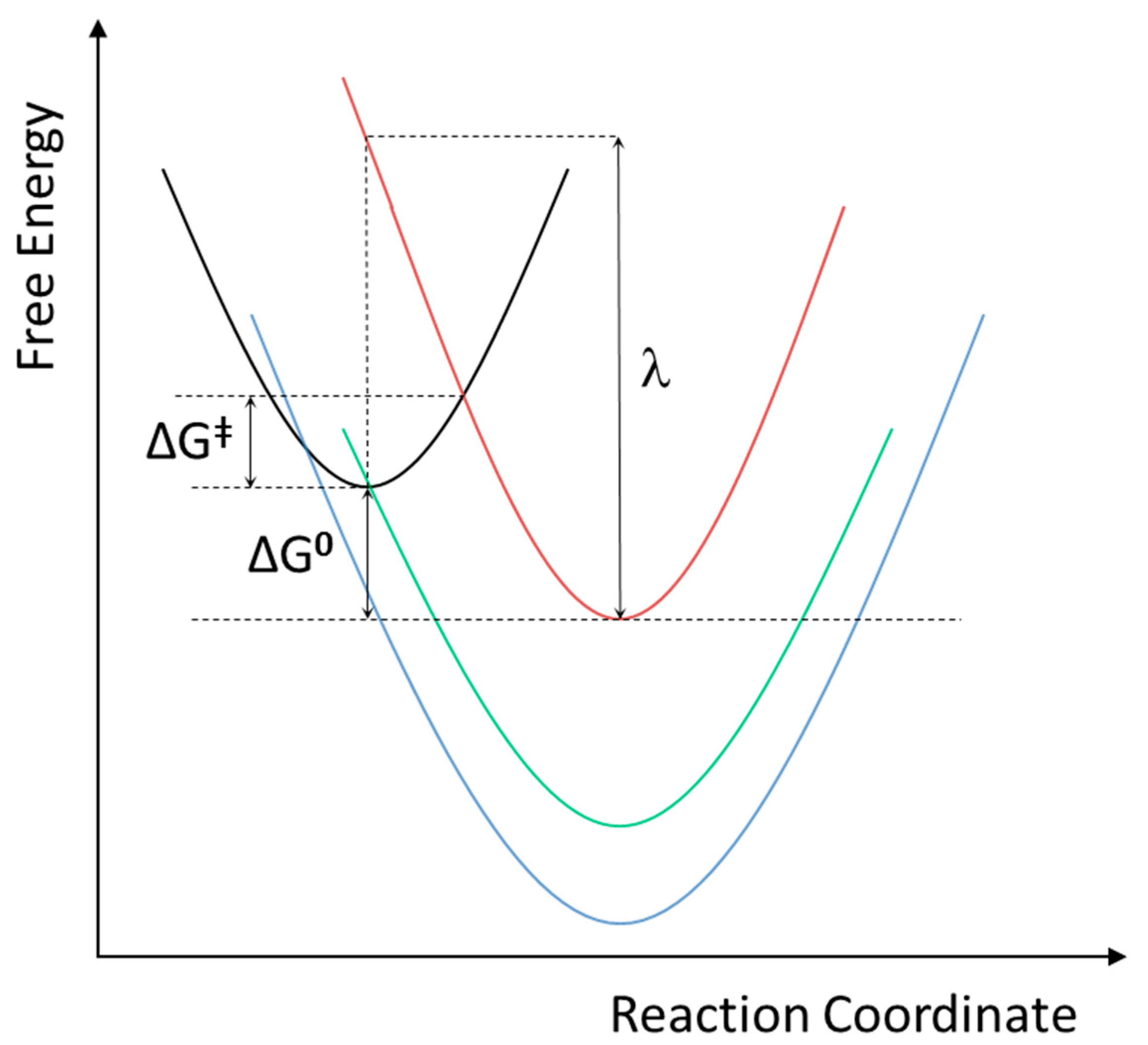
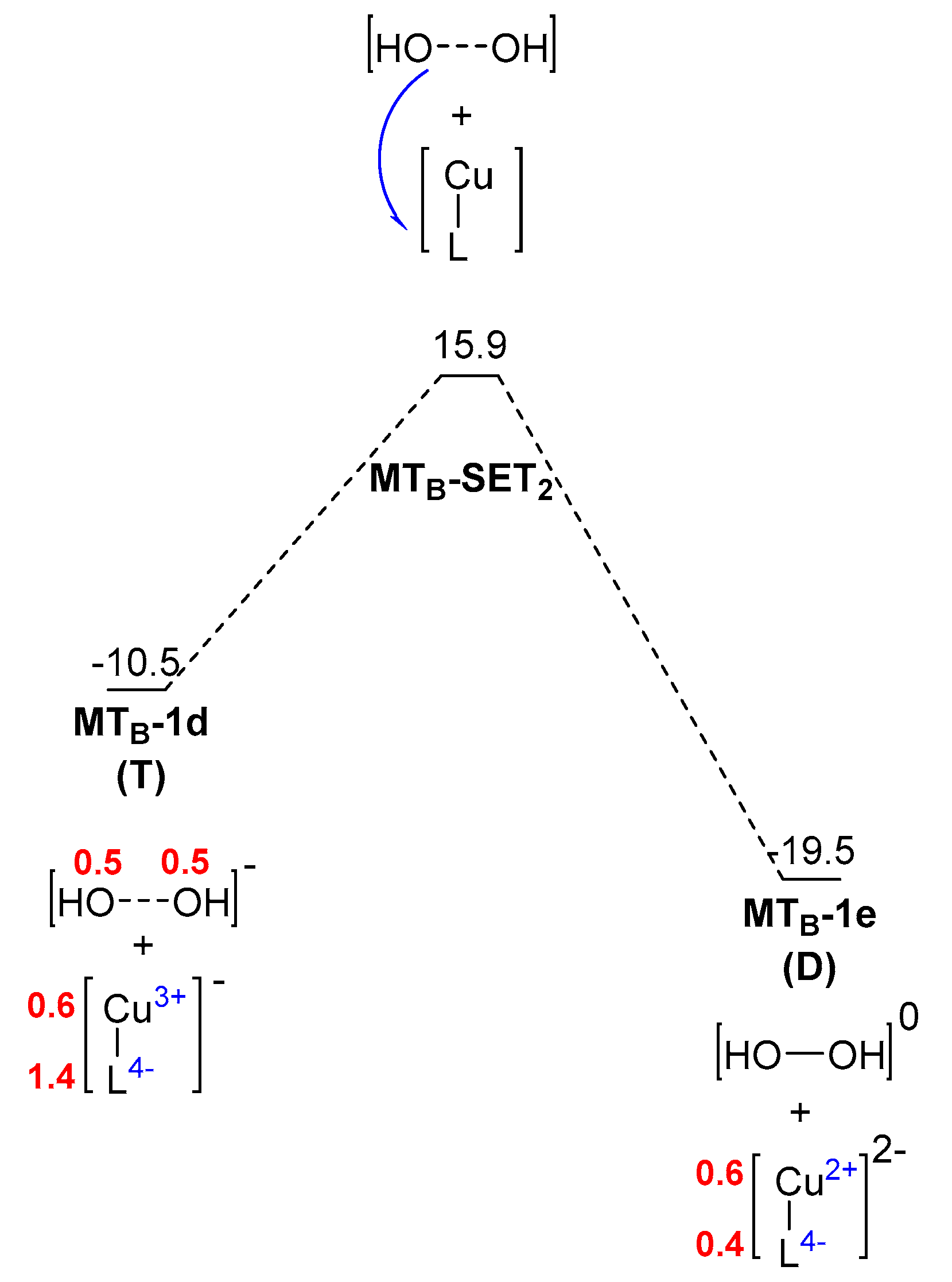
2. Results and Discussion
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nowotny, J.; Sorrell, C.C.; Sheppard, L.R.; Bak, T. Solary-hydrogen: Environmentally safe fuel for the future. Int. J. Hydrogen Energy 2005, 30, 521–544. [Google Scholar] [CrossRef]
- Cox, N.; Pantazis, D.A.; Neese, F.; Lubitz, W. Biological water oxidation. Acc. Chem. Res. 2013, 46, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Bergthorson, J.M. Recyclable metal fuels for clean and compact zero-carbon power. Prog. Energy Combust. Sci. 2018, 68, 169–196. [Google Scholar] [CrossRef]
- Sun, L.; Hammarstrom, L.; Akermark, B.; Styring, S. Towards artificial photosynthesis: Ruthenium–manganese chemistry for energy production. Chem. Soc. Rev. 2001, 30, 36–49. [Google Scholar] [CrossRef]
- Alstrum-Acevedo, J.H.; Brennaman, M.K.; Meyer, T.J. Chemical approeaches to artifical photosynthesis. 2. Inorg. Chem. 2005, 44, 6802–6827. [Google Scholar] [CrossRef] [PubMed]
- Nocera, D.G. The artifical leaf. Acc. Chem. Res. 2012, 45, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, W.J.; Lee, S.-H.A.; Kobayashi, Y.; Hernandez-Pagan, E.A.; Hoertz, P.G.; Moore, T.A.; Moore, A.L.; Gust, D.; Mallouk, T.E. Photoassisted overall water splitting in a visible light-absorbing dye-sensitized photoelectrochemical cell. J. Am. Chem. Soc. 2009, 131, 926–927. [Google Scholar] [CrossRef] [PubMed]
- Mckone, J.R.; Lewis, N.S.; Gray, H.B. Will solar-driven water-splitting devices see the light of day? Chem. Mater. 2014, 26, 407–414. [Google Scholar] [CrossRef]
- Garrido-Barros, P.; Gimbert-Suriñach, C.; Matheu, R.; Sala, X.; Llobet, A. How to make an efficient and robust molecular catalyst for water oxidation. Chem. Soc. Rev. 2017, 46, 6088–6098. [Google Scholar] [CrossRef] [PubMed]
- Kärkaäs, M.D.; Verho, O.; Johnston, E.V.; Åkermark, B. Artificial photosynthesis: Molecular systems for catalytic water oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef] [PubMed]
- Sala, X.; Romero, I.; Rodríguez, M.; Escriche, L.; Llobet, A. Molecular catalysts that oxidize water to dioxygen. Angew. Chem. Int. Ed. 2009, 48, 2842–2852. [Google Scholar] [CrossRef] [PubMed]
- Romain, S.; Vigara, L.; Llobet, A. Oxygen–Oxygen bond formation pathways promoted by ruthenium complexes. Acc. Chem. Res. 2009, 42, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Neudeck, S.; Maji, S.; Lopez, I.; Meyer, S.; Meyer, F.; Llobet, A. New powerful and oxidatively rugged dinuclear Ru water oxidation catalyst: Control of mechanistic pathways by tailored ligand design. J. Am. Chem. Soc. 2014, 136, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Vigara, L.; Ertem, M.Z.; Planas, N.; Bozoglian, F.; Leidel, N.; Dau, H.; Haumann, M.; Gagliardi, L.; Cramer, C.J.; Llobet, A. Experimental and quantum chemical characterization of the water oxidation cycle cataysed by [RuII(damp)(bpy)(H2O)]2+. Chem. Sci. 2012, 3, 2576–2586. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Jurss, J.W.; Brennaman, M.K.; Hoertz, P.G.; Patrocinio, A.O.T.; Murakami Iha, N.Y.; Templeton, J.L.; Meyer, T.J. Making oxygen with ruthenium complexes. Acc. Chem. Res. 2009, 42, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Zong, R.; Thummel, R.P. A new family of Ru complexes for water oxidation. J. Am. Chem. Soc. 2005, 127, 12802–12803. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.; Kunz, V.; Frischmann, P.D.; Würthner, F. A supramolecular ruthenium macrocycle with high catalytic activity for water oxidation that mechanistically mimics photosystem II. Nat. Chem. 2016, 8, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Hull, J.F.; Balcells, D.; Blakemore, J.D.; Incarvito, C.D.; Eisenstein, O.; Brudvig, G.W.; Crabtree, R.H. Highly active and robust Cp* iridium complexes for catalytic water oxidation. J. Am. Chem. Soc. 2009, 131, 8730–8731. [Google Scholar] [CrossRef] [PubMed]
- Lalrempuia, R.; McDaniel, N.D.; Müller-Bunz, H.; Bernhard, S.; Albrecht, M. Water oxidation catalyzed by strong carbene-type donor-ligand complexes of iridium. Angew. Chem. Int. Ed. 2010, 49, 9765–9768. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.M.; Sheehan, S.W.; Hashmi, S.M.; Campos, J.; Hintermair, U.; Crabtree, R.H.; Brudvig, G.W. Electrochemical activation of Cp* iridium complexes for electrode-driven water-oxidation catalysis. J. Am. Chem. Soc. 2014, 136, 13826–13834. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.A.; Bernhard, S.; Albrecht, M. Recent advances in the field of iridium-catalyzed molecular water oxidation. In Molecular Water Oxidation Catalysis; Llobet, A., Ed.; John Wiley & Sons, Ltd.: New York, NY, USA, 2014; pp. 113–134. [Google Scholar]
- McDaniel, N.D.; Coughlin, F.J.; Tinker, L.L.; Bernhard, S. Cyclometalated iridium(III) aquo complexes: Efficient and tunable catalysts for the homogeneous oxidation of water. J. Am. Chem. Soc. 2008, 130, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Limburg, J.; Vrettos, J.S.; Liable-Sands, L.M.; Rheingold, A.L.; Crabtree, R.H.; Brudvig, G.W. A functional model for O–O bond formation by the O2-evolving complex in photosystem II. Science 1999, 283, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Nagano, T.; Takesue, H.; Ye, B.-H.; Tani, F.; Naruta, Y. Characterization of a dinuclear MnV=O complex and is efficient evolution of O2 in the presence of water. Angew. Chem. Int. Ed. 2004, 43, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Limburg, J.; Brudvig, G.W.; Crabtree, R.H. O2 evolution and permanganate formation from high-valent manganese complexes. J. Am. Chem. Soc. 1997, 119, 2761–2762. [Google Scholar] [CrossRef]
- Pushkar, Y.; Davis, K.M.; Palenik, M.C. Model of the oxygen evolving complex which is highly predisposed to O–O bond formation. J. Phys. Chem. Lett. 2018, 9, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Ellis, W.C.; McDaniel, N.D.; Bernhard, S.; Collins, T.J. Fast water oxidation using iron. J. Am. Chem. Soc. 2010, 132, 10990–10991. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, L.D.; Zhou, R.; Zong, R.; Vo, P.; Gagnon, K.J.; Thummel, R.P. Iron complexes of square planar tetradentate polypyridyl-type ligands as catalysts for water oxidation. J. Am. Chem. Soc. 2015, 137, 13260–13263. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, D.J.; Ganesamoorthy, C.; Borau-Garcia, J.; Berlinguette, C.P. Electrochemical evidence for catalytic water oxidation mediated by a high-valent cobalt complex. Chem. Commun. 2011, 47, 4249–4251. [Google Scholar] [CrossRef] [PubMed]
- Rigsby, M.L.; Mandal, S.; Nam, W.; Spencer, L.C.; Llobet, A.; Stahl, S.S. Cobalt analogs of Ru-based water oxidation catalysts: Overcoming thermodynamic instability and kinetic lability to achieve electrocatalytic O2 evolution. Chem. Sci. 2012, 3, 3058–3062. [Google Scholar] [CrossRef]
- Han, Y.; Wu, Y.; Lai, W.; Cao, R. Electrocatalytic water oxidation by a water-soluble nickel porphyrin complex at netural pH with low overpotential. Inorg. Chem. 2015, 54, 5604–5613. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.M.; Goldberg, K.I.; Mayer, J.M. A soluble copper-bipyridine water-oxidation electrocatalyst. Nat. Chem. 2012, 4, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-T.; Chen, Z.; Kang, P.; Meyer, T.J. Electrocatalytic water oxidation with a copper(II) polypeptide complex. J. Am. Chem. Soc. 2013, 135, 2048–2051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, C.; Liu, S.; Wang, J.-L.; Lin, W. A biomimetic copper water oxidation catalyst with low overpotential. J. Am. Chem. Soc. 2014, 136, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Barros, P.; Funes-Ardoiz, I.; Drouet, S.; Benet-Buchholz, J.; Maseras, F.; Llobet, A. Redox non-innocent ligand controls water oxidation overpotential in a new family of mononuclear Cu-based efficient catalysts. J. Am. Chem. Soc. 2015, 137, 6758–6761. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Materna, K.L.; Mercado, B.Q.; Crabtree, R.H.; Brudvig, G.W. Electrocatalytic water oxidation by a copper(II) complex of an oxidation-resistant ligand. ACS Catal. 2017, 7, 3384–3387. [Google Scholar] [CrossRef]
- Liao, R.-Z.; Siegbahn, P.E.M. Quantum chemical modeling of homogeneous water oxidation catalysis. ChemSusChem 2017, 10, 4236–4263. [Google Scholar] [CrossRef] [PubMed]
- Sala, X.; Maji, S.; Bofill, R.; García-Anton, J.; Escriche, L.; Llobet, A. Molecular water oxidation mechanisms followed by transition metals: State of the art. Acc. Chem. Res. 2014, 47, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.J.; Tsai, M.-K.; Muckerman, J.T.; Meyer, T.J. Mechanism of water oxidation by single-site ruthenium complex catalysts. J. Am. Chem. Soc. 2010, 132, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Funes-Ardoiz, I.; Garrido-Barros, P.; Llobet, A.; Maseras, F. Single electron transfer steps in water oxidation catalysis. Redefining the mechanistic scenario. ACS Catal. 2017, 7, 1712–1719. [Google Scholar] [CrossRef]
- de Aguirre, A.; Garrido-Barros, P.; Funes-Ardoiz, I.; Maseras, F. The role of electron-donor substituents in the family of OPBAN-Cu water oxidation catalysts: Effect on the degradation pathways and efficiency. Eur. J. Inorg. Chem. 2019. [Google Scholar] [CrossRef]
- Sameera, W.M.C.; Maseras, F. Transition metal catalysis by density functional theory and density functional theory/molecular mechanics. WIREs Comput. Mol. Sci. 2012, 2, 375–385. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Geng, C.-Y.; Shaik, S.; Neese, F. Electronic structure analysis of multistate reactivity in transition metal catalyzed reactions: The case of C–H bond activation by non-heme iron(IV)–oxo cores. Phys. Chem. Chem. Phys. 2013, 15, 8017–8030. [Google Scholar] [CrossRef] [PubMed]
- Funes-Ardoiz, I.; Nelson, D.J.; Maseras, F. Halide abstraction competes with oxidative addition in the reactions of aryl halides with [Ni(PMenPh(3−n))4]. Chem. Eur. J. 2017, 23, 16728–16733. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.J.; Maseras, F. Steric effects determine the mechanisms of reaction between bis(N-heterocyclic carbene)–nickel(0) complexes and aryl halides. Chem. Commun. 2018, 54, 10646–10649. [Google Scholar] [CrossRef] [PubMed]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current Status of transition-state theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. On the theory of oxidation-reduction reactions involving electron transfer. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry: Theory and experimental (Nobel lecture). Angew. Chem. Int. Ed. Engl. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]
- Fernandez-Alvarez, V.M.; Maseras, F. Computational characterization of the mechanism for the light-driven catalytic trichloromethylation of acylpyridines. Org. Biomol. Chem. 2017, 15, 8641–8647. [Google Scholar] [CrossRef] [PubMed]
- de Aguirre, A.; Funes-Ardoiz, I.; Maseras, F. Four oxidation states in a single photoredox Ni-based catalytic cycle: A computational study. Angew. Chem. Int. Ed. 2019. [Google Scholar] [CrossRef]
- Qi, Z.-H.; Ma, J. Dual role of a photocatalyst: Generation of Ni(0) catalyst and promotion of catalytic C–N bond formation. ACS Catal. 2018, 8, 1456–1463. [Google Scholar] [CrossRef]
- Moia, D.; Vaissier, V.; Lopez-Duarte, I.; Torres, T.; Nazeeruddin, M.K.; O’Regan, B.C.; Nelson, J.; Barnes, P.R.F. The reorganization energy of intermolecular hole hopping between dyes anchored to surfaces. Chem. Sci. 2014, 5, 281–290. [Google Scholar] [CrossRef]
- Vaissier, V.; Barnes, P.; Kirkpatrick, J.; Nelson, J. Influence of polar medium on the reorganization energy of charge transfer between dyes in a dye sensitized film. Phys. Chem. Chem. Phys. 2013, 15, 4804–4814. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parmetrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Francl, M.M.; Petro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Ehlers, A.W.; Böhme, M.; Dapprich, S.; Gobbi, A.; Höllwarth, A.; Jonas, V.; Köhler, K.F.; Stegmann, R.; Veldkamp, A.; Frenking, G. A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc–Cu, Y–Ag and La–Au. Chem. Phys. Lett. 1993, 208, 111–114. [Google Scholar] [CrossRef]
- Roy, L.E.; Hay, P.J.; Martin, R.L. Revised basis sets for the LANL effective core potentials. J. Chem. Theory Comput. 2008, 4, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Moreno, M.; de Graaf, C.; Lopez, N.; Maseras, F.; Poblet, J.M.; Bo, C. Managing the computational chemistry big data problem: The ioChem-BD platform. J. Chem. Inf. Model. 2015, 55, 95–103. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
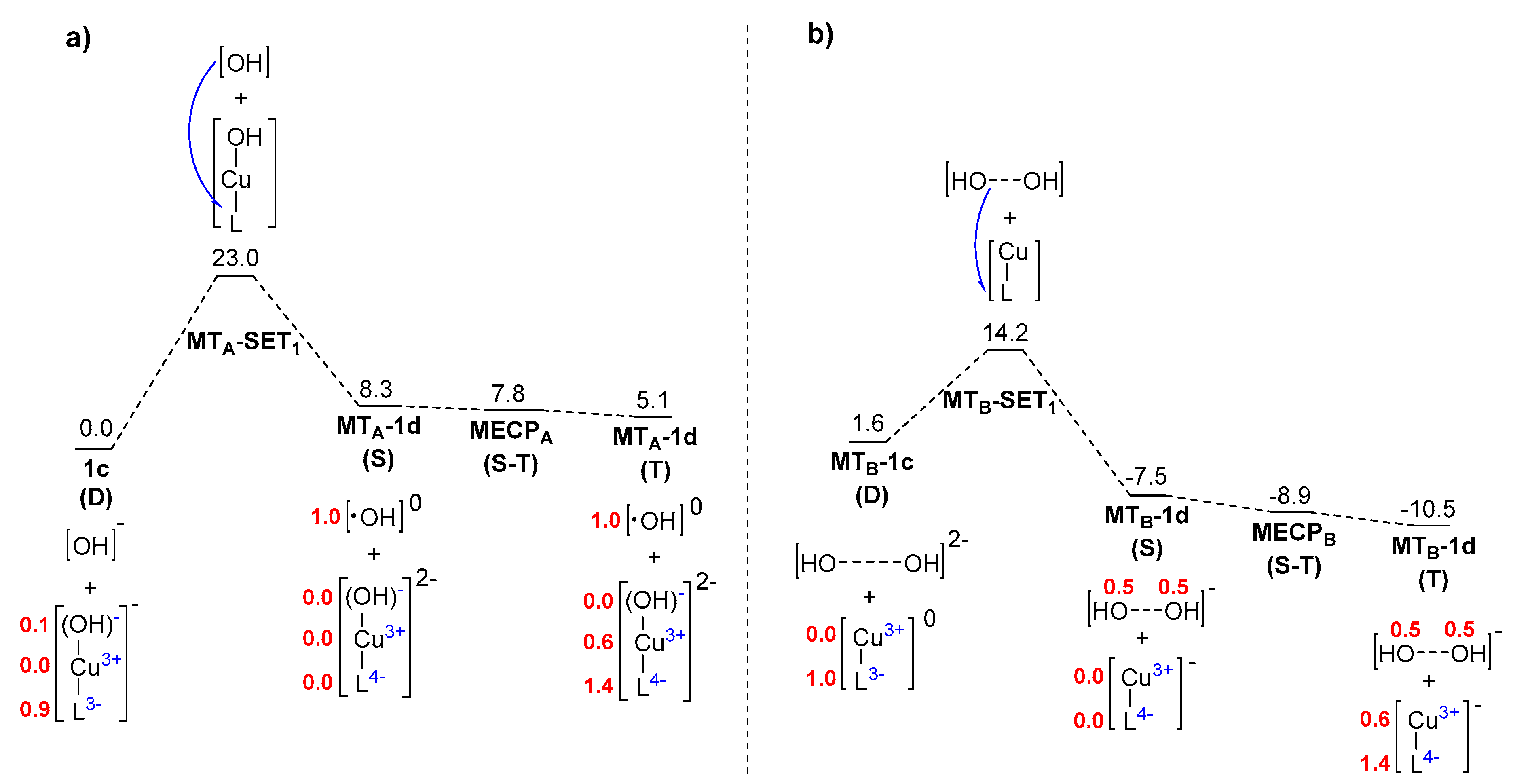{kind=link}
{kind=link}
| Step | ΔG | ΔG‡ | ||
|---|---|---|---|---|
| MTA-SET1 | 74.4 | 0.1 | 8.3 | 23.0 |
| MTB-SET1 | 61.9 | 5.4 | −9.1 | 14.2 |
| Step | ΔG | ΔG‡ | ||
|---|---|---|---|---|
| MTB-SET2 | 62.1 | 60.9 | −8.9 | 26.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Aguirre, A.; Funes-Ardoiz, I.; Maseras, F. Computational Characterization of Single-Electron Transfer Steps in Water Oxidation. Inorganics 2019, 7, 32. https://doi.org/10.3390/inorganics7030032
de Aguirre A, Funes-Ardoiz I, Maseras F. Computational Characterization of Single-Electron Transfer Steps in Water Oxidation. Inorganics. 2019; 7(3):32. https://doi.org/10.3390/inorganics7030032
Chicago/Turabian Stylede Aguirre, Adiran, Ignacio Funes-Ardoiz, and Feliu Maseras. 2019. "Computational Characterization of Single-Electron Transfer Steps in Water Oxidation" Inorganics 7, no. 3: 32. https://doi.org/10.3390/inorganics7030032
APA Stylede Aguirre, A., Funes-Ardoiz, I., & Maseras, F. (2019). Computational Characterization of Single-Electron Transfer Steps in Water Oxidation. Inorganics, 7(3), 32. https://doi.org/10.3390/inorganics7030032