Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates?


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Ruthenium Complexes as Anticancer Agents
3. Case Study: RM175
4. Case Study: RAPTA-C
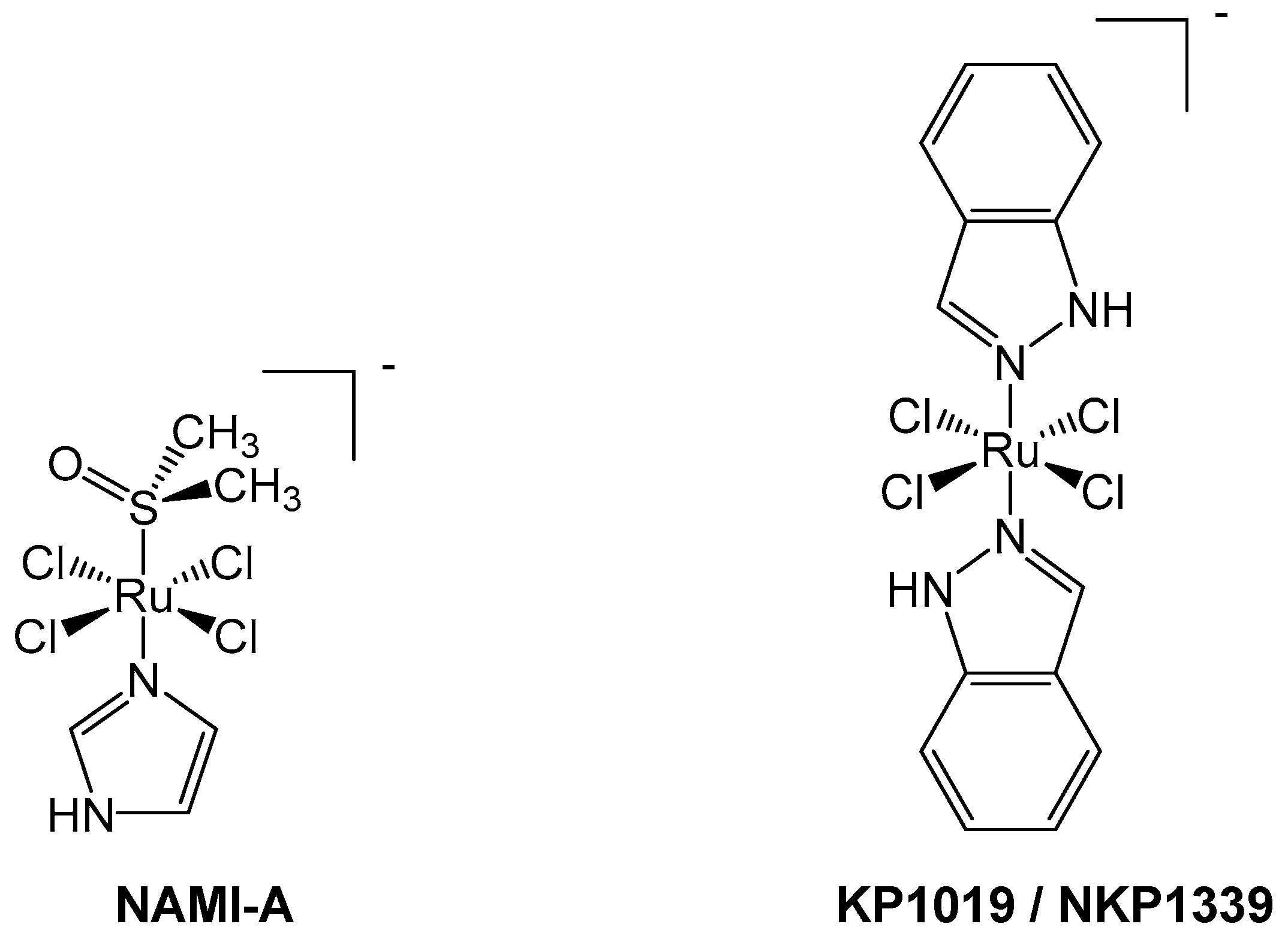
5. Case Study: NAMI-A
6. Case Study: KP1019/NKP1339
7. Current Developments in Ruthenium Anticancer Agents and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Florea, A.M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers (Basel) 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Ai, Z.; Lu, Y.; Qiu, S.; Fan, Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016, 373, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.J.; Eastman, A.; Bostick-Bruton, F.; Reed, E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J. Clin. Invest. 1991, 87, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Schluga, P.; Hartinger, C.G.; Egger, A.; Reisner, E.; Galanski, M.; Jakupec, M.; Keppler, B.K. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalt. Trans. 2006, 1796–1802. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska, J.; Fandzloch, M.; Łakomska, I. The reduction of ruthenium(III) complexes with triazolopyrimidine ligands by ascorbic acid and mechanistic insight into their action in anticancer therapy. Inorg. Chim. Acta 2019, 484, 305–310. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.J.; Habtemariam, A.; McHale, S.E.; Parsons, S.; Sadler, P.J. Catalytic organometallic anticancer complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 11628–11633. [Google Scholar] [CrossRef] [PubMed]
- Kandioller, W.; Balsano, E.; Meier, S.M.; Jungwirth, U.; Goschl, S.; Roller, A.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Hartinger, C.G. Organometallic anticancer complexes of lapachol: Metal centre- dependent formation of reactive oxygen species and correlation with cytotoxicity. Chem. Commun. 2013, 49, 3348–3350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sadler, P.J. Advances in the design of organometallic anticancer complexes. J. Organomet. Chem. 2017, 839, 5–14. [Google Scholar] [CrossRef]
- Fong, J.; Kasimova, K.; Arenas, Y.; Kaspler, P.; Lazic, S.; Mandel, A.; Lilge, L. A novel class of ruthenium-based photosensitizers effectively kills in vitro cancer cells and in vivo tumors. Photochem. Photobiol. Sci. 2015, 14, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.E.; Aird, R.E.; Murdoch, P.D.S.; Chen, H.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I.; et al. Inhibition of cancer cell growth by ruthenium(II) arene complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Masi, A.; Peacock, A.F.A.; Habtemariam, A.; Sadler, P.J.; Sava, G. In vivo tumour and metastasis reduction and in vitro effects on invasion assays of the ruthenium RM175 and osmium AFAP51 organometallics in the mammary cancer model. J. Inorg. Biochem. 2010, 104, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Habtemariam, A.; van der Geer, E.; Fernández, R.; Melchart, M.; Deeth, R.J.; Aird, R.; Guichard, S.; Fabbiani, F.P.; Lozano-Casal, P.; et al. Controlling ligand substitution reactions of organometallic complexes: Tuning cancer cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18269–18274. [Google Scholar] [CrossRef] [PubMed]
- Barry, N.P.E.; Sadler, P.J. Exploration of the medical periodic table: Towards new targets. Chem. Commun. 2013, 49, 5106–5131. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.L.; Schornagel, Q.C.; Tente, R.; Macpherson, J.S.; Aird, R.E.; Guichard, S.; Habtemariam, A.; Sadler, P.J.; Jodrell, D.I. Investigation of the role of Bax, p21/Waf1 and p53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium(II) organometallic agent, RM175. Cancer Chemother. Pharmacol. 2005, 55, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.; Westhorpe, A.; Romero, M.J.; Habtemariam, A.; Gallevo, C.R.; Bark, Y.; Menezes, N.; Sadler, P.J.; Sharma, R.A. Radiosensitisation of human colorectal cancer cells by ruthenium(II) arene anticancer complexes. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Parkinson, J.; Parsons, S.; Coxall, R.; Gould, R.O.; Sadler, P.J. Organometallic ruthenium(II) diamine anticancer complexes: Arene-nucleobase stacking and stereospecific hydrogen-bonding in guanine adducts. J. Am. Chem. Soc. 2002, 124, 3064–3082. [Google Scholar] [CrossRef] [PubMed]
- Aird, R.E.; Cummings, J.; Ritchie, A.A.; Muir, M.; Morris, R.E.; Chen, H.; Sadler, P.J.; Jodrell, D.I. In vitro and in vivo activity and cross resistance profiles of novel ruthenium(II) organometallic arene complexes in human ovarian cancer. Br. J. Cancer 2002, 86, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. P21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Rowland, B.D.; Peeper, D.S. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 2006, 6, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tong, T.; Fan, W.; Fan, F.; Antinore, M.J.; Zhu, X.; Mazzacurati, L.; Li, X.; Petrik, K.L.; Rajasekaran, B.; et al. GADD45-induced cell cycle G2-M arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene 2002, 21, 8696–8704. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(η6-p-cymene)Cl2(pta)] (pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane): A water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 2, 1396–1397. [Google Scholar] [CrossRef]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Phillips, A.D.; Gonsalvi, L.; Romerosa, A.; Vizza, F.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA): Transition metal complexes and related catalytic, medicinal and photoluminescent applications. Coord. Chem. Rev. 2004, 248, 955–993. [Google Scholar] [CrossRef]
- Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T.J.; Sava, G.; Dyson, P.J. In vitro and in vivo evaluation of ruthenium(II)-arene PTA complexes. J. Med. Chem. 2005, 48, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Van Beijnum, J.R.; Casini, A.; Nazarov, A.A.; Wagnières, G.; Van Den Bergh, H.; Dyson, P.J.; Griffioen, A.W. Organometallic ruthenium(II) arene compounds with antiangiogenic activity. J. Med. Chem. 2011, 54, 3895–3902. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Else, R.; Reid, E.; Zeitlin, B.; Aird, R.; Muir, M.; Dodds, M.; Fiebig, H.; Sadler, P.J.; Jodrell, D.I. Anti-tumour activity in non-small cell lung cancer models and toxicity profiles for novel ruthenium(II) based organo-metallic compounds. Biochem. Pharmacol. 2006, 71, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Berndsen, R.H.; Dubois, M.; Müller, C.; Schibli, R.; Griffioen, A.W.; Dyson, P.J.; Nowak-Sliwinska, P. In vivo anti-tumor activity of the organometallic ruthenium(II)-arene complex [Ru(η6-p-cymene)Cl2(pta)] (RAPTA-C) in human ovarian and colorectal carcinomas. Chem. Sci. 2014, 5, 4742–4748. [Google Scholar] [CrossRef]
- Nazarov, A.A.; Risse, J.; Ang, W.H.; Schmitt, F.; Zava, O.; Ruggi, A.; Groessl, M.; Scopelitti, R.; Juillerat-Jeanneret, L.; Hartinger, C.G.; et al. Anthracene-tethered ruthenium(II) arene complexes as tools to visualize the cellular localization of putative organometallic anticancer compounds. Inorg. Chem. 2012, 51, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Furrer, M.A.; Schmitt, F.; Wiederkehr, M.; Juillerat-Jeanneret, L.; Therrien, B. Cellular delivery of pyrenyl-arene ruthenium complexes by a water-soluble arene ruthenium metalla-cage. Dalt. Trans. 2012, 41, 7201–7211. [Google Scholar] [CrossRef] [PubMed]
- Kilpin, K.J.; Clavel, C.M.; Edafe, F.; Dyson, P.J. Naphthalimide-tagged ruthenium–arene anticancer complexes: Combining coordination with intercalation. Organometallics 2012, 31, 7031–7039. [Google Scholar] [CrossRef]
- Bailly, C.; Braña, M.; Waring, M.J. Sequence-Selective Intercalation of Antitumour Bis-Naphthalimides into DNA. Eur. J. Biochem. 2018, 240, 195–208. [Google Scholar] [CrossRef]
- Ang, W.H.; Parker, L.J.; De Luca, A.; Juillerat-Jeanneret, L.; Morton, C.J.; Lo, B.M.; Parker, M.W.; Dyson, P.J. Rational design of an organometallic glutathione transferase inhibitor. Angew. Chemi. Int. Ed. 2009, 48, 3854–3857. [Google Scholar] [CrossRef] [PubMed]
- Chakree, K.; Ovatlarnporn, C.; Dyson, P.J.; Ratanaphan, A. Altered dna binding and amplification of human breast cancer suppressor gene BRCA1 induced by a novel antitumor compound, [Ru(η6-p-phenylethacrynate)Cl2(pta)]. Int. J. Mol. Sci. 2012, 13, 13183–13202. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Biondi, I.; Dyson, P.J.; Bhattacharyya, A. A bifunctional organometallic ruthenium drug with multiple modes of inducing apoptosis. J. Biol. Inorg. Chem. 2011, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, H.U.; Movassaghi, S.; Morrow, S.J.; Kubanik, M.; Hartinger, C.G. Metallomic study on the metabolism of RAPTA-C and cisplatin in cell culture medium and its impact on cell accumulation. Metallomics 2018, 10, 455–462. [Google Scholar] [PubMed]
- Weiss, A.; Ding, X.; van Beijnum, J.R.; Wong, I.; Wong, T.J.; Berndsen, R.H.; Dormond, O.; Dallinga, M.; Shen, L.; Schlingemann, R.O.; et al. Rapid optimization of drug combinations for the optimal angiostatic treatment of cancer. Angiogenesis 2015, 18, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Berndsen, R.H.; Ding, X.; Ho, C.M.; Dyson, P.J.; Van Den Bergh, H.; Griffioen, A.W.; Nowak-Sliwinska, P. A streamlined search technology for identification of synergistic drug combinations. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E. Thirty Years of the Drug Candidate NAMI-A and the Myths in the Field of Ruthenium Anticancer Compounds: A Personal Perspective. Eur. J. Inorg. Chem. 2017, 1549–1560. [Google Scholar] [CrossRef]
- Clarke, M.J. Ruthenium metallopharmaceuticals. Coord. Chem. Rev. 2003, 236, 209–233. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Artner, C.; Holtkamp, H.U.; Hartinger, C.G.; Meier-Menches, S.M. Characterizing activation mechanisms and binding preferences of ruthenium metallo-prodrugs by a competitive binding assay. J. Inorg. Biochem. 2017, 177, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, G.; Alessio, E.; Sava, G.; Pacor, S.; Coluccia, M.; Boccarelli, A. Water-soluble ruthenium(III)-dimethyl sulfoxide complexes: Chemical behaviour and pharmaceutical properties. Met. Based. Drugs 1993, 1, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; Van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Dyson, P.J.; Sava, G. Metal-based antitumour drugs in the post genomic era. Dalt. Trans. 2006, 1929–1933. [Google Scholar] [CrossRef] [PubMed]
- Morbidelli, L.; Donnini, S.; Filippi, S.; Messori, L.; Piccioli, F.; Orioli, P.; Sava, G.; Ziche, M. Antiangiogenic properties of selected ruthenium(III) complexes that are nitric oxide scavengers. Br. J. Cancer 2003, 88, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Oszajca, M.; Kuliś, E.; Stochel, G.; Brindell, M. Interaction of the NAMI-A complex with nitric oxide under physiological conditions. New J. Chem. 2014, 38, 3386–3394. [Google Scholar] [CrossRef]
- Lai, H.; Zhao, Z.; Li, L.; Zheng, W.; Chen, T. Antiangiogenic ruthenium(II) benzimidazole complexes, structure-based activation of distinct signaling pathways. Metallomics 2015, 7, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases Marie. Microbiol. Mol. Biol. Rev. 2011, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Pelillo, C.; Chambery, A.; Sava, G. Influence of components of tumour microenvironment on the response of HCT-116 colorectal cancer to the ruthenium-based drug NAMI-A. J. Inorg. Biochem. 2017, 168, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Gasparoli, L.; Stefanini, M.; Ristori, M.; D’Amico, M.; Alessio, E.; Scaletti, F.; Becchetti, A.; Arcangeli, A.; Messori, L. NAMI-A is highly cytotoxic toward leukaemia cell lines: Evidence of inhibition of KCa 3.1 channels. Dalt. Trans. 2014, 43, 12150–12155. [Google Scholar] [CrossRef] [PubMed]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stühmer, W.; Pardo, L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Grössinger, E.M.; Weiss, L.; Zierler, S.; Rebhandl, S.; Krenn, P.W.; Hinterseer, E.; Schmölzer, J.; Asslaber, D.; Hainzl, S.; Neureiter, D.; et al. Targeting proliferation of chronic lymphocytic leukemia (CLL) cells through KCa3.1 blockade. Leukemia 2014, 28, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of Ion Channels and Transporters in Cell Migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef] [PubMed]
- Bulk, E.; Ay, A.S.; Hammadi, M.; Ouadid-Ahidouch, H.; Schelhaas, S.; Hascher, A.; Rohde, C.; Thoennissen, N.H.; Wiewrodt, R.; Schmidt, E.; et al. Epigenetic dysregulation of KCa3.1 channels induces poor prognosis in lung cancer. Int. J. Cancer 2015, 137, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATMand ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a12716. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Delfino, R.; Casarsa, C.; Sava, G. CDK1 Hyperphosphorylation Maintenance Drives the Time-course of G2-M Cell Cycle Arrest after Short Treatment with NAMI-A in Kb Cells. Anticancer. Agents Med. Chem. 2012, 12, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Pelillo, C.; Mollica, H.; Eble, J.A.; Grosche, J.; Herzog, L.; Codan, B.; Sava, G.; Bergamo, A. Inhibition of adhesion, migration and of α5β1 integrin in the HCT-116 colorectal cancer cells treated with the ruthenium drug NAMI-A. J. Inorg. Biochem. 2016, 160, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Bruno, M.; Boccarelli, A.; Coluccia, M.; Ribatti, D.; Bergamo, A.; Garbisa, S.; Sartor, L.; Sava, G. Inhibition of endothelial cell functions and of angiogenesis by the metastasis inhibitor NAMI-A. Br. J. Cancer 2002, 4, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, G.; Theocharis, A.D.; Karamanos, N.K. Roles of MMP in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Terra, L.F.; Wailemann, R.A.M.; Labriola, L.; Sogayar, M.C. TGF-β1 modulates the homeostasis between MMPs and MMP inhibitors through p38 MAPK and ERK1/2 in highly invasive breast cancer cells. BMC Cancer 2012, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sava, L.B.A.M.G.; Bergamo, A. Effects of the ruthenium-based drug NAMI-A on the roles played by TGF-β1 in the metastatic process. J. Biol. Inorg. Chem. 2015, 20, 1163–1173. [Google Scholar]
- Schoenhacker-Alte, B.; Mohr, T.; Pirker, C.; Kryeziu, K.; Kuhn, P.S.; Buck, A.; Hofmann, T.; Gerner, C.; Hermann, G.; Koellensperger, G.; et al. Sensitivity towards the GRP78 inhibitor KP1339/IT-139 is characterized by apoptosis induction via caspase 8 upon disruption of ER homeostasis. Cancer Lett. 2017, 404, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, S.; Pongratz, M.; Jakupec, M.A.; Heffeter, P.; Berger, W.; Lackinger, L.; Keppler, B.K.; Marian, B. Heterocyclic complexes of ruthenium(III) induce apoptosis in colorectal carcinoma cells. J. Cancer Res. Clin. Oncol. 2005, 131, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Parkinson, J.; Morris, R.E.; Sadler, P.J. Highly selective binding of organometallic ruthenium ethylenediamine complexes to nucleic acids: Novel recognition mechanisms. J. Am. Chem. Soc. 2003, 125, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Dickson, N.R.; Jones, S.F.; Burris, H.A.; Ramanathan, R.K.; Weiss, G.J.; Infante, J.R.; Bendell, J.C.; McCulloch, W.; Von Hoff, D.D. A phase I dose-escalation study of NKP-1339 in patients with advanced solid tumors refractory to treatment. J. Clin. Oncol. 2011, 29, 2607. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z. Increased oxidative stress as a selective anticancer therapy. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Romero-Canelón, I.; Sanchez-Cano, C.; Clarkson, G.J.; Habtemariam, A.; Wills, M.; Sadler, P.J. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 2018, 10, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Soldevila-Barreda, J.J.; Romero-Canelón, I.; Habtemariam, A.; Sadler, P.J. Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design. Nat. Commun. 2015, 6, 6582. [Google Scholar] [CrossRef] [PubMed]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339—A clinically investigated ruthenium-based drug—Involves ER- and ROS-related effects in colon carcinoma cell lines. Invest. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Atil, B.; Kryeziu, K.; Groza, D.; Koellensperger, G.; Körner, W.; Jungwirth, U.; Mohr, T.; Keppler, B.K.; Berger, W. The ruthenium compound KP1339 potentiates the anticancer activity of sorafenib in vitro and in vivo. Eur. J. Cancer 2013, 49, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Thornton, T.M.; Rincon, M. Non-classical p38 map kinase functions: Cell cycle checkpoints and survival. Int. J. Biol. Sci. 2009, 5, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; Castilho, R.F.; Vercesi, A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001, 495, 12–15. [Google Scholar] [CrossRef]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta 2011, 1807, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside—Preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta, Mol. Cell. Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.B.; Huang, W.; Zeleniak, A.E.; Hindoyan, A.; Wu, H.; Donahue, T.R.; Hill, R. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-J.; Wang, W.; Huang, S.-Y.; Hong, Y.; Li, G.; Lin, S.; Tian, J.; Cai, Z.; Wang, H.-M.D.; Ma, D.-L.; et al. Inhibition of the Ras/Raf interaction and repression of renal cancer xenografts in vivo by an enantiomeric iridium(III) metal-based compound. Chem. Sci. 2017, 8, 4756–4763. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Casini, A.; Sava, G.; Dyson, P.J. Organometallic ruthenium-based antitumor compounds with novel modes of action. J. Organomet. Chem. 2011, 696, 989–998. [Google Scholar] [CrossRef]
- Mitrović, A.; Kljun, J.; Sosič, I.; Gobec, S.; Turel, I.; Kos, J. Clioquinol–ruthenium complex impairs tumour cell invasion by inhibiting cathepsin B activity. Dalt. Trans. 2016, 45, 16913–16921. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer activity of metal complexes: Involvement of redox processes. Antioxidants Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Magistrato, A.; Riedel, T.; von Erlach, T.; Davey, C.A.; Dyson, P.J.; Rothlisberger, U. Fighting Cancer with Transition Metal Complexes: From Naked DNA to Protein and Chromatin Targeting Strategies. ChemMedChem 2016, 11, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Adhireksan, Z.; Davey, G.E.; Campomanes, P.; Groessl, M.; Clavel, C.M.; Yu, H.; Nazarov, A.A.; Yeo, C.H.F.; Ang, W.H.; Dröge, P.; et al. Ligand substitutions between ruthenium–cymene compounds can control protein versus DNA targeting and anticancer activity. Nat. Commun. 2014, 5, 3462. [Google Scholar] [CrossRef]
- Wu, B.; Ong, M.S.; Groessl, M.; Adhireksan, Z.; Hartinger, C.G.; Dyson, P.J.; Davey, C.A. A ruthenium antimetastasis agent forms specific histone protein adducts in the nucleosome core. Chem. Eur. J. 2011, 17, 3562–3566. [Google Scholar] [CrossRef] [PubMed]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure-activity relationships for ruthenium and osmium anticancer agents-towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. https://doi.org/10.3390/inorganics7030031
Coverdale JPC, Laroiya-McCarron T, Romero-Canelón I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics. 2019; 7(3):31. https://doi.org/10.3390/inorganics7030031
Chicago/Turabian StyleCoverdale, James P. C., Thaisa Laroiya-McCarron, and Isolda Romero-Canelón. 2019. "Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates?" Inorganics 7, no. 3: 31. https://doi.org/10.3390/inorganics7030031
APA StyleCoverdale, J. P. C., Laroiya-McCarron, T., & Romero-Canelón, I. (2019). Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics, 7(3), 31. https://doi.org/10.3390/inorganics7030031