Abstract
The influence of the environment on the functionality of the oxygen-evolving complex (OEC) of photosystem II has long been a subject of great interest. In particular, various water channels, which could serve as pathways for substrate water diffusion, or proton translocation, are thought to be critical to catalytic performance of the OEC. Here, we address the dynamical nature of hydrogen bonding along the water channels by performing molecular dynamics (MD) simulations of the OEC and its surrounding protein environment in the S1 and S2 states. Through the eigenvector centrality (EC) analysis, we are able to determine the characteristics of the water network and assign potential functions to the major channels, namely that the narrow and broad channels are likely candidates for proton/water transport, while the large channel may serve as a path for larger ions such as chloride and manganese thought to be essential during PSII assembly.
1. Introduction
Photosystem II (PSII) is a 650 kDa complex composed of 20 proteins embedded in the thylakoid membrane of green plant chloroplast and internal membranes of cyanobacteria. PSII sustains aerobic life on Earth by harvesting solar light, initiating the process of photosynthesis, and producing oxygen from water. The water oxidation reaction is catalyzed by the oxygen-evolving complex (OEC), which is a CaMn4O5 cuboidal cluster embedded in the D1 protein subunit. Oxygen is produced through the catalytic Kok cycle. In each turn of the cycle, the OEC evolves through five storage states (S-states) of oxidizing equivalents, with S0 and S4 being the most reduced and oxidized states, respectively [1,2]. The stable dark-adapted S1 state (structure and oxidation states shown in Figure 1) is advanced to the S2 state by a single flash of light and generates a mixture of two redox isomers. Isomer A is an “open” cubane structure where Mn1 and Mn4 have formal oxidation states III and IV, respectively [3]. Isomer B is “closed” cuboid where the oxidation states of Mn1 and Mn4 are switched to IV and III, respectively. S2A exhibits a characteristic multiline EPR signal at g = 2.0 [4], while S2B has a broad EPR band at g = 4.1 [5,6]. A second flash moves the OEC into the S3 state, where all four Mn centers have formal 4+ charges and an additional water is bound between Mn1 and Mn4, setting the complex up for oxygen evolution [7]. A final third flash brings the complex into the S4 state, however this state is highly unstable and almost immediately decays to the S0 state, releasing oxygen in the process. As the S4 state decays faster than it is formed, its structure has not been experimentally characterized, although several theoretical models have been proposed. From the S0 state, the OEC can return to S1, either through a fourth light flash or gradual decay in darkness, although some fraction of OEC centers will remain in the S0 state, even after extensive dark adaptation [8].
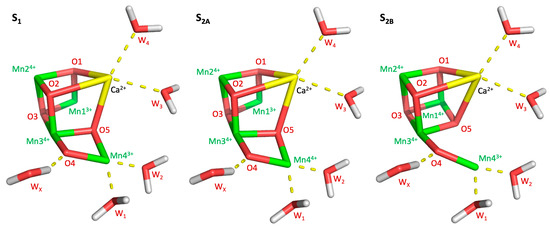
Figure 1.
Manganese oxidation of the S-states examined in this work (S1, S2A, and S2B), as well as the placement of relevant waters.
The varying shape and oxidation states of the OEC make it a challenging system to model. Typically, quantum mechanics (QM) and two-layer QM/MM models are used to study the OEC and its surrounding protein [9,10]. These methods are computationally expensive and limit the size of the system that can be studied. Classical methods, such as molecular mechanics, provide practical approaches for computational modeling, although with significant shortcomings. Force fields require significant parameterization, especially for nonstandard residues and metal-containing systems, such as the OEC. Classical molecular dynamics simulations have been performed on PSII and get around the parameter problem by making the OEC and other metal cofactors static [11,12]. Valence charges are assigned to a non-bonded model of the OEC and the complex and its ligating residues are locked into their crystal structure or QM-calculated coordinates. While the majority of PSII is indeed dynamic in such simulations, the OEC itself is not. One of our ongoing goals has been to derive classical parameters for the OEC and its ligands. Several methods are available to generate parameters for metal complexes, however none have been tested on a system containing as many metal centers and high oxidation states as the OEC. In an effort to make the OEC more dynamic during these simulations, we use a flexible model with ESP-derived partial charges and strong force constants for all metal containing bonds and angles, 500 kcal/mol/Å2 and 100 kcal/mol/rad2, respectively.
Dynamic simulations of the OEC are of major interest because they allow for the study of water passages and the effects of changing S-states on these passages. Previous studies have examined the pathways water takes to and from the OEC [13,14,15]. Molecular dynamics simulations have indicated that the two sites for water binding to the OEC has completely separate sources, with waters bound to the calcium (W3 and W4) originating from the large channel and the water bound to Mn4 (W1 and W2) being supplied by the narrow or broad channels [13]. While the broad and large channels connect near D1-E189, there is little, if any, crossover between the two [14]. While the large channel has been widely accepted as the source for W3 and W4, it is unclear whether W1 and W2 flow from the narrow or broad channel, or even the large channel. Though the broad channel is significantly wider than the narrow channel, as indicated by their names, the narrow actually has fewer and lower permeation barriers than the broad channel [14]. Part of the interest in these channels arises from the implications their purposes have on the mechanism of forming the S-states of the OEC, which requires two new waters to bind to the OEC for every completed turn of the Kok cycle. One of these waters binds during the S2-to-S3 transition, as described above [7]. WX, sharing a hydrogen bond with O4, is the primary candidate for a newly-bound water during this transition, although the mechanism and where it binds is unclear. One mechanism proposes that WX originates from the narrow channel and binds to Mn4 in the S2B isomer as part of a “carousel” mechanism [16]. A second possible mechanism proposes that WX flows up from the large channel and replace W3 on the calcium after that water binds to Mn1 [17]. Studies of the binding of methanol [18] and ammonia [19,20] near or directly to the OEC support the carousel mechanism. Ammonia binds to Mn4 as an additional ligand in the S2 state, and neither ammonia nor methanol shows any interaction with Ca2+. These are strong indicators as to where water binds in the S3, although it does not necessarily show which channel substrate water molecules come from before entering the WX pocket. In addition to supplying waters, channels are also needed for proton shuttling, oxygen evacuation, and OEC reassembly. In the present work, we utilize a flexible model of OEC and network theory to characterize the water dynamics in the surroundings of the complex in the S1 and S2 states. Our analysis clearly identifies three channels of highly diffusive and interconnected waters in direct contact with the OEC complex. Each of these channels display different water mobility and interconnectivity, as well as different sensitivity to the advance from S1 to S2 state. It is shown that restricted mobility might lead to a stronger hydrogen bonding network. In light of these results we postulate different roles played by the identified channels in the OEC activity.
2. Results
2.1. Water Network Dynamics
The mobility of the interstitial water molecules in PSII provides insights on potential diffusion pathways for H2O, H+, or O2. Therefore, we computed the Root Mean Square Fluctuation (RMSF) per water molecule to quantify the mobility of waters in the system. Figure 2 (left column) shows that there are two main channels of highly diffusive waters that connect the Mn4CaO5 complex with the lumen. The narrow channel shows a straight pathway of hydrogen-bonding water molecules with moderate mobility. The second large channel shows a branching point and much higher mobility towards the protein exterior. A third channel bridges the narrow and large channels with intermediate water mobility values. This bridge channel matches the entry of the broad channel described in Reference [18], which splits into two more channels (the broad and a proposed proton-exit channel) between D1–D61 and D1–D65 (not shown here). Interestingly, the structure and mobility of these water channels show sensitivity with respect to the S1 and S2 states studied here, suggesting a potential regulatory role of the water dynamics on OEC activity.
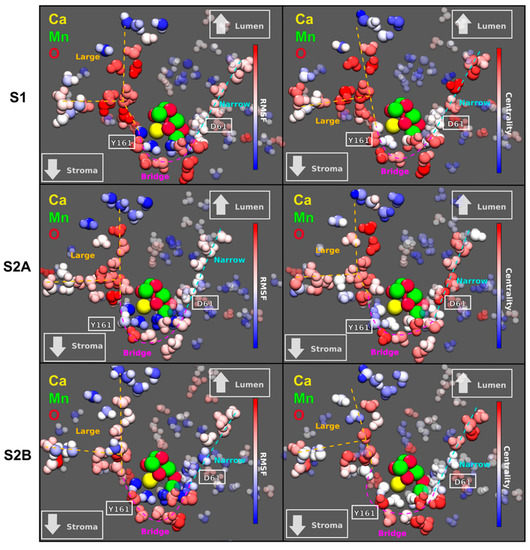
Figure 2.
Left: Root Mean Square Fluctuation (RMSF) per water molecule for three different S-states of the OEC. The color scale is red for highly mobile water molecules and blue for the most static waters. Right: Eigenvector centrality in terms of the mutual information metric computed for the OEC complex in two different states: S1 and S2, having two isomers of S2. The centrality scale goes from red (high centrality) to blue (low centrality). Relative positions of D1–D61 and Y161 are shown in white. The channels colored in cyan, magenta, and orange are named narrow, bridge, and large, respectively. Both the large and the narrow channel have already been recognized in several studies [13,14,15]. The bridge channel, on the other hand, only coincides a fragment of the so-called broad channel.
In order to further analyze the influence of the surrounding waters on the Mn4CaO5 complex, we computed the Eigenvector Centrality (EC) in terms of the Mutual Information (MI) for all the waters in the system [21]. The measure of centrality gives an idea of how much each atom influences the dynamics of neighboring atoms, i.e., how connected each water molecule is with its neighbors. It is expected that higher connectivities can be related with high proton mobility, and therefore, an increased effectiveness in proton transport between the OEC and the exterior of the protein.
This analysis shows that the same three channels that exhibit the highest mobility also show the highest EC within the water network (Figure 2, right column). Again, it is shown that the EC distribution is very sensitive to both the state transition from S1 to S2 and the isomerization S2A to S2B. On the other hand, it is interesting to note that the waters in these two channels manage to solvate the Mn4CaO5 complex, and therefore, long chains of water with great centrality reaching this complex may indicate water-assisted long-range modulation of the reaction taking place in the OEC.
2.2. Diffusion vs. Connectivity
To provide a comprehensive analysis on how the properties of the previously identified water channels change in the different states of the OEC analyzed here, we computed the number of hydrogen bonds per water molecule in each channel (Figure 3). The observed number of hydrogen bonds per water is considerably smaller than the corresponding value in bulk conditions (~4). This is reasonable, considering that the water coordination is restricted inside the protein cavities in comparison with the bulk. Furthermore, several of the hydrogen bond interactions between water molecules are replaced by specific interactions with the amino acids near the OEC. On the other hand, it is clear that the number of hydrogen bonds between water molecules is larger in the narrow channel than in the large or broad cavities, except in the S1 state. This can be explained by the smaller mobility of the water molecules in the narrow channel (Figure 2). The more rigid cavity constrains the waters in positions where the hydrogen bonding is favored in comparison with the large or broad channels. Therefore, while the narrow channel could be more appropriate for proton transport, the large channel would be the most favorable for the diffusion of larger ions through the water network. With respect to changing S-states, the large channel is largely unaffected.
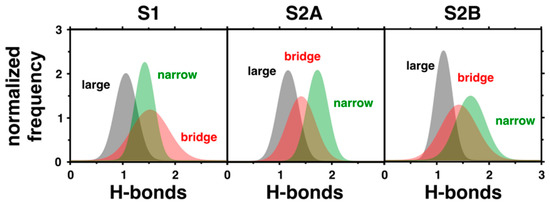
Figure 3.
Hydrogen bonds per water molecule in the three different water channels computed in states S1, S2A, and S2B. The presented histograms account for the normalized frequency of occurrence of different hydrogen bonds per water.
The narrow channel is the most sensitive, with the distribution of hydrogen bonds narrowing and increasing from the S1 to S2A states. In S2B, the mean similarly shifts upward and distribution noticeably broadens compared to the S2A state. Considering the placement of the narrow channel close to Mn4, it is reasonable that the cavity would be so affected by the state transitions since Mn4 is oxidized or changes its coordination in the S2A and S2B states, respectively. Additionally, the bridge channel has its most dramatic change in the S2A state, with its distribution of hydrogen bonds slightly narrowing and shifting downward, although this is nowhere near as dramatic as in the narrow channel. Again, this modification can be attributed to the changes to the OEC in this state, with the oxidation of Mn4, which the bridge channel runs alongside. It is important to note that the great mobility and connectivity between the waters in the intersection of the narrow and bridge channels (Figure 2) indicates that these channels are not isolated from one another and water or protons can flow between them.
Figure 4 (left panel) shows the total root mean square deviation (RMSD) of the atomic positions associated to the water molecules in the three channels. Similar to the RMSF per water, the RMSD accounts for the overall standard deviation of atomic displacements, reflecting the total mobility of the waters in the system. It is clear that water diffusion is more favorable in the large channel. This supports the hypothesis that the large channel is more suitable for the migration of larger ions. On the other hand, the normalized first eigenvalue of the correlation matrix (Figure 4, right panel) is a measure of the overall connectivity between waters in each channel. This value indicates that the bridge channel has the highest connectivity, with the exception of the S2A state where the narrow channel is more connected. This observation supports the idea that the broad channel is part of a proton exit pathway.
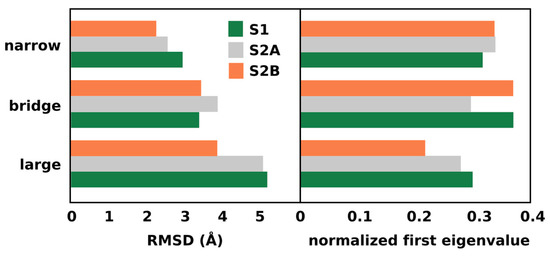
Figure 4.
Total root mean square deviation and normalized first eigenvalue of the water positions in the previously identified water channels.
3. Discussion
We have analyzed the dynamic networks established by interstitial waters adjacent to the OEC of photosystem II by using molecular dynamics simulations. We have identified three channels of highly diffusive and interconnected waters. One of these channels, named the narrow channel, is located in a rigid cavity where water movement is restricted and hydrogen bonding between waters is enhanced. Conversely, in the large channel, waters are much more mobile and hydrogen bonding between waters is more fluctuating. The bridge channel between the two, whose location matches well with the entry of the broad channel, has intermediate character between the other two channels. Therefore, while the structural and dynamical properties of the narrow channel, and the broad channel to a lesser extent, should be ideal to enhance Grotthuss-like proton diffusion, the large channel might be more appropriate for diffusion of larger ions.
Previously, both the narrow and large channels have been identified as possible candidates for supplying substrate waters to the OEC, specifically the WX molecule near O4 [16,17]. Generally, the narrow channel is favored by experiments [22], while the large channel has computational support [17]. Meanwhile, the broad channel has been proposed as a possible exit channel for proton release to the lumen during the Kok cycle [23]. Based on our analysis, the large channel should be ideal for transport of both water and larger ions. This could be key for the periodic reassembly of the OEC and for the delivery of chloride ions. The distant chloride ion, Cl-2, near D1–N184, is located very close to this channel, further indicating that this channel is very likely to be its source. However, the other chloride, Cl-1, near D2–K317, is closer to the broad channel and has no clear path to the large channel. Therefore, although the large channel is the best for delivering ions, it is more likely that the broad channel is the preferred pathway for Cl-1. The channel we have identified as the bridge is part of the broad channel and should be capable of facilitating such a transport.
The narrow channel has been shown by our analysis to be the most rigid of the three near-OEC channels. Though part of the broad channel has been put forward as the proton exit channel, our results suggest that the well-ordered narrow channel is the best pathway for protons. The increased rigidity of the narrow channel in the S2 state could explain the reduced occupancy of the water molecule next to WX in the narrow channel in the 1F (S2) structure observed by Jan Kern et al. [24] and Jimin Wang et al. [25]. If the water molecules are stabilized and unable to move in the S2, water may temporarily be unable to fill this small pocket in the narrow channel until its reappearance in the 3F (S0) structure. This missing water in the S2 state would create a gap in the chain of hydrogen-bonded water molecules in the narrow channel. If this channel is the proton exit path, this gap would be a barrier for exiting protons which would explain why a proton is not released in the S1→S2 transition. However, it was also shown in one structure (3ARC/5V2C) [26,27] that the narrow channel can be blocked by glycerol without apparent inhibition of oxygen evolution. Assuming the presence of this molecule is not an artifact of the experiment, then this channel cannot participate directly in water or proton transport. Instead, the narrow channel could simply be stabilizing the hydrophilic pocket near O4 that WX occupies before binding to the OEC in the S2→S3 transition.
Experimentally, it has been shown that depletion of chloride from PSII prevents the OEC from progressing past the S2 state, indicating that the proton exit pathway is blocked [28,29]. Mutation of D2–K317 to alanine eliminates the chloride dependency, showing that it is the formation of a salt bridge between D1–D61 and K317 that prevents proton release [30]. D61 is located at the junction between the narrow and broad channels, so the aspartate-lysine salt bridge could block either or both channels for a proton released from substrate water on Mn4. As far as the delivery of water molecules to the OEC, our analysis indicates that the broad and large channels are the best candidates, with the narrow channel displaying low water mobility. A recent computational study looked into how water rehydrates a dry PSII and found that the water molecules occupying the WX position near O4 originated from the broad channel, not the large [13]. If WX does enter through the broad channel, then the rigidity of the narrow channel would not rule out the carousel mechanism as the method by which new water molecules bind to the OEC. To summarize, our results indicate that the large channel is the most likely supplier of ions to the OEC cavity, with the exception of Cl-1, which is likely supplied by the broad channel. The narrow channel should be suitable for proton removal, with the broad channel being another possibility. The water mobility of the large and broad channels makes them almost definite suppliers of substrate waters to the OEC. Future analysis will be done using a larger model, such as a complete PSII, in order to determine how far the effects of these channels reach.
4. Materials and Methods
4.1. MD Simulations
Simulations were run using a sphere of residues and cofactors within 25 Å of the OEC. Chain cuts were capped with acetyl (ACE) and N-methylamide (NME) residues and these caps were frozen during simulations. Additionally, heavy atoms within water molecules and cofactors more than 15 Å away from the OEC were also frozen to their crystallographic coordinates. All hydrogen and heavy atoms within 15 Å were free to move. The calcium and manganese centers of the OEC were fit with the ESP charges using the Merz-Singh-Kollman (MK) method [31] in Gaussian09 [32]. ESP charges were fit to a sidechain and backbone model of the OEC and all its directly-bound ligands. This model was calculated with DFT/B3LYP [33,34] using the LanL2DZ [35] basis set and pseudo-potential for calcium and manganese atoms and 6-31(d) [36,37] for all other atoms. Ligand and µ-oxo bridge partial charges were calculated using the RESP [38] method with metal centers present but restrained to their ESP values. A RESP fit of the entire OEC was attempted, however, this resulted in near-zero metal charges and negatively-charged hydrogens. Holding the metal centers to ESP charges solved both of these issues. All metal-containing bonds and angles were held to their QM/MM optimized structure [7,8] with 500 kcal/mol/Å2 and 100 kcal/mol/rad2, respectively. These force constants were used in lieu of those calculated by the Seminario method [39], as the values estimated by the latter method were too weak (<100 kcal/mol/Å2) to hold the system together. The stronger force constants retained the complex structure without completely restraining it in space. The TIP3P force field [40] was used for all water molecules in the system. Bonds and partial charges were calculated for the S1 (III IV IV III), S2A (III IV IV IV), and S2B (IV IV IV III) states. Parameters for metal-containing cofactors were created using the MCPB.py [41,42] script and other cofactors using Antechamber, both available through the AmberTools package [43]. As per the MCPB protocol, metal centers and the atoms bound to them were assigned arbitrary atom types (e.g., M2 or Y6) for which parameters were created. The remaining ligand atoms were assigned their usual Amber atom type, as determined by their amino acid and atom names (e.g., Cγ in an aspartate has Amber atom type CO). Simulations were run in Sander for 100 ns under the Amber 14 [44] force field at 300 K after heating from 0 K for 20 ps, after which the variance of the temperature was less than 3 K, indicating achievement of a stable simulation temperature. Example files containing the charge and bonding parameters for the S1 state, as well as a sample simulation, can be found in the Supplementary Materials.
4.2. Eigenvector Centrality Distribution for Water Networks
To shed light on the collective organization of interstitial waters in PSII, we utilized the eigenvector centrality (EC) methodology recently developed in our group [45]. This technique has successfully identified allosteric pathways in protein residue networks. The EC of a node, , is defined as the sum of the centralities of all nodes that are connected to it by an edge, ,
where the edges are elements of the so-called adjacency matrix , and is the eigenvalue associated to the eigenvector composed by elements. In particular, our approach relies on assigning the functional dynamics to the major collective mode of the system (i.e., the first eigenvector of ), and hence in this context, is the highest eigenvalue of . There are several ways of defining an adjacency matrix, and for insight into the interconnectivity of a water network, the focus was put on the correlated motions of the system. Therefore, we utilized an adjacency matrix weighted by the dynamical correlations between protein residues, the elements of which can be quantified by the generalized correlation coefficient:
where are expressed in terms of the mutual information [21],
being , , and the marginal and joint Shannon entropy [46], for atomic displacements , obtained as ensemble averages over the MD simulation. The EC is a measure of how well connected a node is to other well-connected nodes in the network. In addition, the EC serves as a measure of the connectivity against a fixed scale when normalized, and hence, at variance with other centrality metrics, it can be used to reliably compare different networks.
5. Conclusions
Using a flexible model of the OEC, we conducted an EC analysis on the water channels surrounding the OEC in the S1 and S2 states. Due to its low mobility and high connectivity, we concluded that the narrow channel near O4, which had previously been proposed as a substrate water channel, is more suited to transporting protons rather than molecules such as water. The narrow channel is also the only channel to show significant change between S-states, becoming more rigid in the S2 state, possibly in preparation to shuttle away a proton in the S2→S3 transition. The large and broad channels are mobile enough to easily transport molecules and ions. Because of this and the lack of accessibility by the large channel to the Mn4 binding site, we propose the broad channel as a supplier of substrate water molecules.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/7/3/39/s1. Files to prepare a simulation of the S1 state of the OEC: PDB, mol2, and frcmod files, tLEaP and Sander input, as well as a sample 100 ns simulation (prmtop and mdcrd files).
Author Contributions
Formal analysis, U.N.M.; funding acquisition, V.S.B.; investigation, K.R. and A.T.G.; methodology, K.R.; project administration, V.S.B.; supervision, V.S.B.; visualization, U.N.M.; writing—original draft, K.R. and U.N.M.; writing—review and editing, A.T.G. and V.S.B.
Funding
This research was funded by the U.S. Department of Energy, grant number DESC0001423, with computational resources provided by the National Energy Research Scientific Computing Center.
Acknowledgments
We would like to thank Gary Brudvig and Jimin Wang for their valuable suggestions and input regarding this paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kok, B.; Forbush, B.; McGloin, M. Cooperation of charges in photosynthetic oxygen evolution—I. A linear four step mechanism. Photochem. Photobiol. 1970, 11, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Joliot, P.; Barbieri, G.; Chabaud, R. Model of the system II photochemical centers. Photochem. Photobiol. 1969, 10, 309–329. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Ames, W.; Cox, N.; Lubitz, W.; Neese, F. Two interconvertible structures that explain the spectroscopic properties of the oxygen-evolving complex of photosystem II in the S2 state. Angew. Chem. Int. 2012, 51, 9935–9940. [Google Scholar] [CrossRef] [PubMed]
- Dismukes, G.C.; Siderer, Y. Intermediates of a polynuclear manganese center involved in photosynthetic oxidation of water. Proc. Natl. Acad. Sci. USA 1981, 78, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L.; Sauer, K. EPR detection of a cryogenically photogenerated intermediate in photosynthetic oxygen evolution. Biochim. Biophys. Acta 1984, 767, 21–28. [Google Scholar] [CrossRef]
- Zimmermann, J.L.; Rutherford, A.W. Electron paramagnetic resonance properties of the S2 state of the oxygen-evolving complex of photosystem II. Biochemistry 1986, 25, 4609–4615. [Google Scholar] [CrossRef]
- Askerka, M.; Wang, J.; Vinyard, D.J.; Brudvig, G.W.; Batista, V.S. S3 state of the O2-evolving complex of photosystem II: Insights from QM/MM, EXAFS, and femtosecond X-ray diffraction. Biochemistry 2016, 55, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Vass, I.; Styring, S. pH-dependent charge equilibria between tyrosine-D and the S states in photosystem II: Estimation of relative midpoint redox potentials. Biochemistry 1991, 30, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Askerka, M.; Wang, J.; Brudvig, G.W.; Batista, V.S. Structural changes in the oxygen-evolving complex of photosystem II induced by the S1 to S2 transition: A combined XRD and QM/MM study. Biochemistry 2014, 53, 6860–6862. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Ota, K.; Shibuya, Y.; Noguchi, T. Role of a water network around the Mn4CaO5 cluster in photosynthetic water oxidation: A Fourier transform infrared spectroscopy and quantum mechanics/molecular mechanics calculation study. Biochemistry 2016, 55, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Retegan, M.; Pantazis, D.A. Differences in the active site of water oxidation among photosynthetic organisms. J. Am. Chem. Soc. 2017, 139, 14340–14343. [Google Scholar] [CrossRef] [PubMed]
- Vassiliev, S.; Zaraiskaya, T.; Bruce, D. Molecular dynamics simulations reveal highly permeable oxygen exit channels shared with water uptake channels in photosystem II. Biochim. Biophys. Acta 2013, 1827, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Sakashita, N.; Watanabe, H.C.; Ikeda, T.; Saito, K.; Ishikita, H. Origins of water molecules in the photosystem II crystal structure. Biochemistry 2017, 56, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Vassiliev, S.; Zaraiskaya, T.; Bruce, D. Exploring the energetics of water permeation in photosystem II by multiple steered molecular dynamics simulations. Biochim. Biophys. Acta 2012, 1817, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Ogata, K.; Yuki, T.; Hatakeyama, M.; Uchida, W.; Nakamura, S. All-atom molecular dynamics simulation of photosystem II embedded in thylakoid membrane. J. Am. Chem. Soc. 2013, 135, 15670–15673. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Askerka, M.; Brudvig, G.W.; Batista, V.S. Crystallographic data support the carousel mechanism of water supply to the oxygen-evolving complex of photosystem II. ACS Energy Lett. 2017, 2, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Ugur, I.; Rutherford, A.W.; Kaila, V.R. Redox-coupled substrate water reorganization in the active site of photosystem II—The role of calcium in substrate water delivery. Biochim. Biophys. Acta Bioenergy 2016, 1857, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Retegan, M.; Pantazis, D.A. Interaction of methanol with the oxygen-evolving complex: Atomistic models, channel identification, species dependence, and mechanistic implications. Chem. Sci. 2016, 7, 6463–6476. [Google Scholar] [CrossRef] [PubMed]
- Oyala, P.H.; Stich, T.A.; Debus, R.J.; Britt, R.D. Ammonia binds to the dangler manganese of the photosystem II oxygen-evolving complex. J. Am. Chem. Soc. 2015, 137, 8829–8837. [Google Scholar] [CrossRef] [PubMed]
- Askerka, M.; Vinyard, D.J.; Brudvig, G.W.; Batista, V.S. NH3 binding to the S2 state of the O2-evolving complex of photosystem II: Analogue to H2O binding during the S2→S3 transition. Biochemistry 2015, 54, 5783–5786. [Google Scholar] [CrossRef] [PubMed]
- Kraskov, A.; Stögbauer, H.; Grassberger, P. Estimating mutual information. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2004, 69, 066138. [Google Scholar] [CrossRef] [PubMed]
- Askerka, M.; Brudvig, G.W.; Batista, V.S. The O2-evoling complex of photosystem II: Recent insights from quantum mechanics/molecular mechanics (QM/MM), extended X-ray absorption fine structure (EXAFS), and femtosecond x-ray crystallography data. Acc. Chem. Res. 2017, 50, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ishikita, H.; Saenger, W.; Loll, B.; Biesaidka, J.; Knapp, E.W. Energetics of a possible proton exit pathway for water oxidation in photosystem II. Biochemistry 2006, 45, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Batista, V.S.; Brudvig, G.W. Occupancy of water molecules near the oxygen-evolving complex of photosystem II. Protein Sci. 2019. under review. [Google Scholar]
- Umena, Y.; Kawakami, K.; Shen, J.R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at resolution 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Wiwczar, J.M.; LaFountain, A.M.; Wang, J.M.; Frank, H.A.; Brudvig, G.W. Chlorophyll a with a farnesyl tail in thermophilic cyanobacteria. Photosyn. Res. 2017, 134, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Wincencjusz, H.; vanGorkom, H.J.; Yocum, C.F. The Photosynthetic Oxygen Evolving Complex Requires Chloride for its redox states S2→S3 and S3→S0 transition but not for S0→S1 or S1→S2 transitions. Biochemistry 1997, 36, 3663–3670. [Google Scholar] [CrossRef] [PubMed]
- Rivalta, I.; Amin, M.; Luber, S.; Vassiliev, S.; Pokhrel, R.; Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N.; Bruce, D.; et al. Structural/functional role of chloride in photosystem II. Biochemistry 2011, 50, 6312–6315. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, R.; Service, R.J.; Debus, R.J.; Brudvig, G.W. Mutation of lysine 317 in the D2 subunit of photosystem II alters chloride binding and proton transport. Biochemistry 2013, 52, 4758–4773. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comp. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian09 Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations potentials for the transition-metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–285. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of polarization functions on molecular-orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Seminario, J.M. Calculation of intramolecular force fields from second-derivative tensors. Int. J. Quantum Chem. 1996, 60, 1271–1277. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Fusti-Molnar, L.; Weaver, M.N.; Merz, K.M. Structural survey of zinc-containing proteins and development of the Zinc Amber Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Merz, K.M. MCPB.py: A Python based metal center parameter builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AmberTools18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Negre, C.F.A.; Morzan, U.N.; Hendrickson, H.P.; Pal, R.; Lisi, G.P.; Loria, J.P.; Rivalta, I.; Ho, J.; Batista, V.S. Eigenvector centrality for characterization of protein allosteric pathways. Proc. Natl. Acad. Sci. USA 2018, 115, 12201–12208. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bells Lab Tech. 1948, 27, 623–656. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).