Abstract
Dye-sensitized solar cells (DSSCs) have attracted a substantial interest in the last 30 years for the conversion of solar power to electricity. An important component is the redox mediator effecting the transport of charge between the photoelectrode and the dark counter electrode (CE). Among the possible mediators, metal coordination complexes play a prominent role and at present are incorporated in several types of devices with a power conversion efficiency exceeding 10%. The present review, after a brief introduction to the operation of DSSCs, discusses at first the requirements for a successful mediator. Subsequently, the properties of various classes of inorganic coordination complexes functioning as mediators relevant to DSSC operation are presented and the operational characteristics of DSSC devices analyzed. Particular emphasis is paid to the two main classes of efficient redox mediators, the coordination complexes of cobalt and copper; however other less efficient but promising classes of mediators, notably complexes of iron, nickel, manganese and vanadium, are also presented.
1. Introduction
Dye-sensitized solar cells (DSSCS) have attracted a lot of interest in the last 30 years for the conversion of solar energy to electrical energy. This development was based on earlier research on the dye sensitization of semiconductors, getting back to the late 1880s with the short publication of Moser [1] on dye sensitization of silver halides. Important fundamental research in dye sensitization was performed since 1960 by several research groups, notably by Gerischer and coworkers [2,3]. Research up to the early 1980s has been reviewed by Memming [4]. This research has been associated to some extent with photographic science and technology, as regards the dye sensitization of silver halides, with the essential difference that in photography the reaction needs to take place only once. Contrarily, for energy conversion, the continuous regeneration of the dye is needed. In this respect, several of these studies investigated regeneraton of the phootooxidized or photoreduced dye by a redox species, termed in the earlier literature as cosensitizer or supersensitizer, terms well-known in photography, and in the current dye solar cell literature as redox mediator, in the sense of mediating charge transport between a photoelectrode (PE) and another electrode, either a dark, not needing light for its operation, counter electrode (CE) in the more common type of electricity-producing DSSCs, or another PE. In the present article the term mediator will be the only one used for redox species regenerating the dye, irrespectively on whether it refers to a photoelectrode in a 3-electrode research-type cell or a two-electrode electrochemical photovoltaic cell.
Two developments in the semiconductor sensitization, relevant for the further development of the field, were the sensitization of compact ZnO pellets, produced by sintering porous ZnO at high temperatures and pressures, and the sensitization of single crystal TiO2 electrodes by surface attachment of a ruthenium complex in the absence of a redox mediator. In a series of papers, Tsubomura and associates in the 1970s and early 1980s described the sensitization of sintered ZnO pellets by organic dyes. The incident photon-to-electron efficiency (IPCE), defined as the fraction of incident photons leading to the generation of electric current in the external circuit, reached at best 22% [5], using rose bengal as dye and triiodide/iodide (I3−/I−) as mediator, as compared to more than 80% for contemporary systems. The highest solar-to-electrical power conversion efficiency (PCE) was 2.5% under irradiation with monochromatic green light [6], using rose bengal as dye and I3−/I− as redox mediator, as compared to the recently obtained PCE of 14% for polychromatic solar light. The second noteworthy development was the demonstration of effective attachment of ruthenium coordination complexes with pendant carboxyl groups achieved by Constable, Goodenough, Seddon, and associates [7,8].
An entirely new research front in the field of semiconductor photo-electrochemistry was opened up by the Grätzel group at the Swiss Federal Institute of Technology (EPFL) in Lausanne, which pioneered the study of light induced electron and energy transfer reactions in mesoscopic systems Thus Moser and Grätzel [9] investigated the sensitization of colloidal TiO2 by adsorbed dyes. In their study they demonstrated that the efficient injection of electrons from the photoexcited dye into the TiO2 conduction band is much faster, by orders of magnitude, than the recombination of the latter with the photooxidized dye. Their research laid the scientific basis for the discovery of today’s dye sensitized solar cells (DSCs) using arrays of TiO2 nanoparticles as photoanode. The development of mesoscopic DSCs passed through the use of high-surface area fractal TiO2 electrodes, TiO2 being a stable high-bandgap semiconductor with excellent long-term chemical stability upon irradiation, considered as a model oxide semiconductor for photoelectrochemical and photocatalytic studies. Desilvestro, et al. [10] demonstrated efficient sensitization with RuL3 (L = 2,2′-bipyridye-4,4′-dicarboxylic acid) with hydroquinone as mediator in a 3-electrode cell, with IPCE of 44%. In subsequent research Vlachopoulos et al. [11] obtained for the same dye an IPCE of 70% with iodide and 54% with bromide as mediators. The PCE of a regenerative cell with this type of PE, the bromide/bromine redox mediator and a Pt CE was 12% for monochromatic blue light. This cell was of the agitated beaker type, commonly used in photoelectrochemical research. These results initiated further research in dye-sensitized electrodes and thin-film DSSCs. A broader IPCE spectrum was obtained soon thereafter with dye RuL2(H2O)2 [12]. With the iodide/iodine redox mediator a PCE of 2% was obtained with white light of 700 W·m−2 intensity. The first DSSC patent was submitted by Grätzel and Liska in 1988 [13]. The efficient sensitization of TiO2 according to the Lausanne method was confirmed by Bard and collaborators in the USA [14].
A more efficient thin-layer DSSC version with a PCE reaching 8% was developed in the early 1990s as described in a Nature paper by O’Regan and Grätzel [15]. The DSSC was based on a Ru trimer dye and the I3−/I− redox mediator in ethanol. A novel feature of this system was the introduction of mesoporous colloidal TiO2 electrode, the preparation method of which is less time-consuming and more reproducible than that of the electrode substrates used before, so as to become the standard in DSSC research for the following years. The first DSSC with and a PCE reaching 10% was described in the 1993 publication by Nazeeruddin, et al. [16] with a RuL2(SCN)2 dye exhibiting a better spectral overlap with the visible part of the solar spectrum and the I−/I3− redox mediator in acetonitrile. Shortly thereafter, the efficient operation of cells prepared according to the same approach was independently confirmed by Hagfeldt, et al. [17].
At this initial DSSC research stage I3−/I− was appropriate as mediator, with several advantages. However, when efforts to improve the DSSC performance were undertaken, several drawbacks for this mediator became evident, as later described in this article, so that efforts toward the development of alternative other redox mediators were undertaken. Among these, the wide range of metal coordination complexes has been of primary interest.
Several reviews are dedicated to the physicochemical processes and operation of DSSCs [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44]. A comprehensive account of the status of the field up to the late 1990s is presented in the book by Kalyanasundaram [45].
2. Principles of DSSC Operation
The processes at a DSSC can be divided into two broad categories: useful processes, contributing to generation of photocurrent, and deleterious, limiting the current or the useful cell voltage and contributing to a decrease of the overall performance of the cell. An energy diagram related to the DSSC operation, where both useful and deleterious processes are indicated, is depicted on Figure 1.
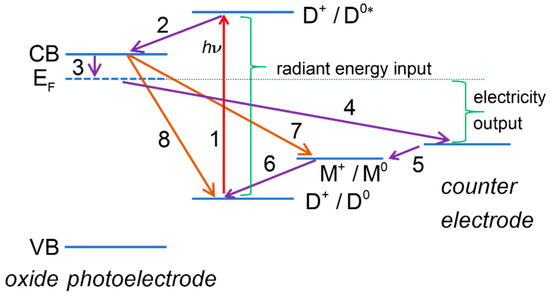
Figure 1.
Energy diagram of a dye-sensitized solar cell. Some of the useful processes: (1) dye photoexcitation; (2) electron injection into the mesoporous oxide semiconductor conduction band; (3) transfer of electrons from mesoporous oxide to transparent conducting oxide support; (4) transfer of electrons to the counter electrode (CE) through the external circuit; (5) electron transfer at the CE; (6) dye regeneration. Some of the deleterious processes: (7) recombination of mesoporous oxide electrons with redox mediator; (8) recombination of mesoporous oxide electrons with oxidized dye.
In this article, only the sensitization of n-type semiconductors (n-DSSCs) will be discussed. Most efficient DSSCs are based on a TiO2 substrate. Therefore, unless otherwise specified, this is the substrate of interest in the present review. Research on DSSCs based on p-type substrates (p-DSSCs), like NiO, and hybrid DSSCs will not be discussed here; their PCEs are inferior to 5% and the related research is more restricted than on DSSCs based on n-type electrodes. The reader is referred to the review by Odobel, et al. [46] and Wood, et al. [47] for further details. Furthermore, only electricity-generating regenerative DSSCs will be considered in this article. Photoelectrolytic cells based on dye-sensitized electrodes, including photoelectrosynthetic or photocatalytic, e.g., with O2 evolution or oxidation of an organic substrate at a dye-sensitized photoanode, despite their considerable interest in fundamental research, will not be included; up to the present time. The energy conversion efficiency of these systems has been considerably lower than these of regenerative DSSCs or of photoelectrolysis systems based on direct semiconductor excitation.
2.1. Useful Processes
2.1.1. Dye Photoexcitation
Light absorption promotes an electron from a higher occupied orbital (HOMO) to a lower unoccupied orbital (LUMO), expressed as
where D0 is the initial form of dye and D0* is the photoexcited state. Light absorption promotes an electron from a higher occupied orbital (HOMO) to a lower unoccupied orbital (LUMO).
The light-harvesting efficiency (LHE), defined as the fraction of incident photons absorbed by the dye, is expressed for incident monochromatic light as
where Rλ is the fraction of reflected light, depending on wavelength λ, Г is the surface molar concentration of the dye (based on the geometric surface area), L is the Avogadro constant (6.022 × 1023 mol−1), and σλ is the attenuation cross section for light absorption by the dye molecules. σλ is related to the molar extinction (attenuation) coefficient ελ of the dye as
The SI units of ελ are m2·mol−1. In terms of the usual molar extinction coefficient units of L·mol−1·cm−1 for ελ, with concentrations expressed in mol per liter (mol·L−1) units, the above equation can be rearranged as
Alternatively, the LHE can be expressed as a function of the volume dye molar concentration c in the electrode layer and the electrode thickness l as following:
where surface and volume concentrations within the oxide layer are related as
Equation (5) is based on the Beer–Lambert law for light absorption by molecules; the underlying theory for which, including the relation between σλ and ελ is discussed in the article by Strong [48] and in the textbook of Skoog and Leary [49].
The surface concentration is related to the molecular dimensions of the dye and to the volume-based specific area SV of the oxide material (the real area which is available for dye adsorption per volume unit).
Consider an oxide electrode in the form of sintered spherical particles of radius r. In this case SV for a single particle is equal to
SV is expressed for the porous material as
where is the porosity of the material, so that for an electrode of thickness l the roughness factor SA i.e., the ratio of real-to-geometrical area, is expressed as
For a typical DSSC electrode of l = 5 μm and εp = 0.5 composed of sintered particles of r = 10 nm the above formula gives SA = 750.
Consider a dye with cross-section σ per molecule upon adsorption on the electrode. In this case, for a flat monolayer (SA = 1) Г is expressed as
For a typical value of σ = 1 nm2 (order of magnitude of area occupied by a typical dye molecule) it is Г = 1.66 × 10−6 mol·m−2. For an electrode of roughness factor SA, it is
or
Alternatively, by combining Equations (12) and (9)
so that on the basis of Equations (13) and (6) the dye volume concentration is expressed as
or, from Equations (14) and (9)
Calculation example:
The surface concentration of a polycrystalline electrode can be measured experimentally by desorbing the dye in an appropriate medium, usually an aqueous or organic alkaline solution, and measuring the UV-VIS spectrum after complete desorption.
Consider a typical charge-transfer metal-free organic dye, e.g., Y123 (discussed in Section 9) with ε = 4.8 × 10−4 M−1·cm−1 at the maximum of the UV-VIS light absorption spectrum. For an electrode of l = 10 μm it is LHE ≈ 1. Contrarily, for a flat electrode (SA = 1) and the same dye, if l = 1 nm (the dye length) is introduced to the above formula, it is c = 1.66 M and LHE = 0.22. This example demonstrates the key importance of optimizing the surface properties of the oxide layer for efficient DSSC operation.
The LHE analysis of this section is useful when comparing the light absorption properties of adsorbed dyes to that of redox mediators in solution, a parasitic process discussed in Section 4.5.
The present analysis is rather simplified, since it neglects the scattering effect of the mesoporous dye-absorbing solid. However, it provides a qualitative insight of the importance of the solid surface structure in achieving efficient light harvesting.
2.1.2. Electron Injection into the Conduction Band of the Semiconductor Oxide
After photoexcitation, electrons are injected from higher unoccupied electron states to the semiconductor and finally relax to the bottom of the conduction band (CB) of the semiconductor oxide. The oxidative quenching mechanism is assumed here, implying that the excited dye undergoes oxidation after photoexcitation.
where D+ is the oxidized state and SC indicates the mesoporous semiconductor phase. The superscript + denotes a net positive charge with respect to the initial form (superscript 0); + and 0 do not necessarily denote the charge of the respective species.
2.1.3. Electron Collection
Following electron injection into the CB of the semiconductor, electrons diffuse through the mesoporous semiconductor layer toward the contact phase (CTSC) of the mesoporous oxide, usually a transparent conducting oxide (TCO) layer deposited on glass or plastic, such as the commonly used F-doped tin oxide (FTO).
Similarly, CTSC can be a metal, as was frequently the case in earlier research before 1990, where TiO2 was deposited on a Ti substrate, or more recently for applications where the mesoporous oxide needs to be deposited on a flexible metal substrate, like Ti or stainless steel foil. In the latter case, the light fist impinges on a transparent, usually TCO-coated, CE.
2.1.4. Dye Regeneration
Electrons are injected from the reduced form M0 of a redox mediator M, at the proximity of the photoelectrode (PE), into D+, so that D0 is regenerated and consequently, M+ is produced. For this process the term hole injection, from D+ to M0, is frequently encountered. The superscripts 0 and + denote relative charges as before.
The overall reaction at the PE is
with the term hv above the arrow denoting the fact that the above reaction takes place under illumination and does not imply absorption of light by the mediator.
The thermodynamic driving force for dye regeneration, equal to the negative of the standard Gibbs free energy ∆GӨREG for this process, is equal to the difference between the redox Fermi level EF of the redox mediator and that of the dye. If all species are in their respective thermodynamic standard states (EFӨ) with all components at unit activities, the Gibbs free energy is
In terms of standard redox potentials
where Q0 is the magnitude of the electron charge (1.602 × 10−19 C). It is possible for both EFӨ and EӨ to be defined versus the same reference system, preferably the redox couple ferrocenium–ferrocene (Fc+/Fc), particularly for nonaqueous DSSC electrolytes. For aqueous redox systems there is a preference for the H+/H2 system (standard hydrogen electrode, SHE). In both cases, the reference energy level for measuring EF values is chosen as the solution Fermi level of electrons equilibrating with the redox system of choice. Alternatively, there is an option of reporting EF values versus vacuum, with the International Union of Pure and Applied Chemistry (IUPAC) recommending the value of −4.45 eV for EFӨ (H+/H2) [50], corresponding to the standard hydrogen electrode (SHE), versus vacuum. Additional explanations on redox potential scales and reference systems are provided in Section 5.
For the dye regeneration reaction, the requirement is
with EFӨ and EӨ being related as
where EF(REF) is the Fermi level of the reference redox system versus vacuum. The components of this redox system can be either in the standard state, as in the standard hydrogen electrode (SHE), or not, as in the saturated calomel and saturated Ag/AgCl Electrode. More details on this equation are provided in Section 5.
2.1.5. Oxidized Mediator Transport from Photoelectrode to CE
After dye regeneration by the mediator, the oxidized species M+ diffuses from the PE to the CE:
2.1.6. Electron Flow through the External Circuit
Electrons are directed from the PE substrate to the CE; during this step electrical energy (I2·REXT) is generated with I as the current and REXT the external resistance:
2.1.7. Electron Transfer at the Counter Electrode
A heterogeneous (electrochemical) electron transfer takes place from CE to the M+ in solution. The CE consists of a substrate with metal-like conductivity, which is either electrocatalytic active itself toward M0 or is coated by an appropriate electrocatalyst (e.g., Pt on FTO).
2.1.8. Reduced Mediator Transport from Counter Electrode to Photoelectrode
The M0 species produced by reduction of M+ at the CE back diffuses to the PE
in order to participate to the oxidized dye regeneration process.
2.2. Deleterious Processes
2.2.1. Deactivation of Excited Dye D0* with Evolution of Heat or Light
The excited dye can either inject an electron into the semiconductor oxide conduction band or be deactivated to the initial dye form together with the emission of light (fluorescence) or heat:
For efficient DSSC operation the latter process should be much slower than electron injection.
2.2.2. Deactivation Excited State by Reaction with a Species in Solution
The species Q causing deactivation can be M0, M+, or another species in the electrolyte:
This type of deactivation is often called quenching. The injection efficiency φINJ of electrons from the photooxidized dye to the SC is expressed as
where kINJ is the first-order rate constant for electron injection, kDEA is the first-order constant of nonradiative or radiative deactivation, and kQ is the second-order quenching constant with a species of concentration CQ.
2.2.3. Recombination Reactions of Semiconductor Electrons
SC electrons can recombine either with the oxidized dye or the oxidized redox mediator. These reactions are closely related to the regeneration reaction:
In fact, if the latter is very fast, the surface concentration of the oxidized dye is so low that recombination with the oxidized mediator predominates.
The competition between electron transport and recombination in the SC layer determines the collection efficiency φCOLL,
where ktr and krec(SC) are the rate constants for transport and recombination, respectively. ktr is a first-order and krec(SC) a pseudo first-order constant (incorporating the concentration of the species, dye or mediator, with which the CB electrons recombine) with respect to electron concentration. If the potential ε(PE) of the dye-coated PE measured versus a reference electrode is sufficiently positive, then krec(SC) is much lower than ktr so that φCOLL is virtually unity.
The above equation is often expressed in terms of the time constants of electron transport and collection [51]
as
If transport process is much faster than recombination
then the last equation can be approximated as [52]
The driving force for the recombination between conduction band electrons and oxidized dye or oxidized redox mediator is equal to the negative of the Gibbs free energy ∆GӨREG of this process. The latter is expressed under standard conditions for the dye or the mediator as
with EF(SC) the (quasi-)Fermi level of injected electrons in the mesoporous semiconductor and both EFӨ and EC being measured versus the same reference level, either the EF of a solution-based redox system or the vacuum level, as already discussed.
Under the conditions of normal DSSC operation the semiconductor conduction band edge is the upper boundary (limit) of EF(SC) so that
In terms of standard redox potentials,
with ECB reported versus the same reference redox system as EӨ.
The above expressions should be compared to Equation (22) for the regeneration driving force stated in the previous section.
This means that, on the one hand, for a more positive standard redox potential of the redox mediator the free energy for the regeneration becomes less negative, resulting to less lost energy for this process; in the absence of recombination reaction, this would imply a larger open-circuit voltage UOC for the solar cell, as explained later. On the other hand, the dark recombination reaction is more favorable, contributing to a larger dark reduction current.
2.2.4. Recombination of TCO Contact Electrons with the Oxidized Dye or the Oxidized Redox Mediator
These reactions are analogous to these described above for semiconductor electrons:
The related driving force is expressed as
Reactions between and dye are of particular significance in the case of the possibility of propagation of the dye reduction by lateral electron transfer, also known as electron hopping [53], depending on the particular dye structure. The D+ molecules at close proximity to the contact, e.g., the TCO substrate, can be reduced by TCO electrons. The TCO layer can adsorb to a small extent, due to its very low roughness factor, dye molecules, which can be reduced by TCO electrons. Subsequently, they can be reoxidized by injecting an electron into (receiving a hole from) an adjacent TiO2-located D+ molecule, which lies a little farther away from the TCO contact, and so on, so that electron hopping can progress far away from the TCO contact.
3. Incident Photon-To-Current Efficiency and Power Conversion Efficiency
If Jph is the photon number flux of incident monochromatic light and Je is the electron flux from the PE toward the CE, then incident photon-to-current efficiency IPCE is defined as
Frequently IPCE is expressed as a percentage of incident photons, as it is the case in the present article, with respect to discussions of experimental results.
The electron particle flux Je is expressed in terms of the current density j as
For monochromatic light, Jph is expressed as the ratio of the incident light intensity P0 to the energy of one mole of photons:
where h is the Planck constant (6.62607 × 10−34 J·s), c is the speed of light (2.99792 × 108 m·s−1) and λ is the wavelength. Therefore, by combining the last two equations, IPCE is derived as
By introducing the values of the various constants, the following useful formula for IPCE calculations is obtained:
The incident photon-to-current conversion efficiency (IPCE%) is expressed as the product of the light-harvesting efficiency (LHE), the electron injection efficiency (φINJ, 0 ≤ φINJ ≤ 1) from the excited dye into the conduction band, and the electron collection efficiency (φCOLL, 0 ≤ φCOLL ≤ 1) at the oxide/conductive support interface as
The absorbed light-to-current efficiency APCE is expressed as
so that
In the case of smooth semiconductor electrodes, like single-crystal TiO2, APCE can be close to unity but LHE is very low, quite often below 0.01, and so is IPCE.
For a sufficiently positive electrode potential (measured versus a reference electrode) of the PE
The solar to electrical power conversion efficiency (PCE) is determined as
where jMPP and UMPP are the current density and the cell voltage at the maximum power point, and PIN is the incident light intensity. For laboratory measurements, simulated Air Mass 1.5 G solar light is generally used. This irradiation is described in the present article as “full sun”. Light attenuated by a neutral density filter will be described, for example, as 50% sun (1/2 sun) or 10% sun (1/10 sun).
For efficient solar power conversion jMPP and UMPP should be as close to the short-circuit current jSC and open-circuit voltage UOC as possible. Because of ohmic and kinetic energy losses at the electrodes and the electrolyte, jMPP and UMPP deviate from jSC and UOC. In this respect, the fill factor (FF), defined as
is below unity. For an efficient cell a FF in the 0.7–0.8 range is expected, with higher values expected at lower light intensities.
If a specific redox mediator is considered, the upper limit of UOC is determined by the difference between its Fermi level and the semiconductor band edge:
For a specific dye, with a mediator of equal Fermi level with that of the dye,
The condition EFӨ(D+/D0) = EFӨ(M+/M0) corresponds to an extreme case in which no driving force is available for the regeneration reaction and, therefore, efficient DSSC operation, is not expected in practice. In fact, redox mediators with
should be excluded for consideration, and mediators with
should be systematically investigated for a chosen dye.
4. Requirements for Efficient Redox Mediators
4.1. Redox Potential Less Positive but Close to That of the Dye
This point has been previously discussed. The minimal driving force needed for regeneration is estimated at around 0.1 eV. A more positive redox potential means that there is less energy lost due to the regeneration, so that a larger photovoltage would be expected. On the other hand, the recombination reactions with electrons, either from the semiconductor or from the contact, are more favorable, resulting to larger dark currents, to the detriment of the photovoltage.
4.2. Long-Term Stability of Both the Reduced and the Oxidized Form
The reduced and oxidized form of the redox mediator should be stable both in the dark and under illumination, during the expected operational time of the DSSC, toward self-decomposition, reactions with the electrolyte components, and, in case of water and oxygen-sensitive species, reactions with these species leaking through the seal from the environment into the electrolyte.
4.3. Solubility
It is expected that solubility should be lower for larger redox-active species. Therefore, small-size species, like triiodide/iodide (I3−/I−), are expected to be preferable in this respect as compared to bulkier species, e.g., Co or Cu-based coordination complexes. In determining the maximal solubility of a species, the overall electrolyte composition should be considered.
4.4. Fast Diffusion
The diffusion coefficient D is related to the size of the mediator and to the properties of the solvent. For dilute solutions, the following equation is often applicable:
where η is the viscosity, r corresponds to the molecular radius, including both the crystallographic radius and the thickness of the solvation shell, with the species assumed to be of spherical shape of radius r, kB is the Boltzmann constant, and T is the absolute temperature. D, being a function of the concentration, can decrease upon concentration increase. However, in addition to molecular diffusion, an additional transport mechanism, electron or atom hopping between reduced and oxidized mediator in solution, should be taken in consideration, especially for mediator concentrations above 0.1 M. In the case of electrolytes containing I3−/I− iodine atom hopping between I− and I3− contributes to charge transport. For metal coordination complexes, electron hopping is the additional transport mechanism. As a result the apparent diffusion coefficient DAPP is higher than D, often called physical diffusion coefficient DPHYS, according to the Dahms–Ruf equation [35,54,55]:
where kEX is the bimolecular rate constant for electron or atom self-exchange, C is the mediator concentration, and δ is the distance between the mediator molecules.
4.5. Low Light Absorption
The electrolyte penetrates into the mesoporous oxide layer. Therefore, absorption of light in the electrolyte means in principle that less light is harvested by the dye. However, since both the dye and the light-absorbing mediator are present in the cell, the important aspect is the competition between the dye and the mediator for light absorption.
For I3−/I− light absorption by I3− occurs in the range 300–500 nm, with two maxima in the ελ versus λ spectrum below 400 nm. For several coordination complexes, like Cu, light absorption is not insignificant, but much lower compared to that attributed to the dye.
Consider redox mediator 1 and dye 2 present in the oxide at concentrations C1 and C2. For the mediator, C1 is related to the concentration in the stock solution and the oxide porosity by
The amount of absorbed light energy Pabs is expressed as
or, in terms of molecular cross sections,
where Iabs is the sum of the amounts Iabs(1) and Iabs(2) absorbed by 1 and 2.
In order to derive the amounts Iabs(1) and Iabs(2), a differential section dl′ is considered in a position l′ < l inside the light-absorbing medium. The absorption of light within dl′ by 1 and 2 is expressed by the relations
where Il′ is the light intensity at l′. Therefore,
so that, at the exit point of the beam (l = l′) after integration the following relation is derived after integration in the interval 0 ≤ l′ ≤ l:
Finally, the fraction Pabs(1)/Pabs(2) of the total absorbed light which is captured by the dissolved mediator is
Correspondingly, the fraction of the incident light absorbed by the mediator (absorptance) is
These equations will be applied in the case of the Cu2+/+bis(2,9-dimethyl-1,10-phenanthroline) (Cu2+/+(dmp2) redox mediator. Cu2+/+(dmp2) is virtually colorless in the visible part of the spectrum. The light absorption will be compared for dye Y123 and Cu+(dmp2) at the maximum of the dye absorption spectrum, at 430 nm. The spectral properties of Cu+(dmp2) have been reported by Saygili, et al. [56]. The concentration of dye in the oxide layer has already been derived in Section 2.1.1. Therefore, by introducing the following variables
the absorptance for the mediator and the dye is
indicating virtually complete light absorption within the solar cell, with less than 1% of light intensity absorbed by the mediator in solution.
If only the mediator is present in the TiO2 layer
so that the unjustified impression is created that the redox mediator is not suitable for efficient DSSCs.
In the absence of the mediator,
Therefore, any considerations about the unsuitability of redox mediators because of light absorption of their solutions should be evaluated with respect to a comparison with the dye spectrum.
The above calculation is preliminary, for precise evaluations the complete spectra of dye and mediator in conjunction with the solar spectrum should be considered. However, such calculations are beyond the scope of the present review.
4.6. Electrode Kinetics at the Counter Electrode
For several redox mediators, the catalytic properties of the CE substrate are of outmost importance, in particular in the case of complex, multi-electron, reactions, like the reduction of I3− to I−, or even for apparently simple one-electron reactions but in fact involving substantial molecular rearrangement between the oxidized and the reduced form. In this respect, relatively expensive CE catalysts, like Pt and Au, should be avoided as much as possible, to the benefit of materials based on earth-abundant elements, including various types of carbon, or conducting polymers. Slow kinetics at the CE would be detrimental to its application in DSSCs, in particular for outdoor applications under light intensity of the order of 100–1000 W·m−2.
4.7. Advantages and Drawbacks of Iodide–Triiodide
This redox mediator has several advantages, which make it still the favorite for DSSC applied research and development as well as a benchmark against which other mediators have to be compared. The small molecular size enables it to be soluble to a wide range of solvents, sometimes at concentrations exceeding 1 M, and to exhibit fast diffusion, aided, at higher concentrations, by iodine atom hopping, as already mentioned. In general, it has a good long-term stability in several solvents. The cost of this mediator is in principle lower to that of several inorganic coordination complexes, depending on the type of the accompanying cation in the iodide salt. For all these reasons, mainstream DSSCs were based on I−/I3− in nonaqueous solvents until recently. The actual PCE record for an iodide-based cell according to Yu, et al. (2010) [57] is 11.7% for full sun (UOP = 0.758 V, jSC = 19.78 mA·cm−2), 12.8% for 1/2 sun (PIN = 532.6 W·m−2, UOP = 0.740 V, jSC = 10.63 mA·cm−2) and 12.1% for 1/4 sun (PIN = 261.4 W·m−2, UOP = 0.721 V, jSC = 5.26 mA·cm−2), with the Ru-dye C106 and an acetonitrile-based electrolyte [58]. Marszalek, et al. (2014) [59] demonstrated excellent stability of DSSCs at 60 °C (2320 h, 92% of the initial PCE = 8% maintained) and 80 °C (1065 h, 80% of the initial PCE = 8% maintained) in an electrolyte based on the addition of a non-volatile solvent into a mixture of ionic liquids.
The main drawback of I3−/I− is the substantial driving force for dye regeneration or, in other terms, the large, by around 0.5V or more, difference between the redox potential (or Fermi level)− and that of the dye. Such a driving force cannot be easily avoided due to the fact that the 2-electron oxidation of I− to I3− is a kinetically complex process, comprising several coupled electrochemical and chemical steps [60,61,62]. As a result, the redox system involved to the electron transfer reaction with the dye, for example I·/I−, with both iodide ion and iodine atom possibly adsorbed so that the redox potential may be different from that of the same species in solution, can have a more positive redox potential, closer to that of the dye, than I3−/I−. The dye regeneration driving force is expected to be lower for redox mediators, especially one-electron metal coordination-based redox systems, so that larger photovoltage values are attainable for the DSSC with alternative mediators.
Other disadvantages are the substantial light absorption of triiodide in the near visible part, 400–500 nm, of the solar spectrum, with the maximum in the ultraviolet, so that the highest tolerable triiodide concentration could not exceed 50–100 mM.
Another disadvantage is the corrosivity of I3−/I− toward several metals of interest as components of the DSSC electrodes, used as electrocatalysts, conducting supports as alternatives to TCO-coated transparent materials, or for the fabrication of current distribution grids in the case of large-area DSSC electrodes [63,64,65].
Finally, I3− is in equilibrium with I− and volatile I2, with the equilibrium concentration of the latter very low but non-negligible in nonaqueous solvents. Therefore, the long-term possibility of iodine diffusing out of the electrolyte, with altered redox mediator concentrations as a result, cannot be overlooked.
The aforementioned problems related to the I3−/I− -containing electrolytes as well as an increase of the UOC in DSSC can be achieved by replacing iodide with transition metal complexes, since ligand engineering offers the advantage of fine-tuning not only the redox potential, but also the physical and chemical properties of the complexes.
The use of outer-sphere redox couples is greatly attractive for DSSC applications as they exhibit fast electron exchanging processes without structural changes (cleavage or reformation of chemical bonds), or formation of intermediate radical species (contrary to the I3−/I− pair), which leads to small reorganization energies so that, in turn, kinetics of electron transfer is mainly determined by solvent reorganization [66]. On the other hand, one of the major disadvantages presented by coordination complexes as redox mediators in DSSC is the high recombination rate of the electrons in the TiO2 conduction band to the redox species in the electrolyte, resulting in low photovoltage and photocurrent with poor overall performance [67]. Development of redox shuttles with coordination complexes should involve high redox potentials (so as to achieve high UOC), fast electron exchange, high stability, and also high steric impediment in order to decrease recombination rates [68]. Following this strategy, at first cobalt complexes and subsequently Cu complexes were introduced as excellent alternatives to I3−/I− in DSSC electrolytes.
5. Redox Potential Definitions and Measurements
The standard electrode potential EӨ is determined from the Nernst equation on the basis of the activities of the redox species and . The electrode potential ε versus a reference electrode [69] is expressed for the redox reaction
as
At first approximation, the activity of a species in solution is equal to the ratio of the concentration to the standard concentration. However, for exact calculations the activity coefficient has to be taken into account. In the case of aqueous solution standard redox potentials have been determined on an elaborate way, by constructing electrochemical cells with low concentrations and extrapolating, on the basis of the Debye–Hückel law, to unit activity. Such measurements have been rarely performed for redox couples in non-aqueous solutions.
In fact, a related quantity, the formal redox potential EӨ′, is easier to measure, in particular by voltammetric techniques (for example cyclic, rotating disk or microelectrode voltammetry) in an excess of supporting electrolytes. In that case, the activity coefficients γ are constant if the concentration of the redox couple of interest is lower than that of the supporting electrolyte, and can be incorporated into the formal redox potential; therefore, in this case the Nernst equation can be accurately used in terms of concentrations. From aqueous electrochemistry, it has been established that standard and formal redox potential are different, the latter being dependent on the supporting electrolyte.
However, in photoelectrochemical studies the equality εӨ′ = εӨ is tacitly assumed, and this will be also the case in the present article.
In voltammetric studies, EӨ′ is derived from the half-wave potential ε1/2. The latter is defined as
where DRed, DOx are the diffusion coefficients, and the coefficient δ depends on the particular method, e.g., δ = 1/2 for cyclic voltammetry at semi-microelectrodes, 2/3 for rotating disk voltammetry, and 1 for microelectrode voltammetry. If DOx and DRed are determined separately, for example from the forward peak current in cyclic voltammetry (CV) at stationary semi-microelectrodes (smaller dimension 0.1 mm–1 cm), in the case of extremely fast electrode kinetics (electrochemically reversible or Nernstian systems), or from the mass-transfer limiting current in rotating disk voltammetry (RDEV) at semi-microelectrodes) or microelectrode voltammetry (MEV), then EӨ′ is directly derived from E1/2.
In fact, for many species DOx and DRed are not much different, and the logarithm of their ratio is close to 0 so that, within a very good approximation it can be assumed that EӨ′ and E1/2 are equal. This is a much better approximation than that of the equality between EӨ and EӨ′.
The difference between chemical and electrochemically reversibility should be considered. A chemically reversible redox mediator in the reduced (oxidized) form can be completely converted to the oxidized (reduced) form by electrolysis if the applied potential is kept sufficiently positive (negative) with respect to EӨ, without side reactions, e.g., decomposition or reactions with the solvent or various electrolyte components. An electrochemically reversible (Nernstian) system is one with extremely fast kinetics, as stated above. For example, in a fast electrochemical experiment, for example a CV measurement at a microelectrode at 1000 V/s, a system can be seen as chemically reversible but as electrochemically quasi-reversible (moderately fast kinetics) or even irreversible (slow kinetics). Contrarily, in a slow experiment, e.g., a CV measurement at 1 mV/s, the system can behave as electrochemically reversible but chemically irreversible, with decomposition of the product of the electrochemical reaction evident. Such considerations are important in investigations of novel redox mediators.
For Nernstian systems E1/2 is determined by CV at stationary semi-microelectrodes as the average of the forward and reverse peak potentials (Ep,f and Ep,r, respectively),
The determination of E1/2 for a Nernstian system is also possible by RDEV or MEV as the mid-potential of the current-potential curve, i.e., at the potential at which the current is the half of the limiting, mass transport-determined value.
The electrochemical reversibility can be ascertained from the shape of the voltammograms. In cyclic voltammetry the criterion is
at 25 °C if the potential Erev at which the scan direction is reversed is more than 300 mV away from Ep,f. For n|Erev − Ep,f| lower than 0.30 V, n|Ep,f − Ep,r| is slightly larger:
For rotating disk and microelectrode voltammetry the electrochemical reversibility (Tomeš criterion) is ascertained by measuring the difference of the potentials corresponding to the current being ¼ and ¾ of the limiting current (E1/4, E3/4). In this respect, at 25 °C it is
If both the oxidized and the reduced form are present in solution at comparable amounts, RDEV and MEV are preferable to CV; the latter requires a zero baseline, achieved if only one of Ox and Red are present in solution, for the accurate determination of E1/2. In this case the baseline for determination of E1/2 or the application of the Tomeš criterion is the line for either the oxidation or the reduction limiting current.
In the case of electrochemically quasi-reversible reactions, the validity of Equation (77) for E1/2 above and, as a result, the evaluation of the redox potential by CV RDEV or MEV, is within reasonable approximation if the peak separation or the difference between E3/4 and E1/4 does not significantly exceed the value for Nernstian reactions, i.e., n|Ep,f − Ep,r| or n|E3/4 − E1/4| is lower than 0.1 V. More details about electrochemical principles and methodology and their application to electrode reactions of species in solution are provided in several textbooks [69,70,71,72].
With respect to the reference electrode for nonaqueous electrochemistry, it has been recommended by IUPAC that the redox potential should be reported versus the Fc+/FC redox system as reference [73]. However, several researchers measure redox potential versus ferrocene and report this versus the aqueous SHE, by using a conversion factor from the literature. The problem is that: (a) this conversion factor is not always mentioned in the publications reporting nonaqueous redox potentials versus Fc+/FC and (b) the difference between the redox potential of Fc+/FC in a non-aqueous solvent and (aqueous) SHE cannot be measured experimentally but only calculated by combining experimental data and theoretical calculations. In fact, there is a large discrepancy in the literature as regards the value of Fc+/FC versus SHE, from 0.34 V to 0.67 V as discussed in some detail by Pavlishchuk and Addison (2000) [74]. For this reason, a detailed quantitative comparison of redox potentials of dyes and redox mediators is not included in the present review.
At this point, some remarks regarding the voltammetric determination of redox potentials in nonaqueous solutions are appropriate. The reference or, quite often, quasi-reference electrode (RE, QRE), should be chosen carefully. The QRE consists of a metal wire (e.g., Pt or Ag), often coated with an insoluble film (e.g., oxide or chloride) and dipped into an inert electrolyte. The metal-electrolyte interfacial voltage of this system is not thermodynamically defined, but can be stable, depending on the experimental conditions, for the time needed to conduct the experiment, or even longer. Such a wire can be dipped directly into the working electrode (WE) solution, as often suggested in the electrochemical literature. However, its interfacial voltage can in same cases exhibit significant fluctuations even at the short term; in particular, it can be influenced by fluctuations of the measuring solution composition during the experiment.
Therefore, a QRE prepared and kept in a sealed bridge tube is a better option, as proposed in a recent publication of Alan Bond and collaborators [75]. They propose a Ag/AgCl QRE in acetonitrile in a supporting electrolyte not containing chloride anions. This electrode reaches a stable potential in a period of a few hours after assembling, and can be used for several successive experiments. Its stability can be ascertained by cyclic voltammetry of Fc+/Fc. An alternative is the non-aqueous Ag/soluble Ag+ salt RE, in the same solvent as that in the WE compartment (e.g., Ag/0.01 M AgNO3 in acetonitrile). However, due care should be taken in order to prevent Ag+ ions leaking into the WE compartment. In this case a double-bridge electrode can be used, with the RE compartment separated from the WE compartment by an intermediate compartment containing an inert electrolyte. This way the possibility of contamination by Ag+ would be lessened. However, a drawback is that the increased resistance of the two separators (e.g., glass frits) between the WE and the RE may cause excessive noise during the measurements.
An often used alternative in conjunction with a bridge configuration is a reference electrode in a different solvent than the one in the WE compartment, for example Ag/AgCl/saturated KCl in water or Ag/AgCl/ LiCl in ethanol. In the former case, precipitation of KCl at the aqueous–nonaqueous junction is a problem, so that KCl may be replaced by chloride salt more soluble in the organic medium, for example NaCl or LiCl. If leaking of water has to be rigorously excluded, the ethanolic variant is preferred. However, a fundamental problem with these electrodes is the presence of a junction potential between two different solvents, which cannot be calculated by thermodynamic methods and, in addition, depends on the details of the reference electrode construction. As a result, these electrodes are in practice QREs, despite the fact that their operation is based on a reversible reaction. Therefore, in all cases the operational reference electrode, even an Ag/soluble Ag+ electrode in the same solvent, should be calibrated with an internal voltammetric standard, preferably with ferrocene.
Initially the case of determination of the ε1/2 of a species in the reduced state by stationary cyclic voltammetry will be considered, with Fc+/Fc as the voltammetric standard. At first, a cyclic voltammogram of the species of interest (M+/M0) is recorded versus the operational RE or QRE without ferrocene Fc+/Fc. Then a small amount of Fc is added and the cyclic voltammogram of the latter is recorded. The difference E1/2(M+/M0) − E1/2(Fc+/Fc) is the quantity of interest. If E1/2 (M+/M0) and ε1/2(Fc+/Fc) do not differ significantly, so that the voltammograms overlap, another species N+/N0 (e.g., cobaltoceniun–cobaltocene) can be used as a voltammetric standard [76]; the redox potential of the latter versus Fc+/Fc is known. In a separate experiment, the redox potentials of M+/M0 and Fc+/Fc can be compared in the same supporting electrolyte as that employed for M+/M0 and N+/N0.
If the species of interest is available in the oxidized state, there is the possibility of a reaction between Fc and M+ if E1/2(M+/M0) > E1/2(Fc+/Fc) upon addition of Fc. In that case, the addition of Fc+ rather than Fc should be the preferred, with the voltammogram scanned initially in the negative direction.
An alternative to the addition of the voltammetric standard in-situ is the use of two different electrochemical cells with the same supporting electrolyte, one containing M+/M0 and another Fc+/Fc0, with the same QRE (or RE) transferred from the one cell to the other; the E1/2(M+/M0) versus Fc+/Fc0 will be simply the difference between E1/2(M+/M0) and E1/2(Fc+/Fc0), both measured against this QRE.
With respect to the interconversion between the electrochemical scale versus a reference electrode (REF), e.g., SHE or Fc+/Fc) and the physical scale (Fermi level versus vacuum), it should be noted at first that the EF values of redox species and electrodes as well as energy band positions in semiconductors can be also expressed in the electrochemical scale (EF/REF, for redox species under standard conditions ). The corresponding relation would be simply
The Fermi level of a redox species with respect to a reference energy level located in vacuum is expressed as
or equivalently
where EӨF/VAC(Ox/Red) and EF/VAC(REF) are the Fermi levels of electrons in an electrolyte equilibrating with the redox system Ox/Red (under standard concentrations) and of the reference redox system (the latter with the species concentrations used for the particular reference redox species, not necessarily under standard conditions), defined with respect to vacuum.
The concept of energy level in vacuum and the resulting definition of EF/VAC(REF) should be further specified. Two definitions are encountered in the electrochemical literature.
In the first definition of the reference state, the electron is located in vacuum far-away from the electrolyte so that it does not experience any chemical interactions. Moreover, upon transferring the electron from vacuum into the interior of the electrolyte no electrical forces are experienced; the electrolyte does not have any free charge or any surface dipoles in contact with vacuum. In this respect, EF/VAC(REF) is in fact equal to the chemical potential of electrons (µe, defined on a per-particle basis) in equilibrium with the reference system.
with μe(REF) related to the chemical potentials (on a per particle basis) per molecule of the oxidized and reduced form μe(Ox, REF) and μe(Red, REF) according to
μe(REF) cannot be measured experimentally but can be determined solely by a combination of some experimental data and theoretical calculations, the latter based on several extra-thermodynamic assumptions. As regards the SHE, in the often-quoted earlier publication by Lohmann (1967) [77] EF/VAC(I)(SHE) has been determined as −4.48 eV. In the more recent publication by Isse and Gennaro (2010) the calculated value is EF/VAC(I)(SHE) = −4.281 eV [78]. For the Fc+/Fc redox system in acetonitrile Namazian, et al. (2010) calculated EF/VAC(I) in these nonaqueous solvents: acetonitrile, 1,2 dichloroethane and dimethylsulfoxide, obtaining the values of −4.988 eV, −4.927 eV, and −5.045 eV, respectively [79].
Similarly, for any redox species, e.g., under standard conditions, it is
In the second definition of the reference state, the electron is located in vacuum very close to electrolyte, at a point where the electron does not experience any electrical forces originating from free charges but only these originating from surface dipoles. Therefore, the work of transferring the electron from this point, called “vacuum at the proximity of electrolyte”, (VAC@EL-REF), into the interior of the electrolyte equilibrating with Ox/Red includes the chemical potential as well as a contribution from surface dipoles, the latter corresponding to a surface potential ΧREF (in fact surface potential difference). ΧREF is defined as the difference between the electrical potentials in the interior of the electrolyte (φEL-REF) and vacuum just outside the electrolyte. (φVAC@EL-REF):
Therefore, the second definition of EF/VAC(EF/VAC(II)) can be stated as following:
In fact, the negative of EF/VAC(II) corresponds to the work function of electrons, and as such can be measured. In the well-known article by Trasatti [50], in which IUPAC recommendations for the definition of the physical scale of electrode potentials are included, EF/VAC(II)(SHE) has been determined as −4.44 eV on the basis of experimental data only. In combination with the calculated value of Χ(SHE) = 0.1 V [50] the value of −4.34 eV is obtained for EF/VAC(I)(SHE), which is close to the aforementioned value by Isse and Genaro and different by 0.1 eV from that of Lohmann.
In the electrochemical literature, EӨF /VAC(II) is more often considered for conversions from the electrochemical to the physical scale; however, in several organic or inorganic physical chemistry studies EӨF /VAC(I) may be more appropriate, since they do not include surface dipole contributions.
Quite often the value of either −EF/VAC(I)/e0 or −EF/VAC(II)/e0 of a species is referred to as absolute redox potential. For example, in the case of ferrocene in acetonitrile, the absolute redox potential (I) would be referred to as +4.988 V in place of a Fermi level of −4.988 eV.
A final point to be discussed is the relation between EӨ (or equivalently EFӨ) and highest occupied molecular orbital (HOMO) energy (EHOMO) of a dye D0 in the ground state (Figure 2a). At first approximation, it is often assumed that EHOMO and EFӨ are equal. However, it should be pointed out that these are different concepts, EHOMO corresponding to an internal energy level and EFӨ to a Gibbs free energy. A better approximation can be obtained by considering the Marcus–Gerischer theory [4,80,81,82], according to which lies in the energy diagram (Figure 2) half-way between the maximum ERed(D+/D0) of the energy distribution function of occupied energy states and that EOx(D+/D0) of the unoccupied energy states, with the difference being equal to twice the reorganization energy λ:
and
under the simplifying assumption that both D0 and D+ have equal reorganization energies.
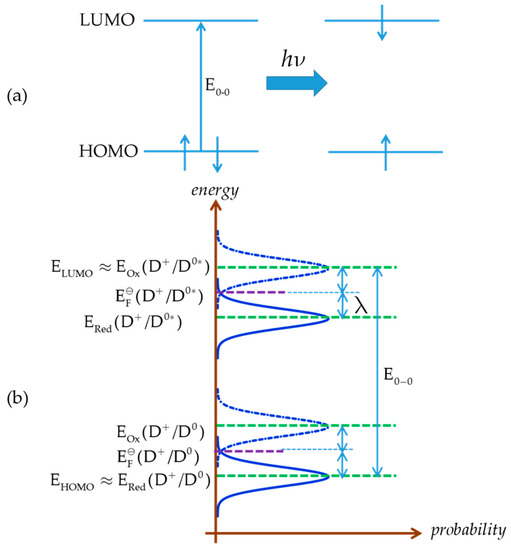
Figure 2.
Energy diagram of the ground and excited state of a dye according to the Marcus–Gerischer theory; (a) principle electron excitation from higher occupied orbital (HOMO) to lower unoccupied orbital (LUMO); (b) energy levels for ground and excited state of dye.
A reasonable approximation is to identify EHOMO of D0 with ERed(D+/D0)
or, in terms of the reorganization energy
If the reorganization energy is very low, which may be the case for large-size dye molecules, the assumption EHOMO ≈ EFӨ(D+/D0) would be reasonable.
The redox potential (Fermi level) of the excited state cannot be readily measured; in some cases this is possible by applying special techniques, e.g., CV at a scan rate of MV/s range [83]. However, it can be indirectly estimated from that of the ground state and the 0–0 photoexcitation energy (E0–0), the latter derived from spectroscopic data, e.g., the intersection of the UV-VIS absorption and emission spectra.
is approximately identified as the energy of the lowest unoccupied molecular orbital (LUMO):
The LUMO is unoccupied and the excited state is occupied with one electron, including a relaxation effect; the electron is at first excited to and then relaxes to . The relaxation effect is equal to 2λ. Upon injection into the semiconductor, the electron energy shifts from to the energy of the conduction band edge.
Therefore,
and
with the same reorganization energy assumed for all of D0, D0* and D+, and, in the case of very low reorganization energy,
E0-0 is approximately related to the LUMO and HOMO energies by
or, on the basis of Equations (92) and (95), as depicted in Figure 2b,
and by combining Equations (90), (94), and (98)
In the case of very low reorganization energy, the above equation is simplified as
or, in terms of redox potentials,
From the above equation the redox potential of the dye excited state can be derived in terms of that of the ground state, determined e.g., by cyclic voltammetry, and the photoexcitation energy:
The determination of from the above equation is often encountered in the DSSC literature.
6. Cobalt Mediators
Co is a transition metal most commonly found in the Co(II) or Co(III) oxidation states. They respectively possess the d7 and d6 electronic structure with a paramagnetic nature for Co(II) and a diamagnetic nature for Co(III). Co-based organometallic complexes tend to form stable compounds with a coordination number of CN = 6, adopting an octahedral geometry. Co is a versatile element that can be used in organic synthesis as a catalyst for a variety of reactions [84,85], for example as a water splitting homogenous catalyst [86,87], and as a potent building block for anticancer activity drugs [88].
Due to its structure, Co ions can form organometallic complexes with either two tridentate ligands (such as terpyridines) or three bidentate ligands (such as 2,2′-bipyridyl or 1,10-phenanthroline). Moreover, d4 to d7 transition metals are known to have their electronic configuration fluctuating between a high spin (HS) or a low spin state (LS), depending on the ligand σ donating/π accepting strength. The latter will mainly influence the ligand-field splitting energy ΔEsplit. In our case we will mainly discuss examples based on aromatic N-donating ligands such as terpyridines and derivatives of 2,2′-bipyridine and 1,10-phenanthroline. A nitrogen-based ligand is usually binding a metal center through its available lone pair of electron located in a p orbital. If the nitrogen atom has an aliphatic nature, as in the case of amines for example, the bond will have a strong σ-donating nature, hence increasing the electron density on the metal center. On the other hand, if the nitrogen atom is embedded in a π-conjugated network, the ligand will display a non-negligible part of π-accepting strength. This is due to the fact that the π* orbitals of these π-conjugated systems are usually relatively low in energy, so that they become accessible for additional π-back bonding [89].
As presented in Figure 3, Co(II) ions have a d7 electronic configuration, split into two distinct spin states: a quartet state, characterized by 3 unpaired electrons (4Co2+ on Figure 3), and a doublet state, in which one electron is left unpaired in the eg orbitals (2Co2+ on Figure 3). The oxidized Co(III) has a d6 electronic configuration as well as two spin states: a singlet state with 6 paired electrons in the t2g orbitals (1Co+3 on Figure 3), and a triplet state where two electrons are unpaired (3Co+3 on Figure 3). Co(II) coordination complexes can be distributed between these two LS and HS states depending on the ligand, whereas for Co(III) the HS state is usually higher in energy, so that the LS state is the preferred ground state.
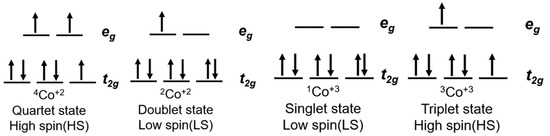
Figure 3.
Electronic configuration of the cobalt(III)/(II) couple as well as ligand field theory diagram.
This difference in spin states can be observed in compounds such as cobalt(II) tris(2,2′-bipyridine) ([Co(bpy)3(II)]) or cobalt(II) bis(2,2′:6′,2″-terpyridine) ([Co(terpy)2(II)]) [90]. The first complex has a favored HS configuration in the ground state, with the LS doublet state not too far away in energy. [Co(bpy)3(II)] will therefore be able to easily cross from one state to another. On the other hand, cobalt(II) bis(2,2′:6′,2″-terpyridine) ([Co(terpy)2(II)]) shows a more stable LS state [91,92]. This is mainly due to the anisotropy induced in the Co–N bond length that reinforces a strong Jahn–Teller effect ultimately stabilizing the singlet state of Co(III).
Redox active Co(III/II) coordination complexes have the ability to transfer one electron following the outer sphere type of mechanism that can be described by Marcus theory. The kinetics of this electron transfer is supposed to be fast, assuming that the bond lengths between the Co center and the ligand remain mostly unchanged, but also that there is no strong influence of the solvent shell reorganization surrounding the complexes [93].
With respect to the energetics of DSSCs, this spin change upon oxidation of Co(II) to Co(III) is expected to have deleterious effects regarding the operational mechanism of the devices. As the photooxidized dye has to be regenerated by the Co(II) coordination complex, the energy required to perform the spin change resulting from this redox reaction is added to the inherent reorganization energy required to rearrange the cobalt–ligand bond length as well as the solvent shell surrounding the organometallic complex.
The first reported use of a Co(III/II) redox mediator in DSSCs was published in 2001 by Nusbaumer, et al. [94]. They studied a Co complex based on the bis [2,6-bis (1′-butylbenzimidazol-2′-yl) pyridine](dbbip) ligand (Figure 4). Dbbip was chosen due to its ability to rapidly transfer an electron upon combination with a Co metal center, notably in vitamin B12 [95]. It was found that [Co(dbbip)2(II)] and [Co(dbbip)2(III)] have absorption maxima in the UV-region, located at 321 nm and 309 nm for the former and at 344 nm for the latter. This was one of the first advantages that Co complexes would have over the I3−/I− redox couple: no parasitic absorption in the visible region of the solar spectrum. This allows the preparation of relatively concentrated mediator solutions without reducing the amount of light absorbed by the dye. These complexes have in acetonitrile (ACN) at 25 °C reversible redox potential of 0.36 V versus SCE or versus 0.60 V versus SHE.
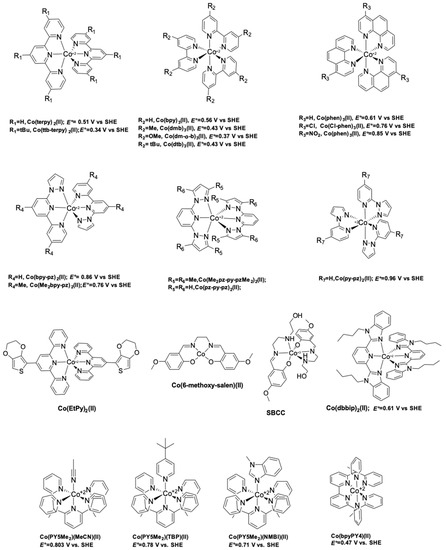
Figure 4.
Chemical structure of polypyridine based cobalt coordination complexes used as redox mediators in DSSCs.
Combined with the Ru dye Z316 (Figure 5) a PCE of 2.2% was produced under simulated AM1.5 G illumination of 1000 W cm−2 (UOC = 0.67 V, jSC = 6.8 mA·cm−2). This low efficiency under full-sun illumination was mainly due to a poor fill factor of 0.46.
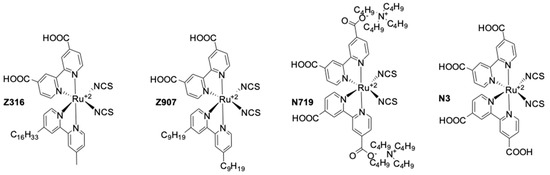
Figure 5.
Ruthenium based dyes used in dye-sensitized solar cells (DSSCs).
This mediator was further investigated in 2003 by the same team [96]. They decided to study the effect of the counter ion on a series of [Co(dbbip)2X2] (X = OTf−, ClO4−, DDS− and PF6−) coordination complexes, (OTf = trifluoromethanesulfonate (CF3SO3), DDS = dodecylsulfate). They noticed almost no influence of the anion on the UOC. However, strong fluctuations regarding the photocurrent were measured. A bulkier counter anion such as DDS− would give low jSC, whereas the smaller PF6− would help providing sufficient jSC. Interestingly, they found that those Co-based organometallic complexes were suffering from important mass-transport issues, especially while increasing the illumination intensity. A diffusion coefficient of 7.72 × 10−6 cm2·s−1 was measured for [Co(dbbip)2(ClO4)2] in acetonitrile at 20 °C. After optimization of the TiO2 layer as well as the electrolyte composition, a PCE of 3.9% (UOC = 0.84 V, jSC = 8.4 mA·cm−2) was measured under full sun, whereas at 10% sun a PCE of 7.9% (UOC = 0.69 V, jSC = 0.24 mA·cm−2) was obtained. This was mainly due to an improvement of the fill factor upon reducing the illumination from full sun (FF = 0.56) to 10% sun (FF = 0.77). These devices were fabricated using the Ru dye Z907 (Figure 5) [96].
An electrochemical study of the [Co(dbbip)2(ClO4)2] complex was conducted by Cameron, et al. [97] in 2004. They discovered that this complex would exhibit slow electron exchange kinetics at the FTO electrode as well as at the Pt CE. A lower exchange current than for the I−/I3− couple was measured, which would ultimately cause important voltage losses in the DSSC. They also measured a very low diffusion coefficient of 1.9 × 10−6 cm2·s−1 in a 40:60 acetonitrile/ethylene carbonate mixture at 20°.
In 2002, a study on 14 different Co complexes was reported by Sapp, et al. [98]. They performed electrochemical measurements as well as device fabrication and characterization. They confirmed the very attractive feature of Co complexes towards light absorption, enabling the preparation of concentrated electrolyte solutions without altering the dye light uptake. Interestingly, it was found that Co-based coordination complexes would show poor electrochemical activity with Pt-based CEs, which was consistent with the report of Cameron, et al. in 2004. The complexes had the generic structure [Co(L)2(ClO4)2], with the ligand (L) being a terpyridine(terpy) (R1 = H), 2,2′-bipyridyl(bpy) (R2 = H), or 1,10-phenanthroline(phen) (R3 = H) derivative (Figure 4). The best efficiency was obtained using a redox mediator based on 4,4′-di-tertbutyl-2,2′-bipyridine (dtb) (R2 = tBu). The measured PCE was 1.3% under full sun (UOC = 0.44 V, jSC = 4.82 mA·cm−2) with the Ru dye N3 (Figure 5).
An important further study on the Co mediator/Ru-dye system was reported in 2012. Masconi, et al. [99] focused on addressing two main questions regarding, on the one hand, the reason for which the performance were so low when Ru-dyes were used in conjunction with Co electrolytes and, on the other hand, the preferred spin state of Co coordination complexes during the oxidized dye regeneration process. In this regard, they performed an experimental as well as computational study of the Co(bpy)3(III/II) redox couple in conjunction with two Ru dyes: N719 and Z907 (Figure 5). At first, they used ab-initio molecular dynamics computations to investigate the interaction between the N719 dye adsorbed on TiO2 surface and the Co(bpy)3(III) complex. The formation of an ion pair between the negatively charged dye and the positively charged Co complex was observed. N719 is known for being charged both in solution and at the TiO2 interface [100,101]. Then, they explained that the formation of this ion pair would bring the Co(III) center very close to the semiconductor surface, therefore causing important recombination between an electron in the TiO2 and the oxidized Co complex. Experimentally, they managed to isolate an ion pair between N719 and Co(bpy)3(III) that was identified by 1H-NMR spectroscopy. Interestingly, upon applying the same procedure to Z907, no product formation could be observed. This indicated that the bulky nonyl(-C9H19) chains at the 4′ of the bpy ligand of Z907 would effectively prevent the formation of such ion pair. The effect of these long-side chains was also noticeable upon device fabrication, where the efficiency under full sun would rise from 1.1% (UOC = 0.578 V, jSC = 3.03 mA·cm−2) with N719 to 2.1% (UOC = 0.649 V, jSC = 4.43 mA·cm−2) upon switching to the bulkier Z907 dye. The second task which Masconi, et al. undertook was investigating about the uncertainty regarding the spin state of Co(II) species during the dye regeneration mechanism. As explained in the early paragraphs, Co(bpy)3(II) has two spin states: a doublet (LS) and a quartet (HS). They used density functional theory (DFT) to compute energy profiles of the electron transfer reaction between the dye and the redox mediator. They reached the result that the reorganizational energy λ is much smaller for the LS state. They obtained values around 0.6 eV for a series of Co complexes, whereas for the HS state, λ would lie at around 1.35 eV.
The first ever high efficiency Ru-dye-based device with a Co-based redox mediator was reported in 2011 by Liu, et al. They highlighted the effectiveness of the nonyl chains of Z907 at improving charge collection and photocurrent generation using electrochemical impedance spectroscopy (EIS) and near infra-red transmittance measurements (NIRTM) [102]. Upon careful optimization of the electrolyte composition and the TiO2 layer thickness, they managed to achieve an impressive PCE of 6.5% (UOC = 0.744 V, jSC = 14.0 mA·cm−2) with Z907 and [Co(bpy)3(III/II)] under full sun.
More recently, organic sensitizers rapidly proved to be promising substitutes for Ru coordination complexes. They offer attractive features such as the absence of rare earth metals, improved loading on the TiO2 surface, as well as much higher molar absorption coefficients, compared to Ru dyes (Figure 5). Figure 6 show examples of organic dyes used in DSSCs. The common feature between all these molecules is the way they are electronically constructed. They all present a “push–pull” directionality that improves the charge transfer from the electron rich donor site to the electron withdrawing accepting site that can be anchored to the TiO2 electrode. These types of dye are called either Donor–π–Acceptor (D–π–A) or Donor–Acceptor–π–Acceptor (D–A–π–A) dyes. In this regard, it was only a matter of time before they would be implemented by several research groups in DSSCs along with the newly discovered cobalt based redox mediator.
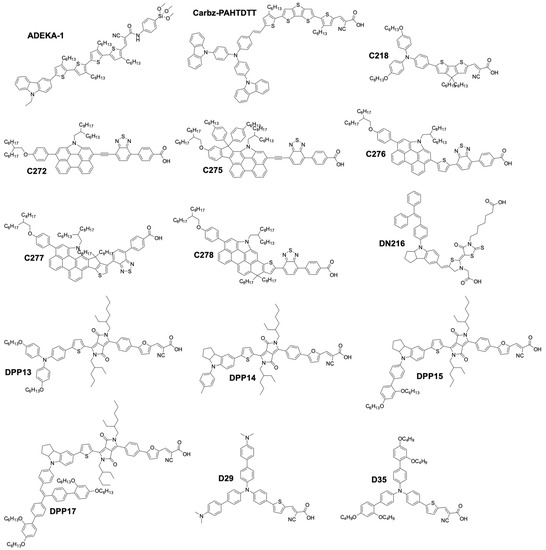
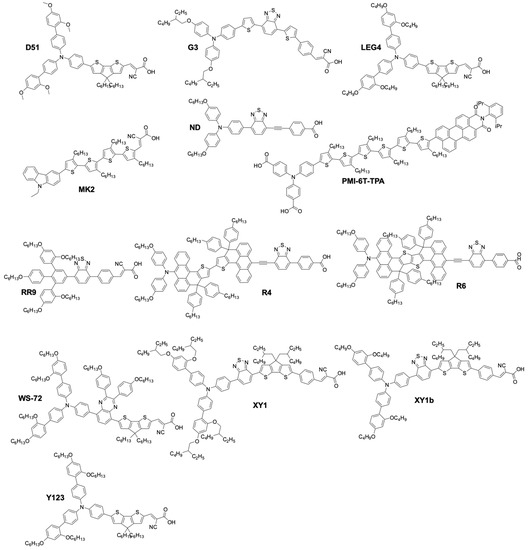
Figure 6.
Chemical structure of organic dyes used in DSSCs.
A major breakthrough came in 2010 with the publication by Feldt, et al. [103] of the first high-efficiency DSSC based on a combination of two organic donor–π–acceptor (D–π–A) charge-transfer type dyes, D35 and D29 (Figure 6), and a series of [Co(L)3(III/II)] complexes where the ligand (L) was 2,2′-bipyridyl, either unsubstituted, or substituted by ligand R2 at the 4,4′ positions (L = 4,4′-dimethyl-2,2′-bipyridyl(dmb), R1 = Me, or L = 4,4′-ditert-butyl-2,2′-bipyridyl (dtb), R1 = tBu). These three ligands would form octahedral complexes upon reaction with a Co salt. The increased bulkiness of the coordination sphere would follow the order [Co(bpy)3(III/II)] < [Co(dmb)3(III/II)] < [Co(dtb)3(III/II)]. The authors found that this steric bulkiness present on the complexes could help reducing the recombination rate between the electrons in the TiO2 and the Co(III) complex, at the cost, however, of some diffusion limitation problems in the electrolyte. It was reported that the best performance was obtained using a bulky dye (D35) and the smallest Co complex: [Co(bpy)3(III/II)]. They obtained an impressive PCE of 6.7% with a UOC of 0.92 V and a jSC of 10.7 mA·cm−2 under full sun. These impressive results were achieved by a judicious choice of the dye/complex combination, rationalized by the evaluation of the conducted DSSC experiments. The mass transport issues were solved by choosing a rather small complex, [Co(bpy)3(III/II)]. On the other side, D35 possesses four repelling n-butoxide(–OC4H9) chains that are exposed to the electrolyte side. They can induce enough steric hindrance to prevent the Co complexes from diffusing too close to the TiO2 electrode. As a result, it improved the electron lifetime in the conduction band and the overall performance.
This impressive value was rapidly improved by Tsao, et al. in 2011 [104]. They increased the efficiency by switching from the previously used dye D35 to the new high extinction coefficient dye Y123. (Figure 6). These two dyes both possess the bulky 4′,4′′′-(phenylazanediyl)bis([1,1′-biphenyl]-2,4-dialkoxy)-type donor earlier described by Feldt, et al. [103], however Y123 uses a cyclopentadithiophene (CPDT) π-bridge which drastically improves light-harvesting properties, ultimately improving the photocurrent by 40% with respect to D35. For a DSSC based on the dye Y123/[Co(bpy)3(III/II)] system a PCE of 8.8% under full sun was obtained (UOC = 0.855 V, jSC = 14.6 mA·cm−2) [104] when used in conjunction with a platinized FTO CE.
Feldt, et al. conducted two further insightful studies on the regeneration and recombination kinetics of Co polypyridine redox mediators in 2011 [105] and 2013 [106]. They synthesized 13 different Co based organometallic complexes with redox potentials ranging from 0.34 and 1.2 V versus SHE and used the Marcus theory to compute the reorganizational energy for a series of dye/electrolyte combinations. In addition to the firstly studied [Co(bpy)3(II)], [Co(terpy)3(II)] (R1 = H) and [Co(phen)3(II)] complexes, the authors used three different types of ligands derived from 6-(1H-pyrazol-1-yl)-2,2′-bipyridine ([Co(bpy-pz)2(II)]), 2,6-di(1H-pyrazol-1-yl)pyridine ([Co(pz-py-pz)2(II)] (R5 = R6 = H), and 2-(1H-pyrazol-1-yl)pyridine ([Co(py-pz)3(III)] (R7 = H)) (Figure 4). The majority of Co complexes were found to be located in the Marcus normal region, with an increasing regeneration rate constant along with an increasing dye regeneration driving force. A high regeneration efficiency was obtained for complexes having a regeneration driving force superior to 0.4 eV. However, this efficiency would rapidly decay for a reorganization energy below 0.2 eV. As explained earlier, the spin crossing that occurs upon oxidation of Co(II) to Co(III) is the main responsible factor for this energy barrier.
The authors also found that even for a redox mediator such as [Co(bpy-pz)2(II)] (R4 = H on Figure 4) having a regeneration driving force as low as 0.25 eV it is possible to regenerate 84% of the D35 dye molecules upon increasing the concentration of the mediator.
The fast development of these Co-based redox mediators brought some interesting results regarding the CE. As mentioned earlier, Pt-based CEs were found to be relatively inefficient towards the catalytic reduction of the Co(III) species. Rapidly, new CE materials such as graphene [107,108,109] or poly(3,4-ethylenedioxythiophene)(PEDOT) [110,111,112] were implemented in DSSCs. The facile deposition of these materials as well as the reduced charge transfer resistance were found to be their most attractive features over expensive and inactive Pt.
As described in their work in 2011 with the Y123/[Co(bpy)3(III/II)] combination, Tsao, et al. managed to improve their previously reported value to reach an impressive PCE of 10.3% under full sun (UOC = 0.910 V, jSC = 15.9 mA·cm−2) and 10.6% under 1/2 sun (UOC = 0.893 V, jSC = 8.1 mA·cm−2) [113]. This was achieved by using electrodeposited PEDOT on a FTO substrate as CE. It was found that by using this conductive polymer, the charge transfer recombination at the CE(RCT), could be effectively reduced to around 0.03 Ω·cm2 compared to a standard Pt CE where RCT = 7.5 Ω cm2, for the [Co(bpy)3(III/II)] redox couple. In this study, two Co coordination complexes were used: [Co(bpy)3(III/II)] and [Co(phen)3(III/II)]. Both candidates showed improved RCT, respectively 0.03 Ω cm2 (7.5 Ω·cm2 with Pt) and <0.03 Ω·cm2 (54 Ω·cm2 with Pt). However, due to the bulkier size of the [Co(phen)3(III/II)] coordination sphere, deleterious mass transport limitations appeared upon increasing the light intensity, causing losses in performance, especially in the fill factor.
Yum, et al. managed to enhance the performance of Co based DSSCs as regards the full-sun photovoltage even further in 2011 [114] by combining the results from Feldt, et al., where photovoltage values superior to 1 V [105] were reported with the [Co(bpy-pz)2(III/II)] redox couple, as well as these from Tsao, et al. [113] on the advantageous use of a thiophene-based conducting polymer as CE electrocatalyst, but with poly(3,4-propylenedioxythiophene) (PProDOT) replacing PEDOT as CE material, and using dye Y123. The relatively positive redox potential of the [Co(bpy-pz)2(III/II)] couple (0.86 V versus SHE) helped generating a sufficiently high photovoltage. In conjunction with this redox couple, the dye Y123 harvested an important portion of the solar photons owing to its particularly high molar extinction coefficient. Finally, the Pt-free PProDOT-based CE helped reducing the losses associated with charge transfer resistance at the CE. Overall, the measured PCE was 10.1% under 1000 W·m−2 (UOC = 0.998 V, jSC = 13.1 mA·cm−2) and 10.7% under 1/2 sun (UOC = 0.977 V, jSC = 7.4 mA·cm−2).
At the interface between organometallic dyes and full organic dyes lies the family of zinc–porphyrins. This building block can be used as a π-bridge in a D-π-A dye and was already reported to work well with the I3−/I− electrolyte [115]. A record efficiency was reported in 2011 by Yella, et al. They synthesized two porphyrin dyes, YD2 and YD2-o-C8 (Figure 4) using a bis(4-alkoxyphenyl) amine donating and a 4-ethynylbenzoic acid accepting group surrounding a zinc porphyrin ring [116].
As shown in Figure 6, the various dyes of this family differ only by the presence of ortho-octyloxy(–OC8H17) side chains on the phenyl ring located at the meso position of the porphyrins. They were attached in order to enhance the electron density on the zinc–porphyrin ring and as repelling agents for the redox mediator. Electron lifetime measurements performed on devices made with the YD2-o-C8 dye and [Co(bpy)3(III)] complex confirmed this hypothesis. The lifetime was considerably improved when these four n-octyloxy groups were present. Moreover, it helped providing a sufficiently high open-circuit voltage of 0.965 V, without depleting the fill factor or the photocurrent, even under 100 mW·cm−2 illumination. A series of devices made with YD2-o-C8 gave an impressive PCE of 11.9% under full sun (UOC = 0.965 V, jSC = 17.3 mA·cm−2). Unfortunately, these porphyrin dyes do not absorb photons between 480 and 630 nm. The authors addressed this issue by co-sensitization with the Y123 dye, which happens to have strong absorption in the 430–600 nm range. This elegant complementarity between these two dyes allowed the fabrication of record-breaking devices with PCEs of 12.5% under full sun (UOC = 0.935 V, jSC = 17.7 mA·cm−2) and 12.7% under 1/2 sun (UOC = 0.910 V, jSC = 9.7 mA·cm−2) with [Co(byp)3(III/II)].
Additional research on the porphyrin/Co system was published three years later by Matthew, et al. in Nature Chemistry [117]. The authors managed to push the efficiencies up to 13% by improving the dye structure. A bulkier triphenylamine-type hydrophobic donor, similar to the one used in D35 and Y123, was attached on dyes SM315 and SM371 (Figure 6). The bulkiness and strong electron donating abilities of the latter has proved to be very well compatible with Co based DSSCs, enabling the fabrication of high efficiency devices. Compared to SM371, a supplementary benzothiadiazole (BTD) electron accepting unit was implemented in SM315. The major improvement came from the spectral response of the dyes. SM315 displayed a broadening of the Soret and Q-band, allowing the dye to harvest more photons in the red and green region of the spectrum. This was an important improvement compared to both SM371 as well as the previous work on the YD2 series of dyes. Device fabrication was performed and a very impressive PCE value of 13.0% under full sun (UOC = 0.91 V, jSC = 18.1 mA·cm−2) was obtained with the state of the art [Co(bpy)3(III/II)] redox couple and SM315.
The highest DSSC efficiency until today has been obtained by Kakiage, et al. [118] with the [Co(phen)3(III/II)] redox mediator. The TiO2 electrode was co-sensitized with two metal-free dyes. One of them was carbazole linked to an alkyl-functionalized thiophene oligomer, with an alkoxysilyl pendant group for anchoring to TiO2 (ADEKA-1, Figure 6). The ADEKA-1 dye is a derivative of the previously developed MK2. The main difference lies in the presence of an alkoxysilyl-anchoring group. The authors speculated that this anchoring group could improve the interaction between the TiO2 and the dye, hence improving the charge injection and ultimately the photocurrent. In combination with LEG4, the spectral response of which is more red-shifted, the authors managed to improve the IPCE by roughly 20% as well as to shift the photocurrent conversion towards shorter wavelength. The PCE was 14.3% under full sun (UOC = 1.01 V, jSC = 18.2 mA·cm−2) and 14.7% under ½ sun (UOC = 0.99 V, jSC = 9.6 mA·cm−2). This attractive feature helped generating a UOC superior to 1 V. Further optimization was performed by replacing the relatively poorly active Pt CE by a new electrocatalytic system based on graphene nanoplatelets and gold deposited on FTO glass. The IPCE exceeds 80% throughout the visible (400–700 nm) range of the spectrum.
One of the attractive features of DSSCs is the possibility to use them as semi-transparent photovoltaic cells, hence making them perfectly suited for building integrated photovoltaics (BIPV). This allows the fabrication of colored panels for BIPV and other integrated PV applications, the only limitations being the dye color palette, the performance, and the stability. The performance is directly related to the dye/electrolyte combination, as altering the dye color and hence its energy levels will restrict the choice in terms of compatible electrolytes for high efficiency devices. So far, it has been shown in this review that the Co-based redox mediators can be synthesized with the possibility to vary the redox potential within a wide range. This enables the possibility to match a Co-coordination complex with a dye having specific energy levels and hence color. Yella, et al. [116] and Matthew, et al. [117] showed the harmonious compatibility between green-colored Zn–porphyrin dyes and the [Co(bpy)3(II/II)] electrolyte. In addition to that, many examples in the literature report the high performance of Co(III/II) complexes with red dyes such as Y123 or D35.
The first blue-colored high efficiency DSSC fabricated using a Co based redox mediator was reported in 2012 by Yum, et al. [119], following up their successful optimization of the Y123/[Co(byp-pz)2(III/II)] system.
These devices used a molecularly engineered D-A-π-A dye bearing the strongly light absorbing diketopyrrolopyrrole (DPP) unit as acceptor and chromophore [119]. This class of compounds is known for their application in optoelectronics as well as in red pigment for car-painting [120]. These impressive, carefully designed dye structures are shown on Figure 6. The (E)-3-(5-(4-(2,5-bis(2-ethylhexyl)-3,6-dioxo-4-(thiophen-2-yl)-2,3,5,6-tetrahydropyrrolo[3,4-c]pyrrol-1-yl)phenyl)furan-2-yl)-2-cyanoacrylic acid motif was kept constant as A–π–A sequence. Only the donor was altered, being either a 4-alkoxy-N-(4-alkoxyphenyl)-N-phenylaniline, with hexyl chains for optimum electron donation as well as efficient repelling of the redox mediator (DPP13), or a derivative of 1,2,3,3a,4,8b-hexahydrocyclopenta[b]indole bearing extra donating/bulky moieties linked to the N atom of the indole (DPP14, DPP15, and DPP17). These dyes were used in combination with the [Co(bpy)3(III/II)] redox couple during the fabrication of DSSCs. The best efficiency obtained was 10.1% PCE under 1000 W·m−2 (UOC = 0.761 V, jSC = 17.9 mA·cm−2) with DPP17 as sensitizer.
More recent work on Co-based highly efficient DSSC was reported by Yao, et al. based on the synthesis of a new class of donor based on 1H-phenanthro [1,10,9,8-cdefg] carbazole unit [121] implemented in dyes C272 and C275 (Figure 6). These two dyes were used with the [Co(phen)3(III/II)] redox couple having a redox potential of 0.63 V versus SHE. Good PCE values under full sun were obtained for C272 and C275, respectively 10.6% (UOC = 0.897 V, jSC = 15.81 mA·cm−2) and 12.5% (UOC = 0.956 V, jSC = 17.03 mA·cm−2). Charge extraction and transient photovoltage decay measurements attributed the enhanced UOC in C275 devices to a longer electron lifetime in the conduction band of TiO2, which might result from more efficient surface insulation by C275 bearing very bulky 4-hexyl-phenyl groups.
Those molecular structures were further altered by the same research group in the same year. New dyes, named C276, C277, and C278 (Figure 6) were synthesized, where this time the bridging ethynyl triple bond was replaced by fused or free thiophene [122]. Three series of devices were fabricated using [Co(bpy)3(III/II)] as redox couple. Two of these aforementioned dyes, C277 and C278 produced cells with PCEs of respectively 11.5% (UOC = 0.820 V, jSC = 19.41 mA·cm−2) and 12.0% (UOC = 0.843 V, jSC = 19.64 mA·cm−2), both measured under full sun. On the other hand, C276-based devices produced PCEs of 9.4% (UOC = 0.818 V, jSC = 15.54 mA·cm−2) under full sun. Compared to the previous system (C272 and C275) the measured open-circuit voltage was lower because of the more negative redox potential of the [Co(bpy)3(III/II)] couple (0.56 V versus SHE), compared to the [Co(phen)3(III/II)] (0.62 V versus SHE). Essentially, C277 and C278 showed a drastically red-shifted IPCE spectrum due to the fused nature of the thiophene-donor segment, improving the spectral response and therefore the photocurrent.
In early 2018, Ren, et al. [123] reported a new highly efficient DSSC based on Co electrolyte and a novel D-π-A blue dye. The latter used a polycyclic aromatic hydrocarbon (PAH), derived from a 9,9,19,19-tetrakis(4-hexylphenyl)-9,19-dihydrobenzo [10′,1′]phenanthro[3′,4′:4,5]thieno [3,2-b]benzo[10,1]phenanthro[3,4-d]thiophene core. This impressive core combined with a bis(4-hexyloxyphenyl)amine donor and the very popular 4-(7-ethynylbenzo[c][1,2,5]-thiadiazol-4-yl)benzoic acid acceptor, gave under full sun a PCE of 12.6% (UOC = 0.850 V, jSC = 19.69 mA·cm−2) and 11.1% (UOC = 0.852 V, jSC = 17.25 mA·cm−2) for respectively R6 and R4 (Figure 6) with [Co(bpy)3(III/II)]. The two dyes only differ by the fusion of the PAH at the center of the dye. This subtle structural change induced a red shift in the absorption spectrum of R6 by 18 nm, improving the jSC by roughly 2 mA·cm−2.
Several efforts have been undertaken to introduce novel Co complexes in DSSC research. In 2015, Koussi-Daoud, et al. [124] reported an interesting ligand based on a terpyridine scaffold functionalized at the 4′ position with a 3,4-ethylenedioxythiophene (EDOT) unit: 4′-(3,4-ethylenedioxythiophene-2,2′:6′,2′′-ter-pyridine(EtPy). The EDOT unit was installed there to facilitate the interaction with the PEDOT CE. The study was conducted using mediators in solution: [Co(EtPy)2(III/II)] together with the popular [Co(phen)3(III/II)]. This ACN electrolyte was tested with two dyes: Ru-based Z907 and organic dye D35. It was found that adding [Co(EtPy)2(III/II)] would help improve PCEs from 1.4% (UOC = 0.70 V, jSC = 3.6 mA·cm−2) to 2.2% (UOC = 0.75 V, jSC = 5.1 mA·cm−2) for Z907 and from 4.2% (UOC = 0.91 V, jSC = 7.3 mA·cm−2) to 5.1% (UOC = 0.92 V, jSC = 8.4 mA·cm−2) for D35; all of those measurements were performed under full sun. This improvement resulted from more efficient electron transfer and reduced recombination. This is a case of a dual mediator system, with the mediator carrying out the regeneration of the photooxidized dye at the PE together with the charge transport between WE and CE, and the co-mediator effective in the CE reaction. This approach is somewhat different from that in the co-mediator system discussed in Section 10 of the present publication, in which case the mediator is effective in charge transport and in the CE reaction and the co-mediator in the regeneration reaction at the PE.
Nasr-Esfahani, et al. [125] undertook in 2014 a study of ligands derived from the 2,2′-ethylenebis(nitrolomethylidene)diphenol-N,N′-ethylenebis(salicylimine) (salen) structure. This ligand is known to form very robust coordination complexes with Co(II), such as the famous oxygen carrier, cosalen also known as salcomine [126]. They prepared two complexes with the following ligands based on the salen structure: 2,2′-((1E,1′E)-(ethane-1,2-diylbis(azaneylylidene)) bis(methaneylylidene))bis(4-methoxyphenol), with the abbreviated name: 6-methoxy-salen, and (E)-2-(((2-((2-hydroxyethyl)amino)ethyl)imino)methyl)-4-methoxyphenol, with the abbreviated name SB (for Schiff base). Unfortunately, due to the irreversible electrochemical behavior of [Co(6-methoxysalen)2(II)], only [Co(SB)2(II)(SBCC)] was used for device fabrication. Compared to the state of the art [Co(bpy)3(III/II)] redox mediator, SBCC managed to improve the open-circuit voltage but, due to poor fill factor and low photocurrent, SBCC-based devices reach a PCE of only 2.53% (UOC = 0.91 V, jSC = 5.2 mA·cm−2) compared to 3.53% (UOC = 0.83 V, jSC = 6.4 mA·cm−2) attained by the authors for the control device based on [Co(bpy)3(III/II)]/D35 under full sun illumination. The improvement in UOC was attributed to the more positive redox potential of SBCC compared to [Co(bpy)3(III/II)]. The overall lower performance compared to [Co(bpy)3(III/II)] was attributed to an increased charge transfer resistance at the CE due to the negatively-charged complex SBCC.
The group led by Udo Bach at the Monash University reported two novel ligand structures in respectively 2012–2013. The first article [127] refers to the synthesis of a pentadentate ligand: 2,6-bis(1,1-di(pyridin-2-yl)ethyl)pyridine coded PY5Me. As explained in the earlier paragraphs, Co(III/II) ions form coordination complexes with CN = 6. In this regard, the authors of the paper hypothesized that the 6th remaining coordination site would be occupied by a coordinating Lewis base that could ultimately tune the redox properties of the organometallic complex. In their study, three complexes were synthesized using two structurally different monodentate Lewis bases: 4-tert-butylpyridine (TBP) and N-methylbenzimidazole (NMBI), as well as acetonitrile (MeCN) as control. The complexes were respectively [Co(PY5Me2)(TBP)(III/II)], [Co(PY5Me2)(NMBI)(III/II)] and [Co(PY5Me2)(MeCN)(III/II)] [94]. Cyclic voltammetry measurements revealed an important shift of the redox potential towards less positive values following the trend: MeCN (0.803 V versus SHE) > TBP (0.780 V versus SHE) > NMBI (0.714 V versus SHE). This trend indicates a strong Lewis basicity coming from the NMBI additive, which ultimately helps reducing the potential by almost 0.1 V versus SHE. Those complexes were tested in devices and along with the dye MK2 (Figure 6). [Co(PY5Me2)(NMBI)(III/II)] outperformed the common [Co(bpy)3(III/II)] system with an efficiency of 8.3% under full sun (UOC = 0.940 V, jSC = 11.8 mA·cm−2). The main improvement was attributed to the more positive redox potential of this compound that allowed the generation of a very high UOC of 0.940 V. The fill factor was also drastically improved, respectively 0.77 for the new complex and 0.67 for Co tris-bipyridyl.
Complex [Co(PY5Me2)(TBP)(III/II)] also performed well in terms of UOC (0.993 V) and fill factor (0.77) but, due to an important decrease of photocurrent, the efficiency achieved was lower than that for [Co(bpy)3(III/II)] with 6.1% under full sun (UOC = 0.993 V, jSC = 8.1 mA·cm−2). The current drop was attributed to faster recombination for electrolytes with more positive redox potentials.
In 2013, the same team [128] managed to improve the stability of Co based DSSCs by tuning the denticity of the ligand. They speculated that by using a hexadentate ligand, the chelating effect would help generating a very thermodynamically stable complex. In this regard, they synthesized the ligand 6,6′-bis(1,1-di(pyridin-2-yl)ethyl)-2,2′-bipyridine (bpyPY4) derived from a 2,2′-bipyridine core. The redox potential of the [Co(bpyPY4)(III/II)] couple was found to be 0.465 V versus SHE, which corresponds to a decrease by approximatively 0.1 V compared to the [Co(bpy)3(III/II)] (0.560 V versus SHE). The devices fabricated with this new Co complex and MK2 as dye produced a PCE of 8.3% under full sun (UOC = 0.757 V, jSC = 14.7 mA·cm−2). Under the same conditions, the devices using [Co(bpy)3(III/II)] produced 7.8% (UOC = 0.826 V, jSC = 13.7 mA·cm−2). The more negative redox potential of [Co(bpyPY4)(III/II)] increased the dye regeneration driving force compared to the [Co(bpy)3(III/II)], ultimately improving the photocurrent by 1 mA·cm−2. Despite the loss of UOC, an improved fill factor of 0.75 was obtained with this new complex, as compared to 0.69 for [Co(bpy)3(III/II)].
The great majority of DSSC studies have involved organic electrolytes as charge-transport media between PE and Ce. However, as regards practical DSSCs, there is a great interest for DSSCs based on aqueous electrolytes, which offer the advantages of being non-toxic and having a low vapor pressure compared to several of their organic analogues. However, for such applications specially designed hydrophobic dyes would be required. The first high-efficiency aqueous dye-sensitized solar cell based on a Co electrolyte was reported in 2013. In this respect, Xiang and co-workers used the combination of MK2 and [Co(bpy)3(III/II)] [129]. Varying amounts of polyethylene glycol (PEG 300) were used to help reducing the phase separation between the hydrophobic dye MK2 and the electrolyte. Their optimization process started by identifying the best proportion of PEG 300 in the electrolyte medium. They found that 1 wt% was the limit before the onset of important mass transport limitations under high sunlight intensity. In a second step they focused on solving the charge resistance issues at the electrolyte-CE interface. It was found that by screen-printing a 13.5 μm thick layer of mesoporous indium tin oxide (ITO) followed by thermal deposition of Pt, the performance would be drastically improved. Ultimately, they obtained aqueous based devices with a PCE of 5.0% under 1000 W·m−2 (UOC = 0.687 V, jSC = 9.8 mA·cm−2), which represents an impressive result for a Co based aqueous DSSC.
Further work was performed on aqueous DSSCs in 2014 by Dong, et al. They improved the performance of the devices by treating the TiO2 electrode with octadecyltrichlorosilane (ODTS) [130]. This compound would generate an insulating layer between the electrolyte and the semiconductor, ultimately reducing the recombination at the semiconductor/electrolyte interface. This hypothesis was confirmed by electrochemical impedance spectroscopy (EIS), showing a much longer electron lifetime compared to that of the ODTS-free devices. Impedance measurements showed an improved charge combination resistance (RCR) increasing from 5.4 Ω to 21 Ω upon treatment of the TiO2 electrode with ODTS. The authors fabricated devices using the well-known [Co(bpy)3(III/II)] cation with a nitrate (NO3−) counter anion in order to improve the aqueous solubility of the latter. They also used 1 wt % of PEG 300, as previously reported, to reduce the phase separation between the dye and the aqueous electrolyte. Overall, these devices produced a PCE of 5.6% under full sun (UOC = 0.821 V, jSC = 10.17 mA·cm−2) with the dye MK2.
In 2016 Ellis, et al. reported a high efficiency aqueous DSSC system using a much more simplified device composition [131]. At first, they proposed Cl− as counterion of the Co complexes. Dong, et al. had reported the use of NO3− counter anions, however, the solubility was still limited and heating was required in order to solubilize the coordination complex. The solubility was then improved from 0.09 M for [Co(bpy)3(NO3)2] to 0.13 M for [Co(bpy)3Cl3] at room temperature. The second issue they addressed was the wetting of the working electrode. They used two dyes, LEG4 and D51 to perform the study. It was speculated that, by using the much shorter pendant methoxy (OCH3) groups present on D51, the water/dye interactions will be improved compared to the butoxy(OC4H9) chains of LEG4. They confirmed this hypothesis by measuring contact angles and found complete wetting for D51 electrodes whereas 105° was obtained for pure water and 48° when using the same electrolyte composition for LEG4. The authors performed the electrochemical characterization of two Co coordination complexes in aqueous electrolyte. They consistently found a less positive redox potential upon replacing the organic solvent by water. A value of 0.33 V versus SHE was measured for [Co(bpy)3Cl2] and [Co(phen)3Cl2] in water, whereas those potentials were found to be respectively 0.56 V versus SHE and 0.63 V versus SHE in acetonitrile. This negative shift of the redox potential was attributed to a much more stabilized charged species in the aqueous electrolyte due the high dielectric constant of water. A first batch of devices was assembled with D51 and two Co coordination complexes: [Co(bpy)3(III/II)] and [Co(phen)3(III/II)]. The performance obtained was respectively 1.4% (UOC = 0.63 V, jSC = 3.4 mA·cm−2) and 3.4% (UOC = 0.75 V, jSC = 6.3 mA·cm−2) both under 1000 W·m−2. These values were further improved to 4.8% for [Co(phen)3(III/II)] under full sun (UOC = 0.78 V, jSC = 8.6 mA·cm−2) by increasing the concentration of Co(III) species to 0.06M in order to reduce some diffusion limitations. Finally, the authors fabricated devices with Cl− as counterion of [Co(bpy-pz)3(III/II)] to improve the UOC. Due to the more positively shifted redox potential of the latter complex, they managed to obtain an impressive PCE of 5.5% (UOC = 0.9 V, jSC = 8.1 mA·cm−2) under full sun.
Relatively few studies have been performed regarding the DSSC long-term stability with Co redox mediators. One of the reasons is that non-volatile electrolytes are required for such studies, and the photocurrents in such media under full sun are low due to mass-transport limitations, resulting in low PCEs.
Jiang, et al. [132] published a study comparing the stability of DSSC with the Ru sensitizer Z907 and the [Co(bpy)3(III/II)] couple dissolved in either acetonitrile (ACN) or 3-methoxypropionitrile (MPN, boiling point 163 °C). In ACN, after 1000 h of full sun irradiation at 20 °C, the PCE decreased to 66% of its initial value of 6.3%. Better stability was obtained with MPN, with the efficiency declining to 91% of its initial value of 4% under the same light-soaking conditions.
One way to improve the stability of a DSSC is the immobilization of a liquid electrolyte into a polymer matrix, so that a quasi-sold state DSSC is obtained. Bella, et al. [133] demonstrated that by immobilization of ACN into a polymer matrix the long-term stability is substantially improved. The particular approach of the authors for in-situ electrolyte immobilization consists of the addition of a monomer precursor, BEMA or PEGMA, as well as a photoinitiator, into the electrolyte and, after assembling the DSSC, exposure to UV irradiation for a short time. A DSSC with dye LEG4 containing the [Co(bpy)3(III/II)] redox mediator in an electrolyte immobilized by this approach, was subjected at first to dark soaking at 60 °C for 1500 h followed by full-sun irradiation for 300 h at 40 °C. At the end of this treatment, the DSSC retained 90% of the initial PCE of 6%. Contrarily, for a similarly prepared DSSC with an ACN electrolyte but not subjected to cross-linking polymerization, the same dark-light soaking treatment leads to the initial PCE drastically declining to around 20% of the initial value. It should be noted that no UV filter was used during this light-soaking procedure, contrarily to the practice for similar stability test on DSSCs, demonstrating the good stability of the particular quasi-polymer solid electrolyte to exposure in the UV part of the solar spectrum.
The structural modification of the mediator itself can play a role in improving the long-term stability. Freitag, et al. [134] studied a novel Co mediator of the hemicage type in an ACN electrolyte toward DSSC stability. In a DSSC cosensitized with the organic dyes C218 and MKA 253, after exposure to full sun at room temperature for 110 h, the PCE increased to around 120% of its initial PCE of 6.6%. In a similar DSSC sensitized with organic dye LEG4, the PCE also increased, to 110% of its initial value of 5.4%. In marked contrast, the performance of cells with the traditional [Co(bpy)3(III/II)] electrolyte drastically declined after the same treatment, to 85% of the initial PCE and 60% for dye LEG4 and dye mixture C281/MKA 253 respectively.
The effect of electrolyte additives on long-term stability was studied by Gao, et al. [135]. Li+ is a frequently used additive to DSSC electrolytes with Co mediators. The authors studied 3 types of DSSCs with the LEG4 dye and three Co(bpy)3-based electrolytes: with Li+ as cation additive, 1-ethyl-methyl-imidazolium (EMI) cation as Li+ replacement, and without additive. The light-soaking treatment consisted of exposing the DSSC to full sun during 1000 h at 60 °C. In presence of Li+ a gradual decrease of PCE was observed, to reach 69% of the initial value of 7.1%, at the end of the 1000 h treatment; the UOC and jSC decreased to 81% and 95% of their initial values of 0.92 V and 12.0 mA·cm−2, respectively. This rapid PCE degradation was attributed to long-term dye desorption in presence of Li+.
For the electrolyte containing the EMI cation additive, an initial increase of PCE was observed to 107% of the initial value of 6.6%, followed by a gradual decline to reach 88% of the initial value, at 5.8%. After 1000 h the UOC decreased to 81% of its initial value of 0.92 V and the jSC increased to 108% of its initial value of 10.9 mA·cm−2.
Similar was the behavior in the absence of any cation additive. The PCE increased at first to 117% of the initial value of 7.3%, and then decreased to 95%. After 1000 h the UOC decreased to 83% of its initial value of 0.92 and the jSC increased to 111% of its initial value of 10.5 mA·cm−2.
The key result of this study is that in the case of the electrolyte containing Li+, for which the PCE is the highest, the long-term stability is the worst. In view of the routine addition of Li+ in DSSC electrolytes, this study deserves particular attention. Another remarkable fact of the stability results of both Gao, et al. [135] and Freitag, et al. [134] is that they were obtained with volatile ACN electrolytes.
In summary, in this section we presented the impressive, mostly recent, research on developing high efficiency DSSCs based on Co electrolyte. Co(III/II)-based redox mediators offer the attractive feature to have a very fast and simple electron transfer mechanism which is, compared to that of the iodine system, much advantageous in terms of losses related to the dye regeneration driving force. This allowed to push the records and drastically improve the DSSCs performance, notably in terms of open-circuit voltages(see Table 1). The electrochemical properties of Co-based coordination complexes can also be finely tuned through rational design and engineering of the coordinating ligand. As we highlighted in the earlier paragraphs, either the electronic density of the ligands or their coordination density can be altered.

Table 1.
Photovoltaic data for the devices employing cobalt redox mediators. The relative efficiency values power conversion efficiency (PCE)rel are calculated as PCErel = PCE/PCEref where PCEref is the PCE of a cell with a reference electrolyte).
The marriage between the dye and the electrolyte is a key point in the good operation of a DSSC. Therefore, we reported the major breakthroughs in terms of fundamental understanding of the dye/Co electrolyte interactions. Notably, we highlighted that bulky donors would help reducing the back recombination reactions and ultimately improving the device performance. Strictly speaking from the dye perspective, we showed that more promising results and performance are to be expected from a system using a combination of an organic dye and a Co electrolyte, rather than a Ru-dye and a Co electrolyte. In this regard, we also presented the large compatibility of those Co coordination complexes with an impressive library of different organic dyes, each bearing different building blocks and properties. Detailed information about photovoltaic performances, associated sensitizer and counter electrode materials can be found in Table 1.
7. Copper Mediators
Cu is a first row transition metal; therefore, its ions are likely to form coordination complexes adopting different geometries. As a d10 system, Cu(I) generally favors adopting a four-coordinate complex in tetrahedral geometry [136]. On the other hand, as a d9 system, Cu(II) complexes can exhibit different stereochemistry such as six-coordinate tetragonal (distorted octahedral) or five-coordinate (square pyramidal or trigonal bi-pyramidal) geometries [137]. As a result of the different geometry preferences of Cu(I) and Cu(II) species, the electron transfer reactions in unconstrained Cu(II/I) systems are expected to require significant reorganizational energies concerning the internal changes in bond lengths and angles as well as the surrounding matrix. Nevertheless, with steric constraints the coordination number and the preferred geometry of Cu species could be altered. Accordingly, the activation energy barrier for electron transfer reactions can be modified [138]. For instance, “blue-Cu” [139] proteins which are involved in many chemical and biochemical catalytic systems in nature show proper redox potentials and electron transfer kinetics owing to their ligand environment [140,141,142].
Cu coordination compounds were utilized as DSSC redox mediators for the first time by Yanagida and coworkers [143]. In this pioneering work, they emphasized the significant features of Cu species to be employed as a redox couple in DSSC applications. According to the Franck–Condon principle as clarified by Marcus [144], in order to achieve fast electron transfer and minimize the required energy for transition between oxidized and reduced species, the geometry of the Cu complexes should be adjusted. Yanagida and co-workers [143] employed [(−)-sparteine-N,N′](maleonitriledithiolato-S,S′)Cu ([Cu(SP)(mmt)]), as a blue Cu model complex, which closely mimics the spectral and redox behavior of the blue Cu proteins involved in the photosynthetic chain in nature. With this redox couple, by achieving a distorted tetragonal geometry, the difference in bond lengths and geometries between Cu(I) and Cu(II) is cleverly minimized and therefore the Cu site is optimized for fast electron transfer and better dye regeneration. Additionally, they studied bis(2,9-dimethy-1,10-phenanthroline)Cu ([Cu(dmp)2](CF3SO3)2) which also exhibits distorted tetragonal geometry due to the steric hindrance effects of methyl groups in the 2,9 positions of phenanthroline ligand. They reported a comparison of the blue Cu model complex and [Cu(dmp)2] to [Cu(phen)2](CF3SO3)2(phen = 1,10-phenanthroline), in which the four N-donor atoms are expected to be nearly coplanar. It was shown that the electron self-exchange rate constant increases in the order: [Cu(phen)2]2+/+ < [Cu(SP)(mmt)]0/− < [Cu(dmp)2]2+/+. With [Cu(dmp)2]2+/+ the structural change between the Cu(II) and Cu(I) complexes was minimized due to the distorted tetragonal geometry. Additionally, they emphasize the importance of choosing electron mediators with sufficiently positive redox potentials, in order to obtain high energy conversion efficiencies with high photovoltage outputs. By employing the N719 dye, the PCE values were reported as 1.4%, 1.3% and 0.12% for [Cu(dmp)2]2+/1+, [Cu(SP)(mmt)]1−/0 and [Cu(phen)2]2+/1+ complexes, respectively, under full-sun illumination. With [Cu(dmp)2]2+/1+, the UOC (0.79 V) was reported to be higher than with the other two complexes and with I3−/I− in accordance with the formal redox potentials of these complexes. (For [Cu(dmp)2]2+/1+; the redox potential was reported as 0.66 V versus Fc+/Fc.)
By following a different strategy, Bignozzi and co-workers [145] also developed new Cu complexes ([Cu(bpy-(COOEt)2)2]2+/1+, [Cu(bpy-(COOnbut)2)2]2+/1+, [Cu(bpy-(COOtbut)2)2]2+/1+, [Cu(PQ)2]2+/1+, [Cu(MeTBPQ)2]2+/1, [Cu(BQ)2]2+/1+) and investigated them as redox mediators in conjunction with Z907-sensitized DSSC devices. In order to minimize the back electron-transfer rates, i.e., the recombination of injected electrons with the oxidized redox species, they preferred Cu complexes having high reorganization energies. Considering the relevant nuclear re-arrangement occurring in tetrahedral Cu(I) oxidation to octahedral or trigonal-bipyramidal Cu(II) species (and vice versa), sluggish electron recombination rates were expected. However, following this strategy resulted in slow dye regeneration processes. With this set of molecules, the IPCE maxima values lied around 35–40%. Among other complexes, [Cu(BQ)2]2+/1+ was reported to have a higher formal redox potential, and therefore smaller driving force for dye regeneration. This complex could however not perform well with Z907 dye. It was also shown that 4,4′-di-substituted bipyridine complexes exhibit very slow electron transfer kinetics, and the bulky substituents in the same position are not advantageous in terms of suppressing the recombination. Correspondingly, both the dye regeneration and the electrochemical response of the CE are negatively affected.
Wang and co-workers [146] re-examined the [Cu(dmp)2]2+/1+ redox shuttle in combination with the C218 dye. By employing the C218 dye, a 7% PCE was reported ((UOC = 0.932 V, jSC = 11.3 mA·cm−2, FF = 0.66). In this work low electron transfer rates on several noble metals, carbon black, and conductive oxides were reported to result in poor fill factors. The same complex [Cu(dmp)2]2+/1+, was studied by Freitag, et al. [147] with an organic dye (LEG4) and a PEDOT-coated CE. In this work, the use of a [Cu(dmp)2]2+/1+ electrolyte led to a higher photovoltaic performance, with a PCE of 8.3% for LEG4-sensitized TiO2 solar cells, with a remarkably high UOC of above 1.0 V under full-sun conditions (jSC = 12.6 mA·cm−2, FF = 0.62). In comparison, the efficiency obtained in the same study was 7.3% with the [Co(bpy)3]3+/2+ mediator (UOC = 0.88 V, jSC = 7.33 mA·cm−2). Additionally, it was unveiled that a small driving force for dye regeneration of only 0.2 eV is sufficient to obtain a virtually unit yield in dye regeneration. By means of electrochemical analysis, it was shown that the Cu complexes have higher diffusion coefficients compared to the Co-based alternatives. Therefore, the Cu-based systems considered exhibit less mass transport limitations compared to Co-based systems. A drawback is the quenching of the dye excited state by the mediator. According to steady-state emission and time-correlated single-photon counting (TC-SPC) measurements, the authors reported bimolecular reductive quenching of the excited LEG4 dye by the [Cu(dmp)2]2+ complex through a dynamic mechanism. Quenching of the reduced dye molecules by the [Cu(dmp)2]2+ complex competes with electron injection and results in a lower photocurrent. It was reported that the quenching of excited LEG4 by the [Cu(dmp)2]1+,[Co(bpy)3]2+, and [Co(bpy)3]3+ complexes followed a static mechanism, however only for the [Cu(dmp)2]2+ complex a dynamic quenching mechanism is engendered from purely collisional encounters between the excited dye and the [Cu(dmp)2]2+ complex. They suggested structural modifications in dye and redox molecules in order to inhibit unwanted quenching processes and hence achieve higher photovoltaic conversion efficiencies. Pradhan et al. [148] studied the gain and loss processes associated with different electrolytes in DSC systems. In this study, a detailed interfacial charge transfer dynamics investigation was performed by using several perturbation techniques employing, Co[(bpy)3]3+/2+ and Cu[(dmp)2]2+/1+ electrolytes.
Cong, et al. [149] reported a novel Cu mediator, [Cu(bpye)2]2+/1+, with the 1,1-bis(2-pyridyl)ethane ligand. They achieved a PCE of 9.0% (UOC = 0.90 V, jSC = 14.1 mA·cm−2) under full sun and of 9.9% (UOC = 0.88 V, jSC = 7.3 mA·cm−2) under 50% sun for this Cu mediator with LEG4 sensitized devices. From cyclic voltammetry (CV) measurements the redox potential of [Cu(bpye)2]2+/1+ was determined as 0.59 V versus SHE, which is 0.030 V higher than that of [Co(bpy)3]3+/2+ (0.56 V versus SHE). The authors reported faster dye regeneration and slower recombination processes, and less charge transport problems with [Cu(bpye)2]2+/1+ in comparison to [Co(bpy)3]3+/2+ as a consequence of the faster electron self-exchange rate of [Cu(bpye)2]2+/1+. They also performed stability tests during 1000 h for DSSC devices employing [Cu(bpye)2]2+/1+ at 40 °C in the light. The efficiency value showed an initial decline of from 9% to 6% and remained stable for around 700 h.
Saygili, et al. [56] reported two Cu bipyridyl complexes, [Cu(dmby)2]2+/1+ (0.97 V versus SHE, dmby = 6,6′-dimethyl-2,2′-bipyridine) and [Cu(tmby)2]2+/1+ (0.87 V versus SHE, tmby = 4,4′,6,6′-tetramethyl-2,2′-bipyridine) as new redox couples for DSSCs. A [Cu(dmby)2]2+/1+ complex was similarly designed as [Cu(dmp)2]2+/1+ by keeping the methyl groups at the two positions adjacent to nitrogen atoms and replacing the phenanthroline with bipyridine. The [Cu(tmby)2]2+/1+ complex has two additional methyl groups at each ligand (4,6-positions), in which the electron-donating effect of the methyl groups shifts the redox potential by 0.1 V in the negative direction. Similarly to [Cu(dmp)2]2+/1+, by maintaining the coordination geometry around the Cu metal center during the change of the Cu oxidation state from I to II, internal reorganization energies were minimized. This allowed the dye regeneration to proceed rapidly at driving forces as low as 0.1 eV. According to density functional theory calculations, the predicted inner-sphere reorganization energies (λin) were found to be about 0.3 eV, which is very low in comparison with those of Co species. For example, for [Co(bpy)3]3+/2+ and similar complexes, the λin values computed with the same approach lie in the range of 0.52–0.63 eV, comparable with the λin values for Co(II) as low spin; λin lies in the range of 1.39–1.78 eV for high-spin Co(II)) [150]. With Y123-sensitized devices, UOC values of over 1.0 V was achieved. The PCE was reported to be higher than 10% under 1000 W·m−2 AM1.5G illumination. In the same work [56], it is also reported that in the case of [Cu(tmby)2]2+ and [Cu(dmby)2]2+ species, more complex voltammograms were obtained, with additional peaks negatively shifted with respect to the redox wave of Cu(I) species. In fact, the authors reported that, upon preparation of the Cu(II) complexes by the reaction of CuCl2 with the corresponding ligand, more complex products than originally assumed are generated. In contrast, the chemical oxidation of Cu(dmp)+ by NOBF4 provided a clean and well-defined cyclic voltammogram. Following this work, Kavan, et al. [151] studied [Cu(dmp)2]2+/1+, [Cu(dmby)2]2+/1+ and [Cu(tmby)2]2+/1+ complexes on various electrodes and in several compositions of electrolyte solutions using cyclic voltammetry and impedance spectroscopy on symmetrical thin-layer cells with identical catalytic electrodes. They showed that the charge-transfer kinetics and the diffusion rate of Cu mediators are highly dependent on the presence of 4-tert-butylpyridine (TBP), a usual additive in DSSC electrolytes. They attributed this dependence on the sensitivity of the coordination sphere of Cu(II)-species on structural and substitutional changes [152]. Moreover, electrochemically clean Cu(II)–bipyridine species were produced by electrochemical oxidation of the parent Cu(I) complexes in order to prevent Cl coordination to Cu(II) species. In order to prepare electrochemically clean Cu(II) species, Ferdowsi, et al. [153] used Cu(II)(TFSI)2 as starting species. With this approach, they obtained electrochemically and optically clean Cu(II/I) redox mediator solutions which exhibit improved charge-transfer rate at the CE (PEDOT) and faster diffusion in the solution. They also studied the effect of four pyridine derivatives: TBP, 2,6-bis-tert-butylpyridine (BTBP), 4-methoxypyridine, and 4-(5-nonyl)pyridine as electrolyte additives. They showed that the electrochemical properties of the redox mediators are strongly affected by the presence and type of these bases. They reported that 2,6-bis-tert-butylpyridine has the smallest influence on the electrochemical behavior of the mediator due to steric hindrance. Owing to the bulky structure of BTBP, the coordination to Cu(II) species is precluded. However, the same base is also ineffective in terms of UOC enhancement through a TiO2 conduction band upshift. The authors showed that there is a correlation of basicity (pKa) of the used pyridine derivatives, charge-transfer rate at the PEDOT surface, and diffusion resistance. They reported that 4-(5-nonyl)pyridine is outperforming all the remaining bases as regards the efficiency of the corresponding solar cells. Saygili et al. [154] investigated the recombination of injected electrons with oxidized redox species and regeneration behavior of Cu[(dmby)2]2+/1+, Cu[(tmby)2]2+/1+, Cu[(eto)2]2+/1+ (eto = 4-ethoxy-6,6′-dimethyl-2,2′-bipyridine), and Cu[(dmp)2]2+/1+ in conjuction with the D5, D35, and D45 dyes. They showed that the DSCs employing D35 dye with Cu[(dmp)2]2+/1+ reached a record value for the open circuit voltage of 1.14 V without compromising the short circuit current density value. In addition with D5 dye, which lacks recombination preventing steric units, open circuit voltage values as high as 1.13 V is possible with Cu[(dmp)2]2+/1+ and Cu[(dmby)2]2+/1+ electrolytes. They showed that the charge recombination stays in Marcus normal regime, in the presence of 4-tert-butylpyridine (TBP). In addition, the recombination resistance and electron lifetime values were higher for the copper redox species in comparison to the reference cobalt redox mediator.
[Cu(dmby)2]2+/1+ and [Cu(tmbp)2]2+/1+ complexes were also studied in comparison to [Co(bpy)3]3+/2+ and I3−/I− by Li, et al. [155]. In conjunction with LEG4 dye, the [Cu(dmby)2]2+/1+ system achieved a UOC of 1.048 V, which is by 0.324 V and 0.204 V higher than that of the I3−/I− and [Co(bpy)3]3+/2+ systems respectively, owing to the more positive redox potential of [Cu(dmby)2]2+/1+. The [Cu(dmby)2]2+/1+ system, with only 0.11 eV driving force for dye regeneration, showed a similar dye regeneration rate as the I3−/I− system and a two times faster rate than the [Co(bpy)3]3+/2+ system. The overall power conversion efficiency reported to be 10.3% at 100 mW·cm−2. The platinized CE resulted in a higher charge-transfer resistance, indicating a poor catalytic activity of [Cu(dmby)2]2+/1+ with Pt [152].
Ferdowsi, et al. [156] compared the photovoltaic performance of the [Cu(tmby)2]2+/1+, I3−/I− and [Co(bpy)3]3+/2+ systems with a novel ND dye (4-{[({7-(4-[bis(4-(hexyloxy)phenyl]amino}phenyl) benzo[c][1,2,5]thia-diazol-4-yl]ethynyl}benzoic acid). With a [Cu(tmby)2]2+/1+ electrolyte the PCE was reported as 7.15%, which was the highest among the three tested redox couples. Lui et al. [157] studied a series of indacenodithiophene (IDT)-based d–p–A organic dyes with varying electron-accepting units by employing Cu[(tmby)2]2+/1+. By control of the electron affinity of the dyes and matching the energy levels of the sensitizer and the electrolyte, high open-circuit voltage values (>1.1 V) and harvesting a large fraction of solar photons was achievable. They reported PCE values of 11.2% and 28.4% under standard AM 1.5 G sunlight and under a 1000 lux fluorescent light tube.
García-Rodríguez, et al. [158], improved the power conversion efficiency of their DSSC devices employing the [Cu(dmp)2]2+/1+ redox couple, by reducing the electrode separation distance using an epoxy resin sealing method, with which they improved the diffusion between WE and CE and accordingly the FF. In the low-frequency range of EIS spectra they observed an inductive behavior for epoxy-sealed devices, which is associated with direct interaction between electrons in the TiO2 mesoporous layer and the PEDOT CE. This inductive behavior is prevented by an insulating ZrO2 interlayer which is applied on the mesoporous TiO2 layer.
Freitag, et al. [159] employed the [Cu(tmby)2]2+/1+ redox couple with TiO2 films cosensitized with D35 and XY1 dyes. They reported the ambient power outputs of these devices as 15.6 and 88.5 mW·cm−2 at 200 and 1000 lux, respectively, under illumination from a model Osram 930 warm-white fluorescent light tube. The PCE was reported as 28.9% and 11.3% under light provided by the 1000 lux fluorescent light tube and full sun, respectively. The spectra of the indoor light source differ from that of sunlight. Good matching between the absorbance of D35/XY1 dye blends and the emission of indoor light source resulted in very high PCE values for ambient lighting. The power output obtained under low light illumination was considered to be sufficient to power or to extend the battery lifetime of a certain range of electronic devices operating in indoor environment. In succeeding research, by directly contacting the PEDOT CE to the photoanode, that is completely avoiding a spacer between the two electrodes, Cao, et al. [160] reported the record PCE of 13.1% under full sun, by employing XY1 and Y123 dye blends with the [Cu(tmby)2]2+/1+ redox mediator. For the same device design, they reported a record power conversion efficiency of 32% under ambient light (1000 lux) which exceeds the performance of today’s best photovoltaics based on silicon or GaAs. These results show that DSSCs are promising candidates for powering a range of electronic devices including e-readers, tablets, and Internet of Things-related appliances.
Hoffeditz, et al. [161] developed a Cu redox shuttle obtained from the 1,8-bis(2′-pyridyl)-3,6-dithiaoctane (PDTO) ligand and the common DSSC additive TBP. In this work they showed that, upon removal of one metal-centered electron, the PDTO ligand surrounding the Cu ions can be replaced with four or more TBP ligands. Thus, in the electrolyte Cu(I) and Cu(II) species exist with different ligand coordination structures, i.e., Cu(PDTO)+ and Cu(TBP)4+x(ACN)y2+. These species are reported to be interconvertible based on transfer of a single electron. The variance of the coordination spheres of Cu(I) and Cu(II) species results in widely differing Cu(II/I) formal potentials and reactivities for the forward as compared to the reverse electron transfer. It is reported that various factors such as charge-transfer resistance, dark current suppression and redox couple potential, and, as a result, the photovoltaic performance of the devices, are highly dependent on the TBP concentration. With increasing TBP concentration both UOC and JSC values are increased, however the FF values are decreased (UOC = 0.88 V, JSC = 4 mA·cm−2, FF = 0.52). Wang et al. [162] studied the effect of TBP as an electrolyte additive. They reported that the TBP substitutes the bidentate ligands on the Cu(II) species to form [Cu(TBP)4]2+. By being a poor electron acceptor, [Cu(TBP)4]2+, provided high voltages and charge collection efficiencies.
Colombo, et al. [163] developed novel [Cu(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2]PF6 and [Cu(2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline)2]PF6 redox couples for DSSC applications. They showed that the Cu(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2]PF6 couple exhibits superior dye regeneration kinetics with respect to simpler phenanthroline-based Cu(I) complexes and a lower dark current with respect to the I3−/I− electrolyte. By using a Fe(II) co-mediator, in combination with the [Ru(4,4′-dicarboxy-2,2′-bipyridine)2(4,4′-dinonyl-2,2′-bipyridine)](PF6)2 dye, they improved the performance of the cells employing [Cu(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2]PF6; these experiments are further discussed in Section 13 dedicated to dual mediator systems. They observed that the ε1/2 of this “kiss lock” complex is significantly shifted to the negative direction (ca. by −0.3V, at −0.02 V versus Fc+/Fc) with respect to that of [Cu(dmp)2]2+/1+, whereas it is essentially identical for [Cu(2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline)2]PF6 and [Cu(dmp)2]2+/1+ (at 0.29 V and 0.30 V versus Fc+/Fc, respectively). In a related publication, Colombo, et al. [164] describe the design of a series of Cu complexes that bear different substituents in the α-positions of phenanthroline. With different ligand substitutions (2-(2-Tolyl)-1,10-phenanthroline, 2-tert-butyl-1,10-phenanthroline, 2-phenyl-1,10-phenanthroline),(2,9-diTolyl-1,10-phenanthroline))) they produced proper candidates for DSSC applications; these complexes feature appropriate redox potentials, distorted tetragonal geometry and effective shield of metal core.
Magni, et al. [152] further studied the redox couple [Cu(I/II)(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2] in comparison to [Cu(I)(dmp)2] and its penta-coordinated oxidized form [Cu(II)(dmp)2Cl]. With these redox couple, they reported an overall 4.4% solar energy conversion efficiency with a π-extended benzothiadiazole dye (G3). For the same experimental conditions devices employing the [Cu(dmp)2]2+/1+ redox couple showed 1.9% efficiency. In this work, they reported that the coordination of a fifth ancillary ligand (Cl− ion or TBP) to Cu(II) species has a detrimental effect on charge transfer processes. They showed that Cl-coordinated Cu(II) (penta-coordinated) species have higher reorganization energy associated with the Cu2+/Cu+ redox process in comparison to tetra-coordinated Cu(II) species. They also reported that the steric hindrance effects of the methyl groups in [Cu(dmp)2]1+ and the two mesityl rings of [Cu(II)(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2]PF6 are different. They speculated that the mesityl rings may induce a smaller conformational modification upon the redox reaction, acting as a “kiss-lock enclosure” that causes a more negative redox potential. This observation is attributed to the destabilization of the electrogenerated Cu(II) species [165], which is expected to reduce the activation barrier for electron transfer.
Benazzi, et al. [166] studied homoleptic Cu redox couples in which the Cu center is coordinated by two 1,10-phenanthrolines bearing various substituents in position 2. ([Cu(2-tolyl-1,10-phenanthroline)2]2+/1+, [Cu(2-phenyl-1,10-phenanthroline)2]2+/1+, and [Cu (2-n-butyl-1,10-phenanthroline)2]2+/1+. They showed that Cu(I) compounds with 2-subsituted ligands exhibit a decreased molar absorption coefficient of the MLCT bands as compared with Cu(I) compounds with di-substituted ligands. This effect becomes stronger if aryl groups are used instead of alkyl ones. They found that the formal redox potential and electron transfer rates are not directly related due to the influence of the geometry of the complex and of its flexibility. Rotational freedom of the internal substituents has also an effect on the electrochemical behavior of the complexes as well as on steric hindrance effects. They also showed that the Cu(I) species with asymmetric substitution exhibit a decreased molar absorption coefficient in comparison to [Cu(dmp)2]+. This effect is expected to reduce the competitive absorption of light by the mediator with respect to dyes. They also reported that DSSCs with [Cu(2-tolyl-1,10-phenanthroline)2]2+/1+ outperform the control cells prepared with [Co(bpy)3]3+/2+ and I3−/I− electrolytes. They used p-extended benzothiadiazole molecules (G3 or G4) as dyes. With these set of redox couples, the dye regeneration efficiency is reported to be higher than 96%, which is comparable or even higher than those obtained with [Co(bpy)3]3+/2+ and I−/I3− reference solutions.
Colombo, et al. [167] studied [Cu(2-mesityl-1,10-phenanthroline)2]12+/+ and other Cu redox mediators, [Cu(dmp)2]2+/1+ and [Cu(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2]1+/2, in conjunction with Zn2+ porphyrin dyes (D1 and D2). It was stated that the dye regeneration constant for Cu+ is 2-fold and 4-fold faster than for Co2+ ([Co(dtb-bpy)3]3+/2+) and I−, respectively, at equimolar concentrations. The best performance was reported for the new [Cu(2-mesityl-1,10-phenanthroline) 2]2+/1+ redox couple at a PCE of 3.7% under full sun (jSC = 5.9 mA·cm−2, UOC = 0.81 V and FF = 0.77) with the D1 dye.
Dragonetti, et al. [168] studied a heteroleptic Cu dye with Cu(II/I)(2-n-butyl-1,10-phenanthroline) and [Cu(dmp)2]2+/1+. Due to a higher molar absorption coefficient, [Cu(dmp)2]2+/1+ showed lower short-circuits photocurrents compared to Cu(II/I)(2-n-butyl-1,10-phenanthroline)2, which is in agreement with a lower IPCE value at 475 nm for [Cu(dmp)2]2+/1+. The highest PCE value is reported as 2% with Cu(II/I)(2-n-butyl-1,10-phenanthroline)2 and a new copper dye D (Jsc = 6.3 mA·cm−2, UOC = 0.61 V and FF = 0.53). Karpacheva, et al. [169] also studied heteroleptic Cu(I)-based dyes in conjunction with homoleptic Cu(II/I) redox shuttles. They obtained a DSSC PCE of 2.06%. The introduction of electron-donating methoxy groups in [Cu(4,4′-dimethoxy-6,6′-dimethyl-2,2′-bi-pyridine)2] results in a lower oxidation potential with respect to [Cu(dmby)2]. It was reported that the PCE improvement with the [Cu(4,4′-dimethoxy-6,6′-dimethyl-2,2′-bi-pyridine)2]2+/1+ redox couple is achieved by a jSC increase in spite of a UOC loss.
Leandri, et al. [170] investigated the reaction of CuCl2 with the dmp ligand. As previously reported [151,152,156,165], it was shown that using CuCl2 as a starting material for Cu(II) synthesis results in a 5-coordinated complex with a less positive half-wave electrode potential in comparison to Cu(I). Demonstrating the importance of the counterion in the redox chemistry of Cu coordination complexes, they suggested using Cu(II) salts with weakly and non-coordinating counterions (such as perchlorate) in order to synthesize Cu(II) species. They showed that the growth of [Cu(dmp)2](ClO4)2 crystals in acetonitrile results in a 5-coordinated complex, [Cu(dmp)2(CH3CN)](ClO4)2, in which a solvent molecule is coordinated to the metal center. They reported that in oxidation/reduction processes the solvent molecule follows coordination/de-coordination behavior. Therefore, the peak potentials obtained in cyclic voltammogram of [Cu(dmp)2(CH3CN)]2+ are identical to the ones of [Cu(dmp)2]1+, if the measurements are performed in acetonitrile.
Pernechele [171] investigated Cu complexes with different monodentate ligands (2-methoxpyridine, 3-methoxypyridine and 4-methoxypyridine), in order to avoid ligand exchange problems observed with TBP. With these redox couples based on methoxypyridines as ligands, the best PCE reported was 9.1% at 1 sun illumination with a UOC value of 0.94 V. The high photovoltage value, was attributed to the higher recombination resistance and negative shift in the conduction band of TiO2.
Michaels et al. [172] introduced new copper complexes with tetradentate 6,6′-bis(4-(S)-isopropyl-2-oxazolinyl)-2,2′-bipyridine ligands, Cu[(oxabpy)]2+/1+, as redox mediators. With the corresponding ligands, the copper complexes exhibit square-planar geometry providing low reorganization energies. The Cu[(oxabpy)]2+/1+ complexes were obtained as viscous gel-like solutions. Nevertheless, an enhancement of charge transport performance was observed in reference to Cu[(tmby)2]2+/1+ redox couple. Following this work, Hu et al. [173] also investigated tetradentate copper complexes with diamine–dipyridine tetradentate ligands (N,N′-dibenzyl-N,N′-bis-(pyridin-2-ylmethyl)ethylenediamine and N,N′-dibenzyl-N,N′-bis(6-methyl-pyridin-2-ylmethyl) ethylenediamine) as redox mediators in DSCs. DSCs with these set of redox couples showed an outstanding long-term stability by maintaining ~90% of the initial efficiency over 500 h under continuous full sun irradiation.
Apart from the conventional liquid electrolyte-based DSSCs, Freitag, et al. [174] demonstrated that by slow solvent evaporation in liquid state devices employing Cu(dmp)2]2+/1+, a solid state DSSC (sDSSC) can be obtained. Normally in liquid state devices with solvent evaporation the devices loses the capability of photovoltaic power conversion, yet for Cu-based electrolytes the device performance is reported to be improved by solvent evaporation. For these solid-state devices, which are colloquially called as “Zombie’cells” (Z-cells or Z-sDSSCs) due to their surprising performance), by employing a LEG4 dye, the authors reported a PCE of 8.2% under full sun (UOC = 1.01 V, jSC = 13.8 mA·cm−2), which is better in comparison to conventional liquid state devices (PCE = 6.0%, UOC = 1.04 V, jSC = 9.4 mA·cm−2) with the same dye and mediator, as well as to conventional solid state devices with Spiro-OMeTAD as solid-state hole conductor (PCE = 5.6%, UOC = 0.90 V, jSC = 9.4 mA·cm−2) with the same dye. The Z-sDSSCs are considered as important candidates to solve the stability problems emerging by poor encapsulation and solvent evaporation.
Following this work, Cao, et al. [175], reported a 11.0% stable Z-sDSSC under simulated AM 1.5 G irradiation based on [Cu(tmby)2]2+/1+, with a jSC of 13.87 mA·cm−2 and a high UOC of 1.08 V). Solid state [Cu(tmby)2]2+/1+ were reported to be superior to the conventional hole transport materials (HTMs) in terms of nanopore filling and conductivity. The authors pointed to the importance of keeping the HTM in the amorphous state in order to achieve high power conversion efficiencies. In case of crystalline HTMs, the holes are likely to get trapped at crystal grain boundaries. This leads to reduced hole mobility and a built-up space charge layer in the Z-sDSSCs owing to the difference in electron and hole mobilities. They also reported better conductivity in solid Cu(II/I) HTM compared to that in the volatile electrolyte of the DSSCs under a photovoltage.
Kavan, et al. [151] studied the Z-cell variety made with symmetrical PEDOT/PEDOT electrodes. They reported that these devices show enhanced charge transfer rates and enhanced diffusion resistance in comparison to their liquid counterparts. These effects were attributed to the fact that in the Z-sDSSCs, the hopping charge-transfer (Dahms–Ruff mechanism) can be considered as the main hole-transport process, whereas the hopping mechanism is less important in the liquid state devices.
Zhang, et al. [176] also employed [Cu(tmby)2]2+/1+ in conjunction with a novel WS-72 dye. Liquid-junction redox electrolyte generates a notable UOC of 1.1 V along with an excellent PCE of 11.6% under simulated AM 1.5 G illumination. The solidification of the liquid-junction devices leads to an even higher PCE of 11.7% (jSC = 13.8 mA·cm−2, UOC = 1.07 V and FF = 0.79). With the systems, both the driving force of electron injection and dye regeneration is reduced through dye molecular engineering and choice of a proper redox mediator, respectively. Moreover, with appropriate dye design, the interfacial electron recombination is reduced.
In addition to DSSCs with a TiO2 substrate, Co and Cu coordination complexes can be used in devices with alternative n-semiconductor substrates, for example n-ZnO, as demonstrated by Ruess, et al. (2018) [177]. In a DSSC with the organic dye DN2016, of the indoline type, a PEDOT-coated CE and the Cu(tmby)2 mediator in acetonitrile, a PCE of 3.8% was obtained (UOP = 0.713 V, JSC = 7.5 mA·cm−2); with the Co(bpy)3 mediator the PCE was 3.6% (UOP = 0.618 V, JSC = 9.3 mA·cm−2).
As observed in Table 2, the most commonly employed CE materials for Cu redox mediators are PEDOT and Pt. Alternatively, Kavan, et al. [178] reported a novel, highly active, optically-transparent composite electrocatalyst, containing Pt, PtOx, graphene oxide and stacked graphene platelet nanofibers (PtGONF), for the [Cu(tmby)2]2+/1+ redox couple. They demonstrated that PtGONF outperforms PEDOT or platinum. Hoffeditz, et al. [161] replaced the Pt CE with a high area, inverse opal Pt electrode, in order to reduce the charge-transfer resistance of the CE.
Table 2.
Photovoltaic data for the devices employing copper redox mediators. The relative efficiency values PCErel are calculated as PCErel = PCE/PCEref where PCEref is the PCE of a cell with a reference electrolyte.
The DSC performance parameters for the copper redox mediators are summarized in Table 2. The ligand structures for copper redox mediators and the corresponding redox potentials are given in Figure 7.
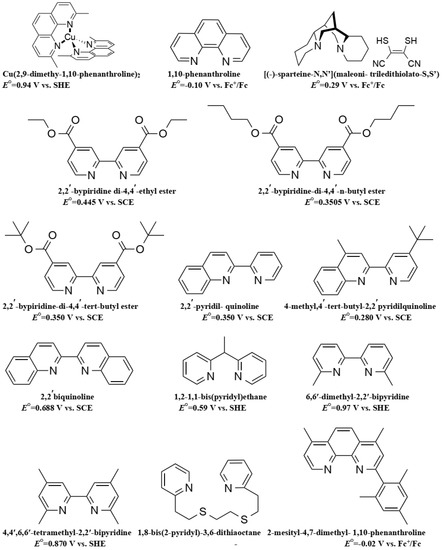
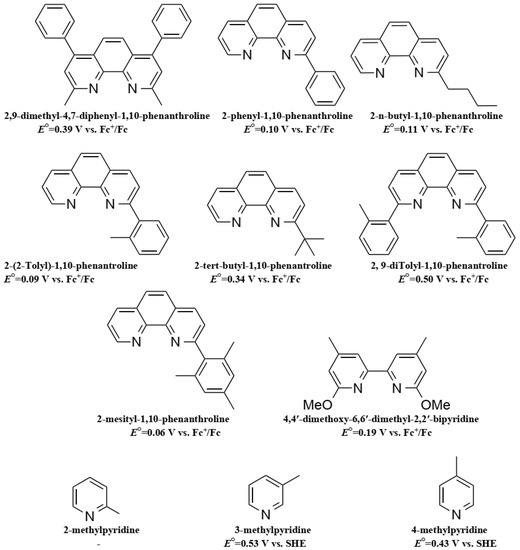
Figure 7.
Copper redox mediators and redox potentials reported for DSSC applications.
8. Iron Mediators
In general, the use of Fe for materials development have attracted a great deal of interest due to its high abundance, low cost and relatively low toxicity. High redox potentials can be obtained for several Fe complexes, which can potentially lead to improved UOC values compare to the ones obtained with iodide-based electrolytes (0.7–0.8 V). To this end, replacing Co by Fe bipyridyl complexes was studied by Rodrigues, et al., in an effort to develop alternatives to Co-based electrolytes, taking advantage of the higher stability of the low-spin (t2g6) Fe(bpy)32+ complex, compared to the high spin (t2g5e2g2) Co(bpy)32+ complex. Additionally, it has been previously reported that the Fe–N bonds in Fe(bpy)32+ are weaker than the Co–N bonds in Co(bpy)32+, which results in a more positive reduction potential (1.37 V versus SHE) for the species Fe(bpy)33+ compared to Co(bpy)33+ (0.56 V versus SHE) [156,179]. Consequently, by selecting a suitable dye with a low HOMO energy level, it could be possible to reach a higher UOC with Fe based-electrolytes than with Co electrolytes. The organic dye RR9 ((E)-2-cyano-3-(4-(7-(2,2′,2′,4,4′-pentakis(hexyloxy)-[1,1′:3′,1′′-terphenyl]-5′yl)benzo[c][1,2,5]thiadiazol-4-yl)phenyl)acrylic acid) with a low-lying energy HOMO level (1.56 V versus SHE) was judiciously synthesized to match the redox potential of Fe(bpy)33+/2+; indeed, the regeneration of the oxidized state of the dye RR9 would only require a driving force of 0.190 eV. Therefore, a negligible potential loss can be expected, and a theoretical maximum UOC of 1.870 V was calculated for this system (Figure 8) [179].
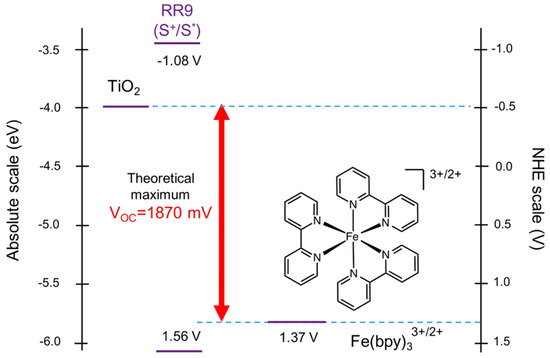
Figure 8.
Energetic scheme of DSSC with RR9 and Fe(bpy)33+/2+ electrolyte. Data from Rodriguez, et al. (2018) [138].
The highest open-circuit voltage achieved so far for a DSSC, of UOC = 1.420 V, was obtained for devices using TiO2 electrodes of 2.7 µm + 4.5 µm (working layer + scattering layer) sensitized with RR9, PEDOT as CE, and a Fe(bpy)33+/2+-based electrolyte. However, a poor overall performance was attained for such devices, with jSC = 2.8 mA·cm−2, FF = 0.47 and PCE = 1.9%. These values can be compared with reference cells based on a Co(bpy)3 electrolyte, using as sensitizer either RR9, with characteristic parameters UOC = 0.680 V, jSC = 3.5 mA·cm−2, FF = 0.72 and PCE = 1.8%, or D35 as reference dye with a PCE = 4.6% ((UOC = 0.760 V, jSC = 8.8 mA·cm−2, FF = 0.67) [179]. Correspondingly, photovoltage transient studies showed that longer electron lifetimes and decreased dark currents resulted for devices with a Fe(bpy)3-based electrolyte compared to devices of [Co(bpy)3]3+/2-based reference electrolyte. The fact that lower recombination rates were obtained for the [Fe(bpy)33+/2+]-based electrolyte was correlated to a shift to the Marcus inverted region, since increasing the driving force beyond a certain value decreases the rate of electron transfer (the difference of the energetic levels of the redox mediator and the TiO2 CB being 1.8 eV). Similar behavior was previously reported by Feldt, et al., for Co complexes where the driving force for recombination is higher than the reorganization energy of the dyes; hence, recombination rates falls within the Marcus inverted region. [106].
Sequential series multijunction (SSM)-DSSCs composed of series-connected separate devices with light absorption at different wavelengths, as discussed by Cheema, et al. [179], can maximize the photovoltage of the integrated system. The authors studied a SSM-DSSC system where one DSSC using the dye RR9 and a [Fe(bpy)3]3+/2+-based electrolyte was connected in series to two devices using Y123 as dye and Co(bpy-z)23+/2+ electrolyte. For the 3-device tandem cell a UOC = 3.340 V was obtained, corresponding to an average of more than 1 V per device, with jSC = 1.9 mA·cm−2, FF = 0.56 and PCE = 3.5%. The low jSC for the system was attributed to competition of light absorption of the dissolved Fe(bpy)33+/2+ complex with the dye.
The performance of the Fe(bpy)3 electrolyte could be further enhanced by ligand engineering; for instance the complex tris-(1,10-phenanthroline)Fe(III/II), Fe(phen)33+/2+ has been previously reported to present high redox potential in nonaqueous solvents, 0.785 V versus Fc+/Fc in (n-TBA)BF4/N-N-dimethylformamide (DMF) versus Fc+/Fc [180]; in comparison, the redox potential of Fe(bpy)3 was 0.802 V versus Fc+/Fc in the same solvent and supporting electrolyte. Consequently, the use of this complex in DSSCs would enable the attainment of a UOC as high as that already discussed for Fe(bpy)3, with the additional advantage that the phenanthroline ligand can provide higher steric protection to the Fe center toward recombination than the bypyridyl ligand; as a result, an increase in the photocurrent and the overall performance can be expected. Future research on this and similarly structured complexes can lead to the development of devices with exceptionally high UOC, a feature being of particular interest in application areas such as portable-energy charging systems, solar fuels production, and miscellaneous high-voltage gadgets, among others.
An additional study by Daenake, et al. included the no-corrosive ferrocyanide complexes as redox mediators for aqueous media DSSC [181]. The redox potential reported for the complex Fe(CN)64−/3− varies from 0.466 V versus SHE to 0.514 V versus SHE [182,183,184], depending on the electrolyte, and it is likely to reach higher UOC values than those obtained with iodide-based electrolytes. The high molar extinction coefficient of the carbazole dye MK-2 makes it appropriate for thin film sensitization. Therefore, it was selected in order to reduce interfacial area for recombination as well as to alleviate mass-diffusion problems of the electrolyte-active species between PE and CE. In order to test the suitability of this complex for DSSC, devices were assembled with a PE comprised of a 1.3 μm active layer and a superposed 5.0 μm of scattering layer sensitized with MK-2 and a Pt-coated CE, and filled with the Fe(CN)63−/4− aqueous electrolyte. The characteristic operational parameters under full sun were UOC = 0.761 V, jSC = 7.21 mA·cm−2, FF = 0.75 and PCE = 4.1%, while reference cells with I3−/I− and the dye MK2 presented characteristic parameters of UOC = 0.769 V, jSC = 11.86 mA·cm−2, FF = 0.66 and PCE = 6.1%. The analysis of charge extraction, intensity modulated photovoltage and photocurrent spectra revealed that the recombination rate for a DSSC with a Fe(CN)64−/3− electrolyte is much faster than for iodide-electrolyte reference cells, by three orders of magnitude, once more confirming the fact that the main disadvantage of this kind of complexes is the high recombination rate. Surface treatment and development of new ligands to replace cyanide may improve the stability of this type of complexes, which, it should be noted, are prone to photolysis and photocatalytic decomposition [181].
Rutkowska, et al., improved the performance of DSSC with ferrocyanate complexes by introducing the Prussian blue-type polynuclear material nickel(II)hexacyanoferrate(III/II) ([NiFe(CN)6]−2/−1, abbreviation NiHCF) with a redox potential of approximately 0.84 V versus SHE, which is substantially more positive than that of I3−/I− or I2/I− (both at 0.54 V versus SHE in water), as well as that of the simple Fe(CN)64−/3− complex (0.36 V versus SHE) [184]. This inorganic material has the advantage of fast and reversible electron transfer reactions in the solid state; likewise, it can be easily adsorbed onto the TiO2 surface, [144] making it an attractive hole conductor material to deposit on top of sensitized-TiO2 electrodes. The NiHCF material had a mixed-valence state, as a mixture of K4NiII[FeII(CN)6] and K4NiII[FeIII(CN)6] with a ratio 1:1 of Fe2+ and Fe3+, resulting in maximized electron-hopping between Fe ions. DSSCs were prepared with TiO2 sensitized with the ruthenium dye N3, followed by the deposition of NiHCF on top of the TiO2-N3 electrode by dipping the electrode into a fresh solution of NiHCF (composed by equal parts of Ni2+ and Fe(CN)63−) and finally sealing with Pt-CE. The obtained results of UOC = 0.790 V, jSC = 8 mA·cm−2, FF = 0.7 and PCE = 4% demonstrated the ability of the material to act as hole transporter.
One of the most interesting alternatives to replace the I3−/I− couple by an Fe-based electrolyte is the well-known redox mediator ferrocenium/ferrocene (Fc+/Fc), which is a non-corrosive, fast electron exchanging outer-sphere redox couple [185]. A key advantage of Fc+/Fc is the more positive redox potential compared to that of I3−/I− (−0.32 V for the latter versus Fc+/Fc in acetonitrile according to a recent detailed study by Bentley, et al. [75]). This can lead to an enhancement of the UOC in a DSSC. An additional advantage is the fast self-exchange electron transfer rate of kex ≈ 8.5 × 106 M−1·s−1 [186], in contrast to most reported values for the exchange rate for the I−/I3− at around kex = 103 M−1·s−1 [187], which are also highly dependent on the concentration of iodine and iodide and also on the reaction for which the rate has been calculated. For the latter redox system it is also well known that the formation of several different high-energy intermediate radical species is involved in the electron transfer between I− and I3− [60,62]. The regeneration of the oxidized state of the dye by ferrocene is a rather fast process (as expected from the kex); nonetheless, recombination processes from the electrons in the conduction band of TiO2 to the ferrocenium species can be quite fast. This process can be suppressed by different methods such as passivation of reactive surface states, shifting the conduction band energy to lower values, and decreasing the electronic coupling between the oxidized/reduced state of Fc+/Fc with the dye. The most common method is surface passivation, which plays a major role in decreasing the recombination rate for devices with Fc+/Fc electrolyte. Several methods of preparing and investigating surface passivation layers are available, including spray pyrolysis, atomic layer deposition (ALD), and silane treatment. Gregg, et al. [188] investigated the effect of two passivation methods, deposition of an insulating polymer and silanization, on the suppression of the recombination current for Fc+/Fc and an organic redox mediator, TPD+/TPD, where TPD is N,N′-di-m-tolyl-N,N′-diphenylbenzidine. Klahr, et al. [189] proposed the passivation of the TiO2 layer by a superposed Al2O3 layer for Co3+/2//Co mediators, an approach which could be extended to other redox mediator in the future. Useful insights about the effectiveness of underlayers in suppressing the dark current are provided in a review article by Ito [190].
Surface electronic states from titanium(IV) ions coordinated to water or a hydroxyl ligand in place of a bridging oxo ligand, can be passivated by treating the TiO2 surface with trimethylaluminium vapor, which can convert these states to full-oxo coordination sites and form an Al2O3 blocking layer that not only reduces the recombination rate, but also enhance the photovoltage of the cells. Hamann, et al. [185] reported the suppression of the shunt processes and the increase in UOC in DSSC with Fc+/Fc-based electrolytes by depositing a 14 nm Al2O3 layer blocking layer by 400 cycles of ALD, thereby increasing the electron-diffusion length (L) almost by a factor of 12 and reducing the recombination rate almost by a factor of 140 compared to the bare electrode.
Another passivation method described by Feldt, et al. [67] is the reaction of trichloromethylsilane (as silanization agent), on the already sensitized TiO2 electrode. It was found that the dark current significantly decreased with an increase of the thickness of the silane blocking layer, and after one and two cycles of silane treatment both the jSC and UOC increased, which indicates an evident decrease in the recombination rate. From the analysis of photoelectrochemical measurements it was deduced that the electron lifetime increased with each cycle of silane treatment, as a result of the deposition of an efficient barrier for interfacial electron-transfer reactions. Nonetheless, the diffusion length was found to be in the order of 0.4 µm, much smaller than the film thickness (~5 µm), greatly affecting the charge collection efficiency [191]. In fact, one of the main drawbacks of this silanization process is the compromise on the dye regeneration rate while aiming to the reduction of the recombination rate. By means of transient absorption spectroscopy it was demonstrated that this type of treatment reduces the regeneration rate of the dye [67].
In a later study by Daenake, et al. [192] the aim was to suppress mass-transport limitations in the Fc+/Fc-containing electrolyte as well as to decrease the active area for recombination by using thin films of TiO2 (2.2 µm of mesoporous layer with a 18 nm underlayer and a 4.4 µm scattering layer) sensitized with the organic dye Carbz-PAHTDTT; Pt was used as CE. Moreover, the electrolyte contained the base TBP in order to passivate TiO2 surface states. The device performance exhibited enhanced parameters UOC = 0.842 V, jSC = 12.2 mA·cm−2, FF = 0.73, and PCE = 7.5%, which are the highest values hitherto reported for Fc-based electrolytes and can be favorably compared to reference cells with parameters UOC = 0.735 V, jSC = 13.3 mA·cm−2, FF = 0.62, and PCE = 6.1% for iodide-based electrolyte. Photocurrent spectroscopy as well as intensity-modulated photovoltage and charge extraction experiments revealed that, indeed, charge recombination is faster for Fc+/Fc than for I3−/I−. Furthermore, it was shown that the addition of chenodeoxycholic acid as co-adsorbent increased the electron lifetime of the system, seemingly by blocking interfacial charge recombination. Congiu, et al. [193] studied the development of Cu2−xS films as CEs for the Fc+/Fc electrolyte, with a lower charge-transfer resistance of RCT = 0.73 Ω·cm2, compared to the commonly used Pt CEs (RCT = 2 Ω·cm2); however, complete devices were not fabricated. Further work with this material could potentially lead to efficiencies higher than 8% with Fc-containing electrolytes. [193].
The synthesis of ferrocene derivatives by the attachment of simple alkyl chains and/or halogen atoms to the cyclopentane ring can result to a shift of the redox potential to more positive values with the possibility of higher UOC. To this aim, Hamann, et al. [185] studied the derivatives Fc-Cl+/Fc-Cl and Fc(Cl)2+/Fc(Cl)2 with redox potentials of 0.17 V and 0.31 V versus Fc+/Fc respectively. Despite the more positive redox potential, the UOC reached by both derivatives was lower than 0.6 V, which could be explained by high reorganization energies (greater than 1.4 eV), as well as shorter electron lifetimes compared to the Fc reference electrolyte.
Additionally, Daenake, et al. [194] studied the derivatives Br2Fc+/0, BrFc+/0, EtFc+/0, Et2Fc+/0 and Me10Fc+/0 as redox shuttles for DSSC (Figure 9), in order to investigate the effect of substituents introduction to the ferrocene moiety in the device performance. The introduction of halogen substituents increased the redox potential to 0.94 V versus SHE for Br2Fc+/0, and 0.80 V versus SHE for BrFc+/0, while the introduction of donor alkyl groups decreased the redox potential compared to the plain Fc+/Fc (0.63 V versus SHE), to values up to 0.57 V versus SHE for EtFc+/0, 0.51 V versus SHE for Et2Fc+/0 and 0.09 V versus SHE for Me10Fc+/0.
Figure 9.
Structure of ferrocene derivatives.
The derivatives BrFc+/0 and Br2Fc+/0 are highly unstable in the presence of TBP, thus all electrolytes were prepared excluding this additive, and tested in devices with the dye Carbz-PAHTDTT and Pt as CE. As expected from the absence of TBP in the electrolyte formulation, lower UOC values than the ones already reported for the Fc+/Fc electrolyte with TBP (0.842 V) were obtained for all derivatives. Characteristic cell performance parameters for the aforementioned derivatives are shown in Table 3.
Table 3.
Characteristic parameters for DSSC with ferrocene derivatives as redox shuttles [194]. IPCE: incident photon-to-electron efficiency.
According to the measurement of regeneration kinetics by transient absorption spectroscopy (TAS) the dye regeneration rate was rather slow for all the derivatives (within 10 ms), where the decay profiles indicated that the regeneration rates increase in the order Br2Fc+/0 < BrFc+/0 < Fc+/Fc ≈ EtFc+/0 ≈ Et2Fc+/0 ≈ Me10Fc+/0, which explained the low performance of the brominated derivatives. Additionally, given the fact that the regeneration rates are very similar for Fc+/Fc, EtFc+/0,Et2Fc+/0 and Me10Fc+/0, the low photocurrents were attributed to diffusion limitations [194]. Intensity-modulated photovoltage spectroscopy revealed that Me10Fc+/0 had the highest electron lifetime, while for Fc+/Fc, EtFc+/0, Et2Fc+/0 the electron lifetimes were very similar; lastly, the brominated derivatives exhibited the lowest lifetimes, which explains the rather low UOC obtained for them, despite larger expected values on the basis of their high redox potential. In general, for all derivatives electron lifetimes were low, which indicates that significant recombination losses were the main cause of their poor overall performance.
Several attempts to use ferrocene moieties attached to organic triphenylamine-based dyes in DSSC have been undertaken [195,196]. However, a very poor performance was obtained due to the fast recombination process, since the Fc moiety is directly attached to the dye and the reductive quenching of the dye by the Fe-containing moiety is a rather fast process, leading to efficiencies lower than 1.5%. Similar quenching behavior has been observed in the earlier days of oxide electrode sensitization research for Fe3+/2+ ions in the attempt to use them as redox mediators in aqueous solution [10].
9. Nickel Mediators
Although several coordination complexes with Ni2+ present high redox potentials, up to 0.96 V versus SHE in CH2Cl2 [197] and higher than 1 V in water [198], for many of them the oxidation process occurs at the ligand instead of the central Ni atom, and this is not a desired feature for a stable redox shuttle in a DSSC. However, Li, et al. [199] introduced the complexes Ni (III)/(IV) bis(dicarbollide) featuring two η5-coordinated deboronated (nido-2) o-carborane ligands (R = H in Figure 10), with a fast and reversible one-electron transfer process, as attractive alternative mediator for DSSC applications [199]. An interesting feature of these compounds is their low corrosivity towards metals used on the DSSC electrodes, either as catalysts or conductive supports.
Figure 10.
Structure of Ni-bis(dicarbollide) mediators. Adapted from [201], with permission from John Wiley and Sons, copyright 2010.
These Ni3+/4+-dicarbollide mediators (Figure 10, no substituents) exhibit low electron-exchange rates since the electron transfer process requires a cis-to-trans conformational rotation (Figure 10), resulting in high reorganization energies [200]; nonetheless, the kinetics of the electron transfer is fast enough to be compared to that of I3−/I−, with the possibility of effective dye regeneration [199]. The redox potential for this mediator is 0.140V more negative than that of Fc+/Fc, which represents a decrease in the expected UOC; however, surface treatment with 1 ALD cycle to form Al2O3 on the TiO2 electrodes sensitized with the N719 dye, in DSSCs with Pt as CE, increased the UOC from 0.580 V to 0.640 V, with jSC = 3.76 mA·cm−2, and PCE = 1.5% [199]. Additional surface treatment leads to a substantial decrease of photocurrent; electron collection is hindered due to the insulating nature of Al2O3. TAS revealed that dye regeneration by this mediator is rather fast (less than 2 µs), nevertheless, charge lifetime measurements showed that electron lifetimes for the Ni-electrolyte are at least 2 orders of magnitude lower compared to I3−/I−, demonstrating that a high recombination rate of the electrons from the CB of TiO2 is one of the main drawbacks of this system.
The performance of this type of mediators was enhanced by Spokoyny, et al. [201], by functionalization of the Ni–bis(dicarbollide) moiety (Figure 10) in the B(9/12) positions with different aryl electron-donating and withdrawing groups in order to tune the redox potentials, as expected the lowest redox potentials were obtained for the complexes with electron-donor groups, with the lowest of 0.37 V versus SHE for the methoxyphenyl group, and the highest redox potentials were obtained for the groups with higher electron-withdrawing effect, up to 0.55 V versus SHE for the 3,5-bis(trifluoromethyl)phenyl group. Correspondingly for devices fabricated with pre-treated electrodes with 1 cycle of ALD to form an Al2O3 blocking layer, using the dye N719, Pt as CE and electrolytes with the Ni-bis(dicarbollide) derivatives, the highest UOC was obtained for the 3,5-bis(trifluoromethyl)phenyl group, with UOC = 0.850 V; the PCE was in the range 0.7 to 2%. Li, et al. [202] improved the photocurrent for devices with Ni–bis(dicarbollide) mediator up to jSC = 6.3 mA·cm−2, by electrode modification using a nanoparticle-and-aerogel framework with high surface area and thickness of 13.6 μm; however, as discussed before, high recombination rates limited the overall device performance to PCE = 2.1% (either with films of 5.8 μm thickness, with UOC = 0.628 V, jSC = 5.3 mA·cm−2, FF = 0.60, or of 13.6 μm thickness, with UOC = 0.586 V, jSC = 6.3 mA·cm−2, and FF = 0.56.
10. Manganese Mediators
Analogously to Fe complexes, the natural abundance of manganese, low toxicity, versatile redox chemistry, and the fact that a Mn cluster plays a crucial role in photosynthesis by regenerating the dye following light induced oxidation, makes the search for Mn-based mediators very interesting, since generally they function as one-electron outer-sphere redox shuttles, and by ligand engineering the redox potential as well the absorption coefficient can be tuned.
As it is well known, the redox chemistry of manganese includes several possible oxidation states, from +2 to +7 [203,204,205,206], and the most relevant states for reversible electron transfer reactions include the pairs Mn(III)/Mn(II) or Mn(IV)/Mn(III). The high oxidation state Mn(IV) can act as a strong Lewis acid and be reduced by solvents or ligands as well as undergo hydrolysis if water is present in the media [203]. In addition, it can easily bind oxygen atoms, as known from the photosynthesis reactions involving the Mn–Ca cluster [207]. On the other hand, the Mn(III) species can act as strong oxidant, undergoing disproportionation to Mn(II) and MnO2. Nonetheless, both states can be stabilized by ligands with donor atoms such as nitrogen and oxygen, with hard Lewis-acid features. As a result, the occurrence of two different metal-center electron-transfer reactions with the pairs Mn(IV/III) and Mn(III/II) is possible, but difficult to distinguish.
Perera, et al., introduced a mediator based on the tris(acetylacetonato)- complex of Mn with oxidation states of manganese (III)/(IV), total charge of the complex 0/1+ ([Mn(acac)3]0/1+), and redox potential of 0.49 V versus SHE, as redox shuttle in DSSCs. The results were obtained with dye MK2, and PEDOT as CE, with characteristic parameters under full sun UOC = 0.733 V, jSC = 8.6 mA·cm−2 and PCE = 4.4%. These values can be compared for cells prepared under the same conditions with a reference I3−/I− that rendered UOC = 0.761 V, jSC = 8.5 mA·cm−2 and PCE = 4.6% [208]. High recombination rates were attained for devices with [Mn(acac)3]0/1+, corresponding to shorter electron lifetimes (by almost 2 orders of magnitude) compared to the [Co(bpy)3]3+/2+ and I3−/I− reference electrolytes, which is in agreement with the low photocurrent and poor performance showed by the devices.
Carli, et al. [209] attempted to enhance the performance of the [Mn(acac)3]0/1+ type of mediator by developing the derivatives [Mn(CF2)3] (CF2 = 4,4-difluoro-1-phenylbutanate-1,3-dione) and [Mn(DBM)3] (dbm = dibenzoylmethanate), (Figure 11). These derivatives exhibited a higher redox potential than [Mn(acac)3]1+/0, with 0.69 V versus SHE for [Mn(III/II)(CF2)3] and 0.41 V versus SHE for [Mn(III/II)(DBM)3], so that they can effectively reduce the oxidized MK2 dye during cell operation. For the Mn(III/IV) species the determined redox potentials were 1.57 V versus SHE for [Mn(CF2)3]3+/4+ and for the species [Mn(DBM)3]3+/2+ 1.20 V versus SHE. Therefore, these higher-valence species are unsuitable for dye regeneration, as regards the usual efficient for electricity generation dyes, due to the mismatch of the redox potential with the dye HOMO level (1.12 V versus SHE). In conclusion, only the Mn3+/2+ species can be effective in dye regeneration, demonstrating that the assignation of the valence states Mn(IV/III) in the previous work from Perera, et al. [208] was inaccurate, so that the total charge of the complex with valence states III/II is [Mn(acac)3]−1/0.
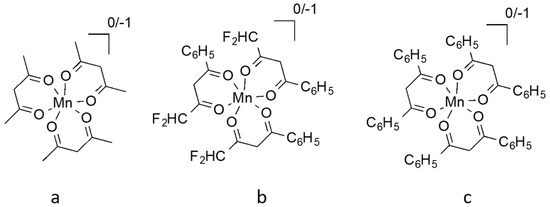
Figure 11.
Structures of manganese complexes (a) [Mn(acac)3], (b) [Mn(CF2)3] and (c) [Mn(DBM)3].
Basic additives such as TBP were excluded from the electrolyte formulation since ESI mass spectra and X-ray structures confirmed that these manganese compounds are susceptible to ligand exchange with TBP to form trans-TBP substituted [Mn(acac)2TBP]+. Hence, dark current inhibition was achieved instead by the co-adsorption of short-chain siloxanes. The highest efficiency was obtained for devices with [Mn(CF2)3]3+/2+ electrolyte, with UOC = 0.80 V, jSC = 4.95 mA·cm−2, and PCE = 2.72%.
Hossain, et al. [210] studied a series of Mn poly(pyrazolyl)borates, with redox potentials more positive than that of Co(bpy)3, in solar cells sensitized with CdS/CdSe quantum dots. The complexes and MnII(pzTp)2, MnII(Tp)2 and MnII(Tp*)2 were expected to show slow reduction kinetics since the one-electron reduction of MnIIIpoly(pyrazolyl)-borates results in spin crossover induced by charge transfer; a transition from low spin to high spin state (t2g4→t2g3eg1; S = 1→5/2) is involved [16]. Even though the overall efficiencies attained by these systems in solar devices are very low, high UOC were obtained, up to 1.01 V, which makes this kind of complexes attractive for further studies in DSSCs. Best performance was obtained for the complex MnII(pzTp)2 with characteristic performance parameters of UOC = 1.08 V, jSC = 0.08 mA·cm−2, and PCE = 0.24%. Electron lifetimes were approximately 2–4 orders of magnitude longer than for the Co(bpy)3 reference mediator at the same UOC, which points to the beneficial effect of using this type of complexes to reduce recombination rates from the electron in the TiO2 CB to the electrolyte mediator. Further work in molecular engineering needs to be done to improve the low photocurrents obtained, which are shown to be diffusion-limited.
Interestingly, little attention has been paid to Mn complexes with polyimine, including polypyridine, ligands [187] which have been reported to present very positive redox potentials, around from 1.1 V to 1.6 V versus NHE for the Mn(III/II) redox pair. These complexes could provide extended steric protection toward recombination compared to [Mn(acac)3]1+/0, and if paired with a suitable dye (low lying HOMO energetic level), could possibly lead to high UOC, as is the case previously discussed for Fe(bpy)3 electrolyte.
11. Vanadium Mediators
Apostolopoulou, et al. [211] investigated the applicability of vanadium complexes as mediators in DSSC, by using the oxidovanadium(IV) reversible redox couple [VIVO (hybeb)]2−/[VVO(hybeb)]− where hybeb4− is a tetradentate diaminodiphenolate(4−) ligand (Figure 12a). The electron self-exchange rate for the [VIVO(hybeb)]2−/[VVO(hybeb)]− couple is fast enough to enable a high dye regeneration rate of kex = 3.9 × 109 M−1·s−1. However, the redox potential of this couple is very negative (−0.047 V versus SHE). Therefore, the low difference between TiO2 CB edge and the Fermi level of the mediator, results to a low UOC of the cell. [211]. For DSSCs with N719 as dye and Pt as CE, characteristic performance parameters of UOC = 0.66 V, jSC = 5.2 mA·cm−2, and PCE = 2% under 1 sun illumination. The complex anion [VIVO(hybeb)]2− is stable in solution and can be easily oxidized to [VVO(hybeb)]−. However, the latter can be easily hydrolyzed, releasing protons to the solution and protonating the phenolic oxygen atom of the [VIVO(hybeb)]2− anion. Hence, dried ACN was used in order to avoid hydrolysis.
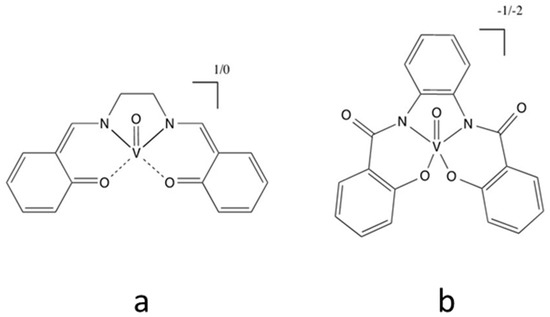
Figure 12.
Structures of (a) [VO(salen)]+/0 and (b) [VO(hybeb)]−1/−2.
Oyaizu, et al. [212] improved the performance of DSSCs with vanadium-based electrolytes by introducing the vanadium-salen [VO(salen)]0/+ complex (salen = N,N′-ethylenebis (salicylideneiminate) (2−), Figure 12b) which have a redox potential of 0.8 V versus SHE and a fast electron exchange rate of kex = 7.4 × 109 M−1·s−1 [170]. A PCE of 5.4% with UOC = 0.74 and jSC = 12.3 mA·cm−2 was obtained for cells with a vanadium-based electrolyte, a D205/D131 co-sensitized system, and a Pt-coated CE under 1 sun illumination.
12. Dual Redox Mediator Systems
In these systems regenerates the dye at the proximity of the PE and mediator transports electrons between PE and CE. The system is often called the co-mediator. As regards the redox potentials, that of should be closer to that of the dye compared to .
The various processes in the DSSC related to the co-mediator system operation are depicted in Figure 13.
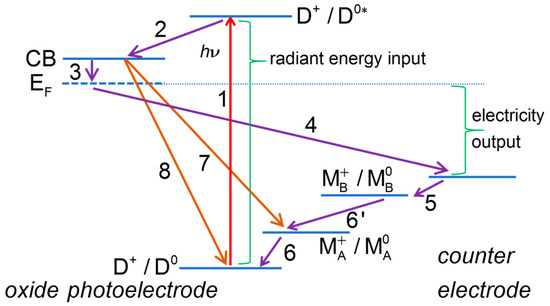
Figure 13.
Energy diagram of a dye-sensitized solar cell with a dual mediator-containing electrolyte. Processes (1–5) and (7–8) are analogous to those in Figure 1. Process (6) corresponds to the reaction between mediator and co-mediator , species interacting with the CE and the photoelectrode, respectively.
Photoelectrode: oxidized dye regeneration by the reduced co-mediator and subsequent reaction between oxidized co-mediator and reduced mediator:
Electrolyte: transport of and between PE and CE:
CE: reduction of .
There are several reasons for which, although is favorable for obtaining a high cell voltage, a co-mediator is needed. A high concentration of may have to be avoided for various reasons: (a) it has a high molar absorption for solar light; (b) it has poor long-term stability; (c) its recombination reactions with electrons from the mesoporous semiconductor or the PE support are too fast; (d) due to the large molecular size, it diffuses slowly from the PE to the CE; (e) its reduction at the CE may be kinetically slow.
With respect to the preparation of the electrolyte added in the DSSC, normally it needs to contain only and . When the DSSC is illuminated for the first time, is oxidized to at the PE and is reduced to at the CE; the DSSC operates as a battery in the charging mode, but without separator between anode and cathode. diffuses away from the CE and reaches the PE where in reacts with so that equilibrium concentration gradients are established. However, in the literature the electrolyte is often prepared by adding , and .
A number of dual mediators where at least one of the components is a metal coordination complex are presented below. In the subsection titles the first mentioned component is and the second is .
12.1. Phenothiazine or Ferrocene as Mediator and Co Complex as Co-Mediator
In their 2006 publication Cazzanti, et al. (2006) [213] present a DSSC with the Z907 sensitizer, a Pt-coated CE, phenothiazine (PTZ) or ferrocene (Fc) as , and Co(DTB)3(II) (DTB = 4,4′-di-tert-butyl-2,2′-bipyridine) as has been investigated. The DSSC with PTZ/Co had a better performance than that in the case of Fc/Co or only Co. The PCE was below 2% under full sun in all cases. For the dual mediator system, the electrolyte added in the DSSC contained only the reduced forms MA0 and MB0. However, a very small amount of was probably present inadvertently in the electrolyte so that the DSSC could operate in the start-up phase, until the establishment of the equilibrium concentrations.
12.2. Iodide as Mediator and Co Complex as Co-Mediator
A DSSC with dye LEG4, a Pt-coated CE, [Co(bpy)3]3+/2 as and I3−/I− as is the subject of the publication by Cong, et al. (2014) [214]. The electrolyte was prepared by the addition of , and I2 in acetonitrile. The dual mediator-based DSSC had a PCE under full sun of 7.5% (UOC = 0.89 V, jSC = 11.8 mA·cm−2), superior in comparison to that of a DSSC with an electrolyte containing only [Co(bpy)3]3+/2+ (PCE = 6.5%, UOC = 0.88 V, jSC = 11.0 mA·cm−2). The redox potential of I3−/I2 is more positive than that of I3−/I− but less than that of the dye so that, during the initial phase of DSSC illumination at the CE, I2 is reduced to I3− and subsequently I3− to I−. The merit of the dual mediator system in this case lies in the fact that the mass transport of is slower than that of , due to the smaller size of the latter.
The system of Xu, et al. [215] was based on Ru dye N719, a Pt-coated CE, and two mediators in an ionic liquid electrolyte. The latter prepared by adding the complex Co(MeIm-bpy)3 (MeIm-bpy = 3,30-(2,20-bipyridine-4,40-diyl-bis(methylene)) bis(1-methyl-1H-imidazol-3-ium)) into a mixture of two ionic liquids, PMII (1-propyl-3-methyl-imidazolim iodide) and, as inert supporting electrolyte, EMINCS (1-ethyl-3-methyl-imidazolium-thiocyanide), together with an oxidizing reagent, NOBF4, needed for the oxidation of the Co(II) complex to Co(III). As in the previous case, Co(III/II) corresponds to and I3−/I− to . The PCE was 7.4% under full sun (UOC = 0.71 V, jSC = 15.1 mA·cm−2) and 8.2% under 1/2 sun (UOC = 0.70 V, jSC = 8.4 mA·cm−2), and was superior to that of a similar DSSC containing solely I3−/I− (at full sun PCE = 6.4%, UOC = 0.64 V, jSC = 13.9 mA·cm−2; at 1/2 sun PCE = 7.0%, UOC = 0.61 V, jSC = 7.8 mA·cm−2). The CE electrocatalyst was Pt.
12.3. Co Complex as Mediator and TEMPO and Co-Mediator
Cong, et al. [214] investigated the dual mediator system of TEMPO, a stable nitroxide (–NO·) radical (2,2,6,6-tetramethylpiperidin-1-yl)oxyl), as and Co(bpy)3(III/II) as . In principle the redox potential of is sufficient for operating a DSSC; however, the fast recombination reaction of the oxidized form of TEMPO (TEMPO+, –N=O+) is a drawback, due to its low electron transfer reorganization energy. Furthermore, the oxidized form TEMPO (–N=O+) is unstable in solution. Therefore, for this particular reason, it is of interest to use the Co complex as co-mediator, despite the fact that both the mass-transport and the electrode kinetics are faster for TEMPO than for the Co complex. A DSSC with the LEG4 dye, a Pt-coated CE and the dual mediator had a PCE of 9.4% at 1/2 sun (UOC = 0.93 V, jSC = 7.2 mA·cm−2) and 8.5% at full sun (UOC = 0.97 V, jSC = 13.6 mA·cm−2). For a DSSC with only the Co mediator the performance was lower, with a PCE of 8.2% at 1/2 sun (UOC = 0.84 V, jSC = 7.0 mA·cm−2) and 7.2% at full sun (UOC = 0.86 V, jSC = 13.0 mA·cm−2). The solution containing the two mediators was prepared by addition of TEMPO to a typical Co(bpy)3-based electrolyte.
12.4. Co Complex as Mediator and p-Anisylamine as Co-Mediator
Hao, et al. [216] considered a dual mediator system similar to the previous one, with the organic redox mediator tris p-anisylamine (TPAA) in place of TEMPO as and Co(bpy)3 as . The problems of using TPAA alone and the advantages of introducing the co-mediator are the same as for the TEMPO/Co system. A DSSC based co-sensitized with two dyes, D35 and Dyenamo blue, and with a Pt-coated CE had a PCE of 10.5% at full sun with the dual mediator system (UOC = 0.92 V, jSC = 15.5 mA·cm−2) and 11.7% under 1/2 sun (UOC = 0.89 V, jSC = 8.2 mA·cm−2). In comparison, a similar DSSC with the Co mediator was less efficient, with a PCE of 8.3% at full sun. (UOC = 0.82 V, jSC = 13.9 mA·cm−2). The solution containing the two mediators was prepared by addition of TPAA to a Co(bpy)3-based electrolyte.
12.5. Fe Complex as Mediator and Cu Complex as Co-Mediator
In the system described by Colombo, et al. [164] [Cu+(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2](PF6)2 (3) was used as and [Fe2+(4,4′-ditertbutyl-2,2′-bipyridine)3 as in a DSSC with the ruthenium sensitizer Z907 (Figure 5) and a PEDOT-coated CE. For the dual mediator system, where the concentrations of , , and were 0.095 M, 0.005 M and 0.010 M the PCE was 1.2% (UOC = 0.51 V, jSC = 4.0 mA·cm−2) as compared 0.9% to a cell with only (either with and concentrations of 0.095 and 0.005 M respectively, where UOC = 0.60 V and jSC = 2.0 mA·cm−2, or with and concentrations of 0.143 and 0.007M, where UOC = 0.58 V and jSC = 2.4 mA·cm−2) and 1.7% with the I3−/I electrolyte (UOC = 0.60 V, jSC = 3.8 mA·cm−2).
12.6. Co Complexes as Mediator and Co-Mediator
In the study by Koussi–Daoud, et al. [124] mentioned in Section 7 the mediator was a Co complex dissolved in solution, effective in both dye regeneration and charge transport between the electrodes, and the co-mediator was a Co complex in which an EDOT pendant group is attached and which acts as a homogeneous catalyst for the reduction of the oxidized form of the mediator at the CE. The reaction scheme is different than that of the systems previously discussed.
Photoelectrode: photooxidation of to
Electrolyte: transport of and MA between PE and CE.
Counter electrode: mediated reduction of to :
This approach may be of interest in future research where the catalytic activity of the CEs with the mediator is not sufficient, so that both a heterogeneous catalyst attached on the CE and a homogeneous catalyst in the electrolyte would be needed.
13. Concluding Remarks
The redox mediator is an essential component of a dye-sensitized solar cell (DSSC). Until the present time relatively little research effort has been devoted to mediators compared to the development of dyes or CEs. The requirements for DSSC mediators have been rather stringent so that only a small number of compounds have been successfully tested. Iodide/triiodide has been the system of choice up to the present point due to efficient dye regeneration, fast diffusion, good long-term stability in solution, and low cost. However, several disadvantages, not the least of which is the large required driving force for dye regeneration, make necessary its replacement. In the last 10 years, cobalt(III/II) coordination complexes have demonstrated good performance with a number of dyes fulfilling special structural requirement toward suppressing the dark recombination reaction between conduction-band electrons and oxidized mediator. At present the 14% solar-to-electrical power conversion efficiency (PCE) record has been obtained with this mediator. This exceptional performance is mainly attributed to the larger cell photovoltage, in agreement with the more positive redox potential of Co(III/II) complexes compared to that of triiodide/iodide. However, there are unresolved issues with this mediator, including slow diffusion in the electrolyte and relatively high cost, so that other mediators, including coordination complexes of several metals, are under considerations. In the last 5 years copper(I/II) complexes have been demonstrated superior performance, with PCE values exceeding 10% in several investigations not only for the typical case of the complex dissolved in a nonaqueous electrolytes but also for a solid-state variant obtained after electrolyte evaporation. The elucidation of the redox chemistry and the optimization of the DSSC devices based on this mediator are still under investigation. Other coordination complexes, notably these incorporating Fe, Ni, Mn and V, have been investigated, it is true, to a rather limited extent, however the performance of DSSCs incorporating such mediators is inferior to that of DSSCs with iodide, cobalt, or copper, with iodide-based electrolytes still of preference for applied research and development; the better demonstrated long-term stability in the dark and under illumination plays in this respect a role. However, it is hoped that ongoing fundamental interdisciplinary research coupling synthetic chemistry, materials science, electrochemistry, and photochemistry–photophysics will lead to efficient, inexpensive and stable redox mediators in the near future, and in this regard inorganic coordination chemistry is promised to play a key role, as amply demonstrated by the recently obtained pioneering results.
Funding
Anders Hagfeldt acknowledges the financial support from the Swiss National Science Foundation (project number 200021 157135/1). Michael Grätzel acknowledges the financial support from H.Glass SA, Villaz-St-Pierre, Switzerland.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
List of frequently used Symbols
| ACN | acetonitrile |
| APCE | absorbed photon-to-current efficiency |
| CB | conduction band |
| CE | counter electrode |
| CTSC | contact phase of the mesoporous oxide |
| CV | cyclic voltammetry |
| DSSC | dye-sensitized solar cell |
| D0, D0* | ground and excited dye of the dye in the reduced form |
| D+ | oxidized dye |
| EF, EFӨ | Fermi level, standard Fermi level |
| e− | electron |
| E | electrode potential |
| FF | fill factor |
| IPCE | incident photon-to-current efficiency |
| Jph | photon number flux |
| Jel | electron number flux |
| jSC | short-circuit current density of DSSC |
| jMPP | current density of DSSC at the maximum power point |
| L | Avogadro constant |
| LHE | light-harvesting efficiency |
| M0, M+ | reduced and oxidized form of redox mediator |
| MEV | microelectrode voltammetry |
| P0 | incident light intensity |
| PCE | solar-to-electrical energy conversion efficiency |
| PE | photoelectrode |
| Q0 | magnitude of electronic charge |
| RDEV | rotating disk electrode voltammetry |
| SC | mesoporous semiconductor phase |
| TAS | transient absorption spectroscopy |
| UOC | open-circuit photovoltage |
| Г | molar surface concentration (based on geometric surface area) |
| ∆GӨR EC | recombination standard free energy |
| ∆GӨR EG | regeneration standard free energy |
| ελ | molar extinction (attenuation) coefficient of a dye |
| Rλ | fraction of reflected light |
| σ | molecule area of a dye |
| σλ | light-absorption molecular cross-section of a dye |
| φCOLL | collection efficiency |
| φINJ | injection efficiency |
References
- Moser, J. Notiz über Verstärkung photoelektrischer Ströme durch optische Sensibilisirung. Monatshefte für Chemie 1887, 8, 373. [Google Scholar] [CrossRef]
- Gerischer, H.; Tributsch, H. Elektrochemische Untersuchungen zur spektralen Sensibilisierung von ZnO-Einkristallen. Berichte der Bunsengesellschaft für Phys. Chemie 1968, 72, 437–445. [Google Scholar] [CrossRef]
- Tributsch, H.; Gerischer, H. Elektrochemische Untersuchungen über den Mechanismus der Sensibilisierung und übersensibilisierung an ZnO-Einkristallen. Berichte der Bunsengesellschaft für Phys. Chemie 1969, 73, 251–260. [Google Scholar] [CrossRef]
- Memming, R. Electron transfer process with excited molöecules at semiconductor electrodes. Prog. Surf. Sci. 2000, 17, 7–73. [Google Scholar] [CrossRef]
- Matsumura, M.; Matsudaira, S.; Tsubomura, H.; Takata, M.; Yanagida, H. Dye Sensitization and Surface Structures of Semiconductor Electrodes. Ind. Eng. Chem. Prod. Res. Dev. 1980, 19, 415–421. [Google Scholar] [CrossRef]
- Tsubomura, H.; Matsumura, M.; Nomzra, Y.; Amamiya, T. Dye sensitised zinc oxide: Aqueous electrolyte: Platinum photocell. Nature 1976, 261, 402–403. [Google Scholar] [CrossRef]
- Anderson, S.; Constable, E.C.; Dare-Edwards, M.P.; Goodenouigh, J.B.; Hamnett, A.; Seddon, K.R.; Wright, R.D. Chemical modification of a titanium (IV) oxide electrode to give stable dye sensitisation without a supersensitiser. Nature 1979, 280, 571–573. [Google Scholar] [CrossRef]
- Dare-Edwards, M.P.; Goodenough, J.B.; Hamnett, A.; Seddon, K.R.; Wright, R.D. Sensitisation of semiconducting electrodes with ruthenium-based dyes. Faraday Discuss. Chem. Soc. 1980, 285. [Google Scholar] [CrossRef]
- Moser, J.; Grätzel, M. Photosensitized electron injection in colloidal semiconductors. J. Am. Chem. Soc. 1984, 106, 6557–6564. [Google Scholar] [CrossRef]
- Desilvestro, J.; Grätzel, M.; Kavan, L.; Moser, J.; Augustynski, J. Highly efficient sensitization of titanium dioxide. J. Am. Chem. Soc. 1985, 107, 2988–2990. [Google Scholar] [CrossRef]
- Vlachopoulos, N.; Liska, P.; Augustynski, J.; Grätzel, M. Very efficient visible light energy harvesting and conversion by spectral sensitization of high surface area polycrystalline titanium dioxide films. J. Am. Chem. Soc. 1988, 110, 1216–1220. [Google Scholar] [CrossRef]
- Liska, P.; Vlachopoulos, N.; Nazeeruddin, M.K.; Comte, P.; Grätzel, M. cis-diaquabis(2,2′-bipyridyl-4,4′-dicarboxylate)-ruthenium(II) sensitizes wide band gap oxide semiconductors very efficiently over a broad spectral range in the visible. J. Am. Chem. Soc. 1988, 110, 3686–3687. [Google Scholar] [CrossRef]
- Gratzel, M.; Liska, P. Photoelectrochemical Cells. U.S. Patent US4927721, 1990. [Google Scholar]
- Dabestani, R.; Bard, A.J.; Campion, A.; Fox, M.A.; Mallouk, T.E.; Webber, S.E.; White, J.M. Sensitization of Titanium Dioxide and Strontium Titanate Electrodes by Ruthenium(II) An Evaluation of Sensitization Efficiency for Component Photoelectrodes in a Muitipanel Device. J. Phys. Chem. 1988, 1872–1878. [Google Scholar] [CrossRef]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Kay, A.; Rodicio, I.; Humphry-Baker, R.; Mueller, E.; Liska, P.; Vlachopoulos, N.; Grätzel, M. Conversion of light to electricity by cis-X2bis(2,2′-bipyridyl-4,4′-dicarboxylate)ruthenium(II) charge-transfer sensitizers (X = Cl−, Br−, I−, CN−, and SCN−) on nanocrystalline titanium dioxide electrodes. J. Am. Chem. Soc. 1993, 115, 6382–6390. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Didriksson, B.; Palmqvist, T.; Lindström, H.; Södergren, S.; Rensmo, H.; Lindquist, S.-E. Verification of high efficiencies for the Grätzel-cell. A 7% efficient solar cell based on dye-sensitized colloidal TiO2 films. Sol. Energy Mater. Sol. Cells 1994, 31. [Google Scholar] [CrossRef]
- Ahmad, S.; Guillén, E.; Kavan, L.; Grätzel, M.; Nazeeruddin, M.K. Metal free sensitizer and catalyst for dye sensitized solar cells. Energy Environ. Sci. 2013, 6, 3439. [Google Scholar] [CrossRef]
- Asghar, M.I.; Miettunen, K.; Halme, J.; Vahermaa, P.; Toivola, M.; Aitola, K.; Lund, P. Review of stability for advanced dye solar cells. Energy Environ. Sci. 2010, 3, 418. [Google Scholar] [CrossRef]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Solar Energy Conversion by Dye-Sensitized Photovoltaic Cells. Inorg. Chem. 2005, 44, 6841–6851. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Recent advances in sensitized mesoscopic solar cells. Acc. Chem. Res. 2009, 42. [Google Scholar] [CrossRef] [PubMed]
- Hagfeld, A.; Grätzel, M. Light-induced redox reactions in nanocrystalline systems. Chem. Rev. 1995, 95, 49–68. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Grätzel, M. Molecular photovoltaics. Acc. Chem. Res. 2000, 33. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Hamann, T.W.; Ondersma, J.W. Dye-sensitized solar cell redox shuttles. Energy Environ. Sci. 2011, 4, 370–381. [Google Scholar] [CrossRef]
- Kavan, L. Electrochemistry and dye-sensitized solar cells. Curr. Opin. Electrochem. 2017, 2, 88–96. [Google Scholar] [CrossRef]
- Li, C.T.; Lin, R.Y.; Lin, J.T. Sensitizers for aqueous-based solar cells. Chem. Asian J. 2017, 12, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Pashaei, B.; Shahroosvand, H.; Abbasi, P. Transition metal complex redox shuttles for dye-sensitized solar cells. RSC Adv. 2015, 5, 94814–94848. [Google Scholar] [CrossRef]
- Bella, F.; Gerbaldi, C.; Barolo, C.; Grätzel, M. Aqueous dye-sensitized solar cells. Chem. Soc. Rev. 2015, 44, 3431–3473. [Google Scholar] [CrossRef] [PubMed]
- Peter, L.M. The Grätzel Cell: Where Next? J. Phys. Chem. Lett. 2011, 2, 1861–1867. [Google Scholar] [CrossRef]
- Peter, L.M. Dye-sensitized nanocrystalline solar cells. Phys. Chem. Chem. Phys. 2007, 9, 2630–2642. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, S.; Yu, Y.; Manseki, K. Iodine/iodide-free dye-sensitized solar cells. Acc. Chem. Res. 2009, 42, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Vlachopoulos, N.; Gorlov, M.; Kloo, L. Liquid electrolytes for dye-sensitized solar cells. Dalton Trans. 2011, 40, 10289–10303. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lan, Z.; Lin, J.; Huang, M.; Huang, Y.; Fan, L.; Luo, G. Electrolytes in dye-sensitized solar cells. Chem. Rev. 2015, 115, 2136–2173. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N. CuI versus RuII: Dye-sensitized solar cells and beyond. ChemSusChem 2008, 1, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Housecroft, C.E.; Constable, E.C. The emergence of copper(I)-based dye sensitized solar cells. Chem. Soc. Rev. 2015, 44, 8386–8398. [Google Scholar] [CrossRef] [PubMed]
- Bella, F.; Galliano, S.; Gerbaldi, C.; Viscardi, G. Cobalt-based electrolytes for dye-sensitized solar cells: Recent advances towards stable devices. Energies 2016, 9, 384. [Google Scholar] [CrossRef]
- Bignozzi, C.A.; Argazzi, R.; Boaretto, R.; Busatto, E.; Carli, S.; Ronconi, F.; Caramori, S. The role of transition metal complexes in dye sensitized solar devices. Coord. Chem. Rev. 2013, 257, 1472–1492. [Google Scholar] [CrossRef]
- Caramori, S.; Cristino, V.; Boaretto, R.; Argazzi, R.; Di Carlo, A.; Bignozzi, C.A. New Components for Dye-Sensitized Solar Cells. Int. J. Photoenergy 2010, 458614. [Google Scholar] [CrossRef]
- Cong, J.; Yang, X.; Kloo, L.; Sun, L. Iodine/iodide-free redox shuttles for liquid electrolyte-based dye-sensitized solar cells. Energy Environ. Sci. 2012, 5, 9180. [Google Scholar] [CrossRef]
- Johansson, E.M.J.; Sandell, A.; Siegbahn, H.; Rensmo, H.; Mahrov, B.; Boschloo, G.; Figgemeier, E.; Hagfeldt, A. Interfacial properties of photovoltaic TiO2/dye/PEDOT–PSS heterojunctions. Synth. Met. 2005, 149, 157–167. [Google Scholar] [CrossRef]
- Freitag, M.; Boschloo, G. The revival of dye-sensitized solar cells. Curr. Opin. Electrochem. 2017, 2, 111–119. [Google Scholar] [CrossRef]
- Giribabu, L.; Bolligarla, R.; Panigrahi, M. Recent Advances of Cobalt(II/III) Redox Couples for Dye-Sensitized Solar Cell Applications. Chem. Rec. 2015, 15, 760–788. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, K. Dye-Sensitized Solar Cells; EPFL Press: Lausanne, Switzerland, 1990; ISBN 9781439808665. [Google Scholar]
- Odobel, F.; Pellegrin, Y.; Gibson, E.A.; Hagfeldt, A.; Smeigh, A.L.; Hammarström, L. Recent advances and future directions to optimize the performances of p-type dye-sensitized solar cells. Coord. Chem. Rev. 2012, 256, 2414–2423. [Google Scholar] [CrossRef]
- Wood, C.J.; Summers, G.H.; Clark, C.A.; Kaeffer, N.; Braeutigam, M.; Carbone, L.R.; D’Amario, L.; Fan, K.K.; Farré, Y.; Narbey, S.; et al. Comprehensive comparison of dye-sensitized NiO photocathodes for solar energy conversion. Phys. Chem. Chem. Phys. 2016, 18, 10727–10738. [Google Scholar] [CrossRef] [PubMed]
- Strong, F.C. Theoretical Basis of Bouguer-Beer Law of Radiation Absorption. Anal. Chem. 1952, 24, 338–342. [Google Scholar] [CrossRef]
- Skoog, D.A.; Leary, J.J. Principles of Instrumental Analysis; Saunders College Pub: Fort Worth, TX, USA, 1992; ISBN 9780030233432. [Google Scholar]
- Trasatti, S. International Union of Pure and Applied Chemistry Commission on Electrochemistry* the Absolute Electrode Potential: An Explanatory Note. Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar] [CrossRef]
- Van de Lagemaat, J.; Park, N.-G.; Frank, A.J. Influence of Electrical Potential Distribution, Charge Transport, and Recombination on the Photopotential and Photocurrent Conversion Efficiency of Dye-Sensitized Nanocrystalline TiO2 Solar Cells: A Study by Electrical Impedance and Optical Modulation Techniques. J. Phys. Chem. B 2000, 104, 2044–2052. [Google Scholar] [CrossRef]
- Bertoluzzi, L.; Ma, S. On the methods of calculation of the charge collection efficiency of dye sensitized solar cells. Phys. Chem. Chem. Phys. 2013, 15, 4283–4285. [Google Scholar] [CrossRef] [PubMed]
- Bonhôte, P.; Gogniat, E.; Tingry, S.; Barbé, C.; Vlachopoulos, N.; Lenzmann, F.; Comte, P.; Grätzel, M. Efficient Lateral Electron Transport inside a Monolayer of Aromatic Amines Anchored on Nanocrystalline Metal Oxide Films. J. Phys. Chem. B 1998, 102, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Blauch, D.N.; Saveant, J.M. Dynamics of electron hopping in assemblies of redox centers. Percolation and diffusion. J. Am. Chem. Soc. 1992, 114, 3323–3332. [Google Scholar] [CrossRef]
- Thorsmølle, V.K.; Rothenberger, G.; Topgaard, D.; Brauer, J.C.; Kuang, D.-B.; Zakeeruddin, S.M.; Lindman, B.; Grätzel, M.; Moser, J.-E. Extraordinarily Efficient Conduction in a Redox-Active Ionic Liquid. ChemPhysChem 2011, 12, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Saygili, Y.; Söderberg, M.; Pellet, N.; Giordano, F.; Cao, Y.; Munoz-García, A.B.; Zakeeruddin, S.M.; Vlachopoulos, N.; Pavone, M.; Boschloo, G.; et al. Copper Bipyridyl Redox Mediators for Dye-Sensitized Solar Cells with High Photovoltage. J. Am. Chem. Soc. 2016, 138, 15087–15096. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, Y.; Yi, Z.; Zu, N.; Zhang, J.; Zhang, M.; Wang, P. High-efficiency dye-sensitized solar cells: The influence of lithium ions on exciton dissociation, charge recombination, and surface states. ACS Nano 2010, 4, 6032–6038. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhou, D.; Shi, Y.; Si, X.; Wang, Y.; Wang, P. Stable and efficient dye-sensitized solar cells: Photophysical and electrical characterizations. Energy Environ. Sci. 2010, 3, 1722–1725. [Google Scholar] [CrossRef]
- Marszalek, M.; Arendse, F.D.; Decoppet, J.-D.; Babkair, S.S.; Ansari, A.A.; Habib, S.S.; Wang, M.; Zakeeruddin, S.M.; Grätzel, M. Ionic liquid-sulfolane composite electrolytes for high-performance and stable dye-sensitized solar cells. Adv. Energy Mater. 2014, 4, 1301235. [Google Scholar] [CrossRef]
- Boschloo, G.; Hagfeldt, A. Characteristics of the iodide/triiodide redox mediator in dye-sensitized solar cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.M.; Giaimuccio, J.M.; Meyer, G.J. Evidence for iodine atoms as intermediates in the dye sensitized formation of I−I bonds. J. Am. Chem. Soc. 2008, 130, 17252–17253. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.G.; Farnum, B.H.; Ardo, S.; Meyer, G.J. Iodide Chemistry in Dye-Sensitized Solar Cells: Making and Breaking I−I Bonds for Solar Energy Conversion. J. Phys. Chem. Lett. 2010, 1, 3132–3140. [Google Scholar] [CrossRef]
- Olsen, E.; Hagen, G.; Eric Lindquist, S. Dissolution of platinum in methoxy propionitrile containing LiI/I2. Sol. Energy Mater. Sol. Cells 2000, 63, 267–273. [Google Scholar] [CrossRef]
- Miettunen, K.; Etula, J.; Saukkonen, T.; Jouttijärvi, S.; Halme, J.; Romu, J.; Lund, P. Insights into corrosion in dye solar cells. Prog. Photovoltaics Res. Appl. 2014, 23, 1045–1056. [Google Scholar] [CrossRef]
- Miettunen, K.; Halme, J.; Lund, P. Metallic and plastic dye solar cells. Wiley Interdiscip. Rev. Energy Environ. 2013, 2, 104–120. [Google Scholar] [CrossRef]
- Gennett, T.; Milner, D.F.; Weaver, M.J. Role of solvent reorganization dynamics in electron-transfer processes. Theory-experiment comparisons for electrochemical and homogeneous electron exchange involving metallocene redox couples. J. Phys. Chem. 1985, 89, 2787–2794. [Google Scholar] [CrossRef]
- Feldt, S.M.; Cappel, U.B.; Johansson, E.M.J.; Boschloo, G.; Hagfeldt, A. Characterization of Surface Passivation by Poly(methylsiloxane) for Dye-Sensitized Solar Cells Employing the Ferrocene Redox Couple. J. Phys. Chem. C 2010, 114, 10551–10558. [Google Scholar] [CrossRef]
- Stergiopoulos, T.; Falaras, P. Minimizing Energy Losses in Dye-Sensitized Solar Cells Using Coordination Compounds as Alternative Redox Mediators Coupled with Appropriate Organic Dyes. Adv. Energy Mater. 2012, 2, 616–627. [Google Scholar] [CrossRef]
- Bard, A.J.L.; Faulkner, L.R. Electrochemical Methods, Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2001; ISBN 04710433729. [Google Scholar]
- Bagotsky, V.S. Fundamentals of Electrochemistry, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2005; ISBN 9780471741992. [Google Scholar]
- Koryta, J.J.; Dvořák, J.; Kavan, L.; Dvorak, J.; Kavan, L. Principles of Electrochemistry; John Wiley & Sons: Chichester, UK, 1993; ISBN 9781119966845. [Google Scholar]
- Oldham, K.B.; Myland, J.C.; Bond, A.M. Electrochemical Science and Technology: Fundamentals and Applications; John Wiley & Sons: Chichester, UK, 2011; ISBN 9781119966845. [Google Scholar]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potentials in nonaqueous solvents (Recommendations 1983). Pure Appl. Chem. 1984, 56, 461–466. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorganica Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Bentley, C.L.; Bond, A.M.; Hollenkamp, A.F.; Mahon, P.J.; Zhang, J. Voltammetric Determination of the Iodide/Iodine Formal Potential and Triiodide Stability Constant in Conventional and Ionic Liquid Media. J. Phys. Chem. C 2015, 119, 22392–22403. [Google Scholar] [CrossRef]
- Zhang, J.; Bond, A.M. Practical considerations associated with voltammetric studies in room temperature ionic liquids. Analyst 2005, 130, 1132–1147. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, F. Notizen: Fermi-Niveau und Flachbandpotential von Molekülkristallen aromatischer Kohlenwasserstoffe. Zeitschrift für Naturforsch. A 1967, 22, 843–844. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A. Absolute Potential of the Standard Hydrogen Electrode and the Problem of Interconversion of Potentials in Different Solvents. J. Phys. Chem. B 2010, 114, 7894–7899. [Google Scholar] [CrossRef] [PubMed]
- Namazian, M.; Lin, C.Y.; Coote, M.L. Benchmark Calculations of Absolute Reduction Potential of Ferricinium/Ferrocene Couple in Nonaqueous Solutions. J. Chem. Theory Comput. 2010, 6, 2721–2725. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.R. Electrochemistry at Semiconductor and Oxidized Metal Electrodes; Plenum Press: New York, NY, USA, 1980; ISBN 0-306-40524-5. [Google Scholar]
- Willig, F.; Gerischer, H. Reaction of Excited Dye Molecules at Electrodes. Top. Appl. Chem. 1976, 61, 31–84. [Google Scholar] [CrossRef]
- Swierk, J.R.; Mallouk, T.E. Design and development of photoanodes for water-splitting dye-sensitized photoelectrochemical cells. Chem. Soc. Rev. 2013, 42, 2357–2387. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.J.; Keyes, T.E. Redox Properties of Ground and Electronically Excited States: [Ru(bpy)2Qbpy]2+ Monolayers. J. Phys. Chem. B 1998, 102, 10004–10012. [Google Scholar] [CrossRef]
- Tilly, D.; Dayaker, G.; Bachu, P. Cobalt mediated C–H bond functionalization: Emerging tools for organic synthesis. Catal. Sci. Technol. 2014, 4, 2756–2777. [Google Scholar] [CrossRef]
- Gosmini, C.; Moncomble, A. Cobalt-catalyzed cross-coupling reactions of aryl halides. Isr. J. Chem. 2010, 50, 568–576. [Google Scholar] [CrossRef]
- Yamazaki, H.; Shouji, A.; Kajita, M.; Yagi, M. Electrocatalytic and photocatalytic water oxidation to dioxygen based on metal complexes. Coord. Chem. Rev. 2010, 254, 2483–2491. [Google Scholar] [CrossRef]
- Losse, S.; Vos, J.G.; Rau, S. Catalytic hydrogen production at cobalt centres. Coord. Chem. Rev. 2010, 254, 2492–2504. [Google Scholar] [CrossRef]
- Bura, T.; Blaskovits, J.T.; Leclerc, M. Direct (Hetero)arylation Polymerization: Trends and Perspectives. J. Am. Chem. Soc. 2016, 138, 10056–10071. [Google Scholar] [CrossRef] [PubMed]
- Josephsen, J.; Schaffer, C. The Position of 2,2-Bipyridine and 1,10-Phenanthroline in the Spectrochemical Series. Acta Chem. Scand. A 1977, 31, 813–824. [Google Scholar] [CrossRef]
- Krivokapic, I.; Zerara, M.; Daku, M.L.; Vargas, A.; Enachescu, C.; Ambrus, C.; Tregenna-Piggott, P.; Amstutz, N.; Krausz, E.; Hauser, A. Spin-crossover in cobalt(II) imine complexes. Coord. Chem. Rev. 2007, 251, 364–378. [Google Scholar] [CrossRef]
- Kremer, S.; Henke, W.; Reinen, D.; Kremer, S.; Henke, W.; Reinen, D. High-Spin-Low-Spin Equilibria of Cobalt2+ in the Terpyridine Complexes Co(terpy)2X2·nH2O. Inorg. Chem. 1982, 21, 3013–3022. [Google Scholar] [CrossRef]
- Hayami, S.; Komatsu, Y.; Shimizu, T.; Kamihata, H.; Lee, Y.H. Spin-crossover in cobalt(II) compounds containing terpyridine and its derivatives. Coord. Chem. Rev. 2011, 255, 1981–1990. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Nusbaumer, H.; Moser, J.E.; Zakeeruddin, S.M.; Nazeeruddin, M.K.; Grätzel, M. CoII(dbbip))22+ complex rivals tri-iodide/iodide redox mediator in dye-sensitized photovoltaic cells. J. Phys. Chem. B 2001, 105, 10461–10464. [Google Scholar] [CrossRef]
- Hodgkin, D.C.; Kamper, J.; MacKay, M.; Pickworth, J.; Trueblood, K.N.; White, J.G. Structure of vitamin B12. Nature 1956, 178, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Nusbaumer, H.; Zakeeruddin, S.M.; Moser, J.-E.; Grätzel, M. An alternative efficient redox couple for the dye-sensitized solar cell system. Chem. Eur. J. 2003, 9, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.J.; Peter, L.M.; Zakeeruddin, S.M.; Grätzel, M. Electrochemical studies of the Co(III)/Co(II)(dbbip)2 redox couple as a mediator for dye-sensitized nanocrystalline solar cells. Coord. Chem. Rev. 2004, 248, 1447–1453. [Google Scholar] [CrossRef]
- Sapp, S.A.; Elliott, C.M.M.; Contado, C.; Caramori, S.; Bignozzi, C.A. Substituted Polypyridine Complexes of Cobalt(II/III) as Efficient Electron-Transfer Mediators in Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2002, 124, 11215–11222. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, E.; Yum, J.-H.; Kessler, F.; Gómez García, C.J.; Zuccaccia, C.; Cinti, A.; Nazeeruddin, M.K.; Grätzel, M.; De Angelis, F. Cobalt electrolyte/dye interactions in dye-sensitized solar cells: A combined computational and experimental study. J. Am. Chem. Soc. 2012, 19438–19453. [Google Scholar] [CrossRef] [PubMed]
- Nazeeruddin, K.; Amirnasr, M.; Comte, P.; Mackay, J.R.; McQuillan, A.J.; Houriet, R.; Grätzel, M. Adsorption studies of counterions carried by the sensitizer cis-dithiocyanato(2,2′-bipyridyl-4,4′-dicarboxylate) ruthenium(II) on nanocrystalline TiO2 films. Langmuir 2000, 16, 8525–8528. [Google Scholar] [CrossRef]
- Buscaino, R.; Baiocchi, C.; Barolo, C.; Medana, C.; Grätzel, M.; Nazeeruddin, M.K.K.; Viscardi, G. A mass spectrometric analysis of sensitizer solution used for dye-sensitized solar cell. Inorg. Chim. Acta 2008, 361, 798–805. [Google Scholar] [CrossRef]
- Liu, Y.; Jennings, J.R.; Huang, Y.; Wang, Q.; Zakeeruddin, S.M.; Grätze, M. Cobalt redox mediators for ruthenium-based dye-sensitized solar cells: A combined impedance spectroscopy and near-IR transmittance study. J. Phys. Chem. C 2011, 115, 18847–18855. [Google Scholar] [CrossRef]
- Feldt, S.M.; Gibson, E.A.; Gabrielsson, E.; Sun, L.; Boschloo, G.; Hagfeldt, A. Design of Organic Dyes and Cobalt Polypyridine Redox Mediators for High-Efficiency Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2010, 132, 16714–16724. [Google Scholar] [CrossRef] [PubMed]
- Tsao, H.N.; Yi, C.; Moehl, T.; Yum, J.H.; Zakeeruddin, S.M.; Nazeeruddin, M.K.; Grätzel, M. Cyclopentadithiophene bridged donor–acceptor dyes achieve high power conversion efficiencies in dye-sensitized solar cells based on the tris-cobalt bipyridine redox couple. ChemSusChem 2011, 4, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Feldt, S.M.; Wang, G.; Boschloo, G.; Hagfeldt, A. Effects of driving forces for recombination and regeneration on the photovoltaic performance of dye-sensitized solar cells using cobalt polypyridine redox couples. J. Phys. Chem. C 2011, 115, 21500–21507. [Google Scholar] [CrossRef]
- Feldt, S.M.; Lohse, P.W.; Kessler, F.; Nazeeruddin, M.K.; Grätzel, M.; Boschloo, G.; Hagfeldt, A. Regeneration and recombination kinetics in cobalt polypyridine based dye-sensitized solar cells, explained using Marcus theory. Phys. Chem. Chem. Phys. 2013, 15, 7087–7097. [Google Scholar] [CrossRef] [PubMed]
- Kavan, L.; Yum, J.H.; Nazeeruddin, M.K.; Grätzel, M. Graphene nanoplatelet cathode for Co(III)/(II) mediated dye-sensitized solar cells. ACS Nano 2011, 5, 9171–9178. [Google Scholar] [CrossRef] [PubMed]
- Roy-Mayhew, J.D.; Boschloo, G.; Hagfeldt, A.; Aksay, I.A. Functionalized graphene sheets as a versatile replacement for platinum in dye-sensitized solar cells. ACS Appl. Mater. Interfaces 2012, 4, 2794–2800. [Google Scholar] [CrossRef] [PubMed]
- Carli, S.; Casarin, L.; Syrgiannis, Z.; Boaretto, R.; Benazzi, E.; Caramori, S.; Prato, M.; Bignozzi, C.A. Single Walled Carbon Nanohorns as Catalytic Counter Electrodes for Co(III)/(II) Electron Mediators in Dye Sensitized Cells. ACS Appl. Mater. Interfaces 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.; Vlachopoulos, N.; Häggman, L.; Perruchot, C.; Jouini, M.; Boschloo, G.; Hagfeldt, A. PEDOT counter electrodes for dye-sensitized solar cells prepared by aqueous micellar electrodeposition. Electrochim. Acta 2013, 107, 45–51. [Google Scholar] [CrossRef]
- Pringle, J.M.; Armel, V.; MacFarlane, D.R. Electrodeposited PEDOT-on-plastic cathodes for dye-sensitized solar cells. Chem. Commun. 2010, 46, 5367–5369. [Google Scholar] [CrossRef] [PubMed]
- Carli, S.; Casarin, L.; Bergamini, G.; Caramori, S.; Bignozzi, C.A. Conductive PEDOT Covalently Bound to Transparent FTO Electrodes. J. Phys. Chem. C 2014, 118, 16782–16790. [Google Scholar] [CrossRef]
- Tsao, H.N.; Burschka, J.; Yi, C.; Kessler, F.; Nazeeruddin, M.K.; Grätzel, M. Influence of the interfacial charge-transfer resistance at the counter electrode in dye-sensitized solar cells employing cobalt redox shuttles. Energy Environ. Sci. 2011, 4, 4921. [Google Scholar] [CrossRef]
- Yum, J.-H.; Baranoff, E.; Kessler, F.; Moehl, T.; Ahmad, S.; Bessho, T.; Marchioro, A.; Ghadiri, E.; Moser, J.-E.; Yi, C.; et al. A cobalt complex redox shuttle for dye-sensitized solar cells with high open-circuit potentials. Nat. Commun. 2012, 3, 631. [Google Scholar] [CrossRef] [PubMed]
- Bessho, T.; Zakeeruddin, S.M.; Yeh, C.Y.; Diau, E.W.G.; Grätzel, M. Highly efficient mesoscopic dye-sensitized solar cells based on donor–acceptor-substituted porphyrins. Angew. Chemie Int. Ed. 2010, 49, 6646–6649. [Google Scholar] [CrossRef] [PubMed]
- Yella, A.; Lee, H.-W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.-G.; Yeh, C.-Y.; Zakeeruddin, S.M.; Grätzel, M. Porphyrin-sensitized solar cells with cobalt (II/III)-based redox electrolyte exceed 12 percent efficiency. Science 2011, 334, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Grätzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J.-I.J.; Hanaya, M. Highly-efficient dye-sensitized solar cells with collaborative sensitization by silyl-anchor and carboxy-anchor dyes. Chem. Commun. 2015, 51, 15894–15897. [Google Scholar] [CrossRef] [PubMed]
- Yum, J.-H.; Holcombe, T.W.; Kim, Y.; Rakstys, K.; Moehl, T.; Teuscher, J.; Delcamp, J.H.; Nazeeruddin, M.K.; Grätzel, M. Blue-coloured highly efficient dye-sensitized solar cells by implementing the diketopyrrolopyrrole chromophore. Sci. Rep. 2013, 3, 2446. [Google Scholar] [CrossRef] [PubMed]
- Grzybowski, M.; Gryko, D.T. Diketopyrrolopyrroles: Synthesis, Reactivity, and Optical Properties. Adv. Opt. Mater. 2015, 3, 280–320. [Google Scholar] [CrossRef]
- Yao, Z.; Zhang, M.; Wu, H.; Yang, L.; Li, R.; Wang, P. Donor/Acceptor Indenoperylene Dye for Highly Efficient Organic Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2015, 137, 3799–3802. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Zhang, M.; Li, R.; Yang, L.; Qiao, Y.; Wang, P. A metal-free n-annulated thienocyclopentaperylene dye: Power conversion efficiency of 12% for dye-sensitized solar cells. Angew. Chemie Int. Ed. 2015, 54, 5994–5998. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Sun, D.; Cao, Y.; Tsao, H.N.; Yuan, Y.; Zakeeruddin, S.M.; Wang, P.; Grätzel, M. A Stable Blue Photosensitizer for Color Palette of Dye-Sensitized Solar Cells Reaching 12.6% Efficiency. J. Am. Chem. Soc. 2018, 140, 2405–2408. [Google Scholar] [CrossRef] [PubMed]
- Koussi-Daoud, S.; Schaming, D.; Fillaud, L.; Trippé-Allard, G.; Lafolet, F.; Polanski, E.; Nonomura, K.; Vlachopoulos, N.; Hagfeldt, A.; Lacroix, J.-C. 3,4-Ethylenedioxythiophene-based cobalt complex: An efficient co-mediator in dye-sensitized solar cells with poly(3,4-ethylene dioxythiophene) counter-electrode. Electrochim. Acta 2015, 179, 237–240. [Google Scholar] [CrossRef]
- Nasr-Esfahani, M.; Zendehdel, M.; Yaghoobi Nia, N.; Jafari, B.; Khosravi Babadi, M. Fabrication and characterization of a new dye sensitized solar cell with a new Schiff base cobalt complex as a redox mediator. RSC Adv. 2014, 4, 15961–15967. [Google Scholar] [CrossRef]
- Appleton, T.G. Natural oxygen-carrier and storage proteins contain a transition metal to which the oxygen reversibly coordinates. J. Chem. Educ. 1977, 54, 443. [Google Scholar] [CrossRef]
- Kashif, M.K.; Axelson, J.C.; Du, N.W.; Forsyth, C.M.; Chang, C.J.; Long, R.; Spiccia, L.; Bach, U. A New Direction in Dye-Sensitized Solar Cells Redox Mediator Development: In Situ Fine-Tuning of the Cobalt(II)/(III) Redox Potential through Lewis Base Interactions. J. Am. Chem. Soc. 2012, 134, 16646–16653. [Google Scholar] [CrossRef] [PubMed]
- Kashif, M.K.; Nippe, M.; Duffy, N.W.; Forsyth, C.M.; Chang, C.J.; Long, J.R.; Spiccia, L.; Bach, U. Stable dye-sensitized solar cell electrolytes based on cobalt(II)/(III) complexes of a hexadentate pyridyl ligand. Angew. Chem. Int. Ed. Engl. 2013, 52, 5527–5531. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Huang, F.; Cheng, Y.-B.; Bach, U.; Spiccia, L. Aqueous dye-sensitized solar cell electrolytes based on the cobalt(II)/(III) tris(bipyridine) redox couple. Energy Environ. Sci. 2013, 6, 121–127. [Google Scholar] [CrossRef]
- Dong, C.; Xiang, W.; Huang, F.; Fu, D.; Huang, W.; Bach, U.; Cheng, Y.B.; Li, X.; Spiccia, L. Controlling interfacial recombination in aqueous dye-sensitized solar cells by octadecyltrichlorosilane surface treatment. Angew. Chemie Int. Ed. 2014, 53, 6933–6937. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.; Jiang, R.; Ye, S.; Hagfeldt, A.; Boschloo, G. Development of high efficiency 100% aqueous cobalt electrolyte dye-sensitised solar cells. Phys. Chem. Chem. Phys. 2016, 18, 8419–8427. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Anderson, A.; Barnes, P.R.F.; Xiaoe, L.; Law, C.; O’Regan, B.C. 2000 hours photostability testing of dye sensitised solar cells using a cobalt bipyridine electrolyte. J. Mater. Chem. A 2014, 2, 4751–4757. [Google Scholar] [CrossRef]
- Bella, F.; Vlachopoulos, N.; Nonomura, K.; Zakeeruddin, S.M.; Grätzel, M.; Gerbaldi, C.; Hagfeldt, A. Direct light-induced polymerization of cobalt-based redox shuttles: An ultrafast way towards stable dye-sensitized solar cells. Chem. Commun. 2015, 51, 16308–16311. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Yang, W.; Fredin, L.A.; D’Amario, L.; Karlsson, K.M.; Hagfeldt, A.; Boschloo, G. Supramolecular Hemicage Cobalt Mediators for Dye-Sensitized Solar Cells. ChemPhysChem 2016, 17, 3845–3852. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yang, W.; Pazoki, M.; Boschloo, G.; Kloo, L. Cation-Dependent Photostability of Co(II/III)-Mediated Dye-Sensitized Solar Cells. J. Phys. Chem. C 2015, 119, 24704–24713. [Google Scholar] [CrossRef]
- Hathaway, B.J. Copper. Coord. Chem. Rev. 1981, 35, 211–252. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Billing, D.E. The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion. Coord. Chem. Rev. 1970, 5, 143–207. [Google Scholar] [CrossRef]
- Karlin, K.D.; Yandell, J.K. Redox behavior of blue copper model complexes. Redox potentials and electron-transfer kinetics of some copper(II)–copper(I) complexes with nitrogen and thioether donors. Inorg. Chem. 1984, 23, 1184–1188. [Google Scholar] [CrossRef]
- Gross, E.L. Plastocyanin: Structure and function. Photosynth. Res. 1993, 37, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Henrick, K.; McMillin, D.R. Crystal and molecular structures of bis(4,4′,6,6′-tetramethyl-2,2′-bipyridyl)copper(I) perchlorate, bis(4,4′,6,6′-tetramethyl-2,2′-bipyridyl) copper(II) diperchlorate, and bis(4,4′,6,6′-tetramethyl-2,2′-bipyridyl)copper(II) diperchlorate dihydrate. Inorg. Chem. 1982, 21, 1881–1886. [Google Scholar] [CrossRef]
- Rorabacher, D.B. Electron Transfer by Copper Centers. Chem. Rev. 2004, 104, 651–698. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, N. Photoactive mono- and polynuclear Cu(I)–phenanthrolines. A viable alternative to Ru(II)–polypyridines? Chem. Soc. Rev. 2001, 30, 113–124. [Google Scholar] [CrossRef]
- Hattori, S.; Wada, Y.; Yanagida, S.; Fukuzumi, S. Blue copper model complexes with distorted tetragonal geometry acting as effective electron-transfer mediators in dye-sensitized solar cells. J. Am. Chem. Soc. 2005, 127, 9648–9654. [Google Scholar] [CrossRef] [PubMed]
- Kubota, L.T.; Gushikem, Y. Cyclic voltammetry studies of copper and nickel hexacyanoferrate immobilized on a silica gel surface coated with titanium(IV) oxide. J. Electroanal. Chem. 1993, 362, 219–225. [Google Scholar] [CrossRef]
- Brugnati, M.; Caramori, S.; Cazzanti, S.; Marchini, L.; Argazzi, R.; Bignozzi, C.A. Electron Transfer Mediators for Photoelectrochemical Cells Based on Cu(I) Metal Complexes. Int. J. Photoenergy 2007, 2007, 1–10. [Google Scholar] [CrossRef]
- Bai, Y.; Yu, Q.; Cai, N.; Wang, Y.; Zhang, M.; Wang, P. High-efficiency organic dye-sensitized mesoscopic solar cells with a copper redox shuttle. Chem. Commun. 2011, 47, 4376–4378. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Giordano, F.; Yang, W.; Pazoki, M.; Hao, Y.; Zietz, B.; Grätzel, M.; Hagfeldt, A.; Boschloo, G. Copper phenanthroline as a fast and high-performance redox mediator for dye-sensitized solar cells. J. Phys. Chem. C 2016, 120, 9595–9603. [Google Scholar] [CrossRef]
- Pradhan, S.C.; Hagfeldt, A.; Soman, S. Resurgence of DSCs with copper electrolyte: A detailed investigation of interfacial charge dynamics with cobalt and iodine based electrolytes. J. Mater. Chem. A 2018, 6, 22204. [Google Scholar] [CrossRef]
- Cong, J.; Kinschel, D.; Daniel, Q.; Safdari, M.; Gabrielsson, E.; Chen, H.; Svensson, P.H.; Sun, L.; Kloo, L. Bis(1,1-bis(2-pyridyl)ethane)copper(I/II) as an efficient redox couple for liquid dye-sensitized solar cells. J. Mater. Chem. A 2016, 4, 14550–14554. [Google Scholar] [CrossRef]
- Sun, Z.-Z.; Zheng, K.-M.; Li, Q.-S.; Li, Z.-S. Rational design of Co-based redox mediators for dye-sensitized solar cells by density functional theory. RSC Adv. 2014, 4, 31544–31551. [Google Scholar] [CrossRef]
- Kavan, L.; Saygili, Y.; Freitag, M.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. Electrochemical Properties of Cu(II/I)-Based Redox Mediators for Dye-Sensitized Solar Cells. Electrochim. Acta 2017, 227, 194–202. [Google Scholar] [CrossRef]
- Magni, M.; Giannuzzi, R.; Colombo, A.; Cipolla, M.P.; Dragonetti, C.; Caramori, S.; Carli, S.; Grisorio, R.; Suranna, G.P.; Bignozzi, C.A.; et al. Tetracoordinated Bis-phenanthroline Copper-Complex Couple as Efficient Redox Mediators for Dye Solar Cells. Inorg. Chem. 2016, 55, 5245–5253. [Google Scholar] [CrossRef] [PubMed]
- Ferdowsi, P.; Saygili, Y.; Zakeeruddin, S.M.; Mokhtari, J.; Grätzel, M.; Hagfeldt, A.; Kavan, L. Alternative bases to 4-tert-butylpyridine for dye-sensitized solar cells employing copper redox mediator. Electrochim. Acta 2018, 265, 194–201. [Google Scholar] [CrossRef]
- Saygili, Y.; Stojanovic, M.; Michaels, H.; Tiepelt, J.; Teuscher, J.; Massaro, A.; Pavone, M.; Giordano, F.; Zakeeruddin, S.M.; Boschloo, G.; et al. Effect of Coordination Sphere Geometry of Copper Redox Mediators on Regeneration and Recombination Behavior in Dye-Sensitized Solar Cell Applications. ACS Appl. Energy Mater. 2018, 1, 4950–4962. [Google Scholar] [CrossRef]
- Li, J.; Yang, X.; Yu, Z.; Gurzadyan, G.G.; Cheng, M.; Zhang, F.; Cong, J.; Wang, W.; Wang, H.; Li, X.; et al. Efficient dye-sensitized solar cells with [copper(6,6[prime or minute]-dimethyl-2,2[prime or minute]-bipyridine)2]2+/1+ redox shuttle. RSC Adv. 2017, 7, 4611–4615. [Google Scholar] [CrossRef]
- Richert, S.A.; Tsang, P.K.S.; Sawyer, D.T. Ligand-centered Redox Processes for MnL3, FeL3, and CoL3 Complexes (L = Acetylacetonate, 8-quinolinate, Picolinate, 2,2′-bipyridyl, 1,10-phenanthroline) and For Their Tetrakis(2,6-dichlorophenyl)porphinato Complexes [M(POR)]. Inorg. Chem. 1989, 28, 2471–2475. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Stojanovic, M.; Dar, M.I.; Pechy, P.; Saygili, Y.; Hagfeldt, A.; Zakeeruddin, S.M.; Grätzel, M. Electron-Affinity-Triggered Variations on the Optical and Electrical Properties of Dye Molecules Enabling Highly Efficient Dye-Sensitized Solar Cells. Angew. Chem. 2018, 130, 14321–14324. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, R.; Jiang, R.; Canto-Aguilar, E.J.; Oskam, G.; Boschloo, G. Improving the mass transport of copper-complex redox mediators in dye-sensitized solar cells by reducing the inter-electrode distance. Phys. Chem. Chem. Phys. 2017, 19, 32132–32142. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Teuscher, J.; Saygili, Y.; Zhan, X.; Giordano, F.; Liska, P.; Hua, J.; Zakeeruddin, S.M.; Moser, J.-E.; Grätzel, M.; et al. Dye-sensitized solar cells for efficient power generation under ambient lighting. Nat. Photonics 2017, 11, 372–378. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, Y.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. Direct Contact of Selective Charge Extraction Layers Enables High-Efficiency Molecular Photovoltaics. Joule 2018, 2, 1108–1117. [Google Scholar] [CrossRef]
- Hoffeditz, W.L.; Katz, M.J.; Deria, P.; Cutsail, G.E., III; Pellin, M.J.; Farha, O.K.; Hupp, J.T.; Cutsail, G.E.; Pellin, M.J.; Farha, O.K.; et al. One Electron Changes Everything. A Multispecies Copper Redox Shuttle for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2016, 120, 3731–3740. [Google Scholar] [CrossRef]
- Wang, Y.; Hamann, T.W. Improved performance induced by in situ ligand exchange reactions of copper bipyridyl redox couples in dye-sensitized solar cells. Chem. Commun. 2018, 54, 12361. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Dragonetti, C.; Magni, M.; Roberto, D.; Demartin, F.; Caramori, S.; Bignozzi, C.A. Efficient Copper Mediators Based on Bulky Asymmetric Phenanthrolines for DSSCs. ACS Appl. Mater. Interfaces 2014, 6, 13945–13955. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Ossola, R.; Magni, M.; Roberto, D.; Jacquemin, D.; Castellano, C.; Demartin, F.; Dragonetti, C. Intriguing C–H···Cu interactions in bis-(phenanthroline)Cu(I) redox mediators for dye-sensitized solar cells. Dalt. Trans. 2018, 47, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- Magni, M.; Colombo, A.; Dragonetti, C.; Mussini, P. Steric vs electronic effects and solvent coordination in the electrochemistry of phenanthroline-based copper complexes. Electrochim. Acta 2014, 141, 324–330. [Google Scholar] [CrossRef]
- Benazzi, E.; Magni, M.; Colombo, A.; Dragonetti, C.; Caramori, S.; Bignozzi, C.A.; Grisorio, R.; Suranna, G.P.; Cipolla, M.P.; Manca, M.; et al. Bis(1,10-phenanthroline) copper complexes with tailored molecular architecture: From electrochemical features to application as redox mediators in dye-sensitized solar cells. Electrochim. Acta 2018, 271, 180–189. [Google Scholar] [CrossRef]
- Colombo, A.; Di Carlo, G.; Dragonetti, C.; Magni, M.; Orbelli Biroli, A.; Pizzotti, M.; Roberto, D.; Tessore, F.; Benazzi, E.; Bignozzi, C.A.; et al. Coupling of Zinc Porphyrin Dyes and Copper Electrolytes: A Springboard for Novel Sustainable Dye-Sensitized Solar Cells. Inorg. Chem. 2017, 56, 14189–14197. [Google Scholar] [CrossRef] [PubMed]
- Dragonetti, C.; Magni, M.; Colombo, A.; Melchiorre, F.; Biagini, P.; Roberto, D. Coupling of a Copper Dye with a Copper Electrolyte: A Fascinating Springboard for Sustainable Dye-Sensitized Solar Cells. ACS Appl. Energy Mater. 2018, 1, 751–756. [Google Scholar] [CrossRef]
- Karpacheva, M.; Malzner, F.J.; Wobill, C.; Büttner, A.; Constable, E.C.; Housecroft, C.E. Cuprophilia: Dye-sensitized solar cells with copper(I) dyes and copper(I)/(II) redox shuttles. Dye. Pigment. 2018, 156, 410–416. [Google Scholar] [CrossRef]
- Leandri, V.; Daniel, Q.; Chen, H.; Sun, L.; Gardner, J.M.; Kloo, L. Electronic and Structural Effects of Inner Sphere Coordination of Chloride to a Homoleptic Copper(II) Diimine Complex. Inorg. Chem. 2018, 57, 4556–4562. [Google Scholar] [CrossRef] [PubMed]
- Pernechele, R. New Copper Redox Couples for Liquid Dye-Sensitized Solar Cells, Degree Project in Chemical Science and Engineering; KTH Royal Institute of Technology: Stockholm, Sweden, 2017. [Google Scholar]
- Michaels, H.; Benesperi, I.; Edvinsson, T.; Muñoz-Garcia, A.B.; Pavone, M.; Boschloo, G.; Freitag, M. Copper Complexes with Tetradentate Ligands for Enhanced Charge Transport in Dye-Sensitized Solar Cells. Inorganics 2018, 6, 53. [Google Scholar] [CrossRef]
- Hu, M.; Shen, J.; Yu, Z.; Liao, R.; Gurzadyan, G.G.; Yang, X.; Hagfeldt, A.; Wang, M.; Sun, L. Efficient and Stable Dye-Sensitized Solar Cells Based on a Tetradentate Copper(II/I) Redox Mediator. ACS Appl. Mater. Interfaces 2018, 10, 30409–30416. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Daniel, Q.; Pazoki, M.; Sveinbjörnsson, K.; Zhang, J.; Sun, L.; Hagfeldt, A.; Boschloo, G. High-efficiency dye-sensitized solar cells with molecular copper phenanthroline as solid hole conductor. Energy Environ. Sci. 2015, 8, 2634–2637. [Google Scholar] [CrossRef]
- Cao, Y.; Saygili, Y.; Ummadisingu, A.; Teuscher, J.; Luo, J.; Pellet, N.; Giordano, F.; Zakeeruddin, S.M.; Moser, J.-E.; Freitag, M.; et al. 11% efficiency solid-state dye-sensitized solar cells with copper(II/I) hole transport materials. Nat. Commun. 2017, 8, 15390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, Y.; Bahng, H.W.; Cao, Y.; Yi, C.; Saygili, Y.; Luo, J.; Liu, Y.; Kavan, L.; Moser, J.E.; et al. Comprehensive control of voltage loss enables 11.7% efficient solid-state dye-sensitized solar cells. Energy Environ. Sci. 2018, 11, 1779–1787. [Google Scholar] [CrossRef]
- Ruess, R.; Nguyen, T.H.Q.; Schlettwein, D. Metal Complexes as Redox Shuttles in Dye-Sensitized Solar Cells Based on Electrodeposited ZnO: Tuning Recombination Kinetics and Conduction Band Energy. J. Electrochem. Soc. 2018, 165, H3115–H3121. [Google Scholar] [CrossRef]
- Kavan, L.; Krysova, H.; Janda, P.; Tarabkova, H.; Saygili, Y.; Freitag, M.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. Novel highly active Pt/graphene catalyst for cathodes of Cu(II/I)-mediated dye-sensitized solar cells. Electrochim. Acta 2017, 251, 167–175. [Google Scholar] [CrossRef]
- Rodrigues, R.R.; Cheema, H.; Delcamp, J.H. A High-Voltage Molecular-Engineered Organic Sensitizer-Iron Redox Shuttle Pair: 1.4 V DSSC and 3.3 V SSM-DSSC Devices. Angew. Chemie Int. Ed. 2018, 57, 5472–5476. [Google Scholar] [CrossRef] [PubMed]
- Braterman, P.S.; Song, J.I.; Peacock, R.D. Electronic absorption spectra of the iron(II) complexes of 2,2′-bipyridine, 2,2′-bipyrimidine, 1,10-phenanthroline, and 2,2′:6′,2″-terpyridine and their reduction products. Inorg. Chem. 1992, 31, 555–559. [Google Scholar] [CrossRef]
- Daeneke, T.; Uemura, Y.; Duffy, N.W.; Mozer, A.J.; Koumura, N.; Bach, U.; Spiccia, L. Aqueous dye-sensitized solar cell electrolytes based on the ferricyanide-ferrocyanide redox couple. Adv. Mater. 2012, 24, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Horvat-Radošević, V.; Kvastek, K.; Križekar, D. Kinetics of the [Fe(CN)6]3−/[Fe(CN)6]4− Redox Couple Reaction on Anodically Passivated Fe80B20. Croat. Chem. Acta 1997, 70, 537–561. [Google Scholar]
- Ameur, Z.O.; Husein, M.M. Electrochemical Behavior of Potassium Ferricyanide in Aqueous and (w/o) Microemulsion Systems in the Presence of Dispersed Nickel Nanoparticles. Sep. Sci. Technol. 2013, 48, 681–689. [Google Scholar] [CrossRef]
- Rutkowska, I.A.; Andrearczyk, A.; Zoladek, S.; Goral, M.; Darowicki, K.; Kulesza, P.J. Electrochemical characterization of Prussian blue type nickel hexacyanoferrate redox mediator for potential application as charge relay in dye-sensitized solar cells. J. Solid State Electrochem. 2011, 15, 2545–2552. [Google Scholar] [CrossRef]
- Hamann, T.W.; Farha, O.K.; Hupp, J.T. Outer-Sphere Redox Couples as Shuttles in Dye-Sensitized Solar Cells. Performance Enhancement Based on Photoelectrode Modification via Atomic Layer Deposition. J. Phys. Chem. C 2008, 112, 19756–19764. [Google Scholar] [CrossRef]
- Penner, R.M.; Heben, M.J.; Longin, T.L.; Lewis, N.S. Fabrication and Use of Nanometer-Sized Electrodes in Electrochemistry. Science 1990, 250, 1118–1121. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Stanbury, D.M. Kinetics and Mechanism of the One-Electron Reduction of Iodine by [RuII(NH3)5 isn]2+ in Aqueous Solution. Inorg. Chem. 1998, 37, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Gregg, B.A.; Pichot, F.; Ferrere, S.; Fields, C.L. Interfacial Recombination Processes in Dye-Sensitized Solar Cells and Methods to Passivate the Interfaces. J. Phys. Chem. B 2001, 105, 1422–1429. [Google Scholar] [CrossRef]
- Klahr, B.M.; Hamann, T.W. Performance Enhancement and Limitations of Cobalt Bipyridyl Redox Shuttles in Dye-Sensitized Solar Cells. J. Phys. Chem. C 2009, 113, 14040–14045. [Google Scholar] [CrossRef]
- Ito, S. Investigation of Dyes for Dye-Sensitized Solar Cells: Ruthenium-Complex Dyes, Metal-Free Dyes, Metal-Complex Porphyrin Dyes and Natural Dyes. In Solar Cells—Dye-Sensitized Devices; Kosyachenko, L.A., Ed.; InTech: Rijeka, Croatia; Shanghai, China, 2011; pp. 19–48. ISBN 978-953-307-735-2. [Google Scholar]
- Ondersma, J.; Hamann, T. Impedance Investigation of Dye-Sensitized Solar Cells Employing Outer-Sphere Redox Shuttles. J. Phys. Chem. C 2010, 638–645. [Google Scholar] [CrossRef]
- Daeneke, T.; Kwon, T.-H.; Holmes, A.B.; Duffy, N.W.; Bach, U.; Spiccia, L. High-efficiency dye-sensitized solar cells with ferrocene-based electrolytes. Nat. Chem. 2011, 3, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Congiu, M.; Nunes-Neto, O.; De Marco, M.L.; Dini, D.; Graeff, C.F.O. Cu2−xS films as counter-electrodes for dye solar cells with ferrocene-based liquid electrolytes. Thin Solid Films 2016, 612, 22–28. [Google Scholar] [CrossRef]
- Daeneke, T.; Mozer, A.J.; Kwon, T.-H.; Duffy, N.W.; Holmes, A.B.; Bach, U.; Spiccia, L. Dye regeneration and charge recombination in dye-sensitized solar cells with ferrocene derivatives as redox mediators. Energy Environ. Sci. 2012, 5, 7090–7099. [Google Scholar] [CrossRef]
- Cariello, M.; Ahn, S.; Park, K.-W.; Chang, S.-K.; Hong, J.; Cooke, G. An investigation of the role increasing π-conjugation has on the efficiency of dye-sensitized solar cells fabricated from ferrocene-based dyes. RSC Adv. 2016, 6, 9132–9138. [Google Scholar] [CrossRef]
- El-Zohry, A.M.; Cong, J.; Karlsson, M.; Kloo, L.; Zietz, B. Ferrocene as a rapid charge regenerator in dye-sensitized solar cells. Dye. Pigment. 2016, 132, 360–368. [Google Scholar] [CrossRef]
- Takaichi, J.; Morimoto, Y.; Ohkubo, K.; Shimokawa, C.; Hojo, T.; Mori, S.; Asahara, H.; Sugimoto, H.; Fujieda, N.; Nishiwaki, N.; et al. Redox chemistry of nickel(II) complexes supported by a series of noninnocent β-diketiminate ligands. Inorg. Chem. 2014, 53, 6159–6169. [Google Scholar] [CrossRef] [PubMed]
- Zilbermann, I.; Maimon, E.; Cohen, H.; Meyerstein, D. Redox chemistry of nickel complexes in aqueous solutions. Chem. Rev. 2005, 105, 2609–2625. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Spokoyny, A.M.; She, C.; Farha, O.K.; Mirkin, C.A.; Marks, T.J.; Hupp, J.T. Ni(III)/(IV)Bis(dicarbollide) as a Fast, Noncorrosive Redox Shuttle for Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2010, 132, 4580–4582. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, M.F.; Zink, J.I.; Skelton, J.M.; Bayer, M.J.; Liu, C.; Livshits, E.; Baer, R.; Neuhauser, D. Electrical or Photocontrol of the Rotary Motion of a Metallacarborane. Science 2004, 303, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Spokoyny, A.M.; Li, T.C.; Farha, O.K.; Machan, C.W.; She, C.; Stern, C.L.; Marks, T.J.; Hupp, J.T.; Mirkin, C.A. Electronic Tuning of Nickel-Based Bis(dicarbollide) Redox Shuttles in Dye-Sensitized Solar Cells. Angew. Chemie Int. Ed. 2010, 49, 5339–5343. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Fabregat-Santiago, F.; Farha, O.K.; Spokoyny, A.M.; Raga, S.R.; Bisquert, J.; Mirkin, C.A.; Marks, T.J.; Hupp, J.T. SiO2 Aerogel Templated, Porous TiO2 Photoanodes for Enhanced Performance in Dye-Sensitized Solar Cells Containing a Ni(III)/(IV) Bis(dicarbollide) Shuttle. J. Phys. Chem. C 2011, 115, 11257–11264. [Google Scholar] [CrossRef]
- Yamaguchi, K.S.; Sawyer, D.T. The Redox Chemistry of Manganese(III) and -(IV) Complexes. Isr. J. Chem. 1985, 25, 164–176. [Google Scholar] [CrossRef]
- Morrison, M.M.; Sawyer, D.T. Redox chemistry of the polyimine complexes of manganese(II), -(III), and -(IV) in acetonitrile. Inorg. Chem. 1978, 17, 333–337. [Google Scholar] [CrossRef]
- Arora, C.L. Lecture demonstration of the various oxidation states of manganese. J. Chem. Educ. 1977, 54, 302–303. [Google Scholar] [CrossRef]
- Kolb, D. Oxidation states of manganese. J. Chem. Educ. 1988, 65, 1004–1005. [Google Scholar] [CrossRef]
- Krewald, V.; Neese, F.; Pantazis, D.A. Resolving the Manganese Oxidation States in the Oxygen-evolving Catalyst of Natural Photosynthesis. Isr. J. Chem. 2015, 55, 1219–1232. [Google Scholar] [CrossRef]
- Perera, I.R.; Gupta, A.; Xiang, W.; Daeneke, T.; Bach, U.; Evans, R.A.; Ohlin, C.A.; Spiccia, L. Introducing manganese complexes as redox mediators for dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2014, 16, 12021–12028. [Google Scholar] [CrossRef] [PubMed]
- Carli, S.; Benazzi, E.; Casarin, L.; Bernardi, T.; Bertolasi, V.; Argazzi, R.; Caramori, S.; Bignozzi, C.A. On the stability of manganese tris(β-diketonate) complexes as redox mediators in DSSCs. Phys. Chem. Chem. Phys. 2016, 18, 5949–5956. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Rigsby, M.A.; Duncan, C.T.; Milligan, P.L.; Lord, R.L.; Baik, M.; Schultz, F.A. Synthesis, Structure, and Properties of Low-Spin Manganese(III)–Poly(pyrazolyl) borate Complexes. Inorg. Chem. 2007, 46, 2596–2603. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulou, A.; Vlasiou, M.; Tziouris, P.A.; Tsiafoulis, C.; Tsipis, A.C.; Rehder, D.; Kabanos, T.A.; Keramidas, A.D.; Stathatos, E. Oxidovanadium(IV/V) complexes as new redox mediators in dye-sensitized solar cells: A combined experimental and theoretical study. Inorg. Chem. 2015, 54, 3979–3988. [Google Scholar] [CrossRef] [PubMed]
- Oyaizu, K.; Hayo, N.; Sasada, Y.; Kato, F.; Nishide, H. Enhanced bimolecular exchange reaction through programmed coordination of a five-coordinate oxovanadium complex for efficient redox mediation in dye-sensitized solar cells. Dalt. Trans. 2013, 42, 16090–16095. [Google Scholar] [CrossRef] [PubMed]
- Cazzanti, S.; Caramori, S.; Argazzi, R.; Elliott, C.M.; Bignozzi, C.A. Efficient non-corrosive electron-transfer mediator mixtures for dye-sensitized solar cells. J. Am. Chem. Soc. 2006, 128, 9996–9997. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Hao, Y.; Sun, L.; Kloo, L. Two redox couples are better than one: Improved current and fill factor from cobalt-based electrolytes in dye-sensitized solar cells. Adv. Energy Mater. 2014, 4, 1301273. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, H.; Chen, X.; Yan, F. Imidazolium functionalized cobalt tris(bipyridyl) complex redox shuttles for high efficiency ionic liquid electrolyte dye-sensitized solar cells. J. Mater. Chem. A 2013, 1, 11933–11941. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, W.; Zhang, L.; Jiang, R.; Mijangos, E.; Saygili, Y.; Hammarström, L.; Hagfeldt, A.; Boschloo, G. A small electron donor in cobalt complex electrolyte significantly improves efficiency in dye-sensitized solar cells. Nat. Commun. 2016, 7, 13934. [Google Scholar] [CrossRef] [PubMed]
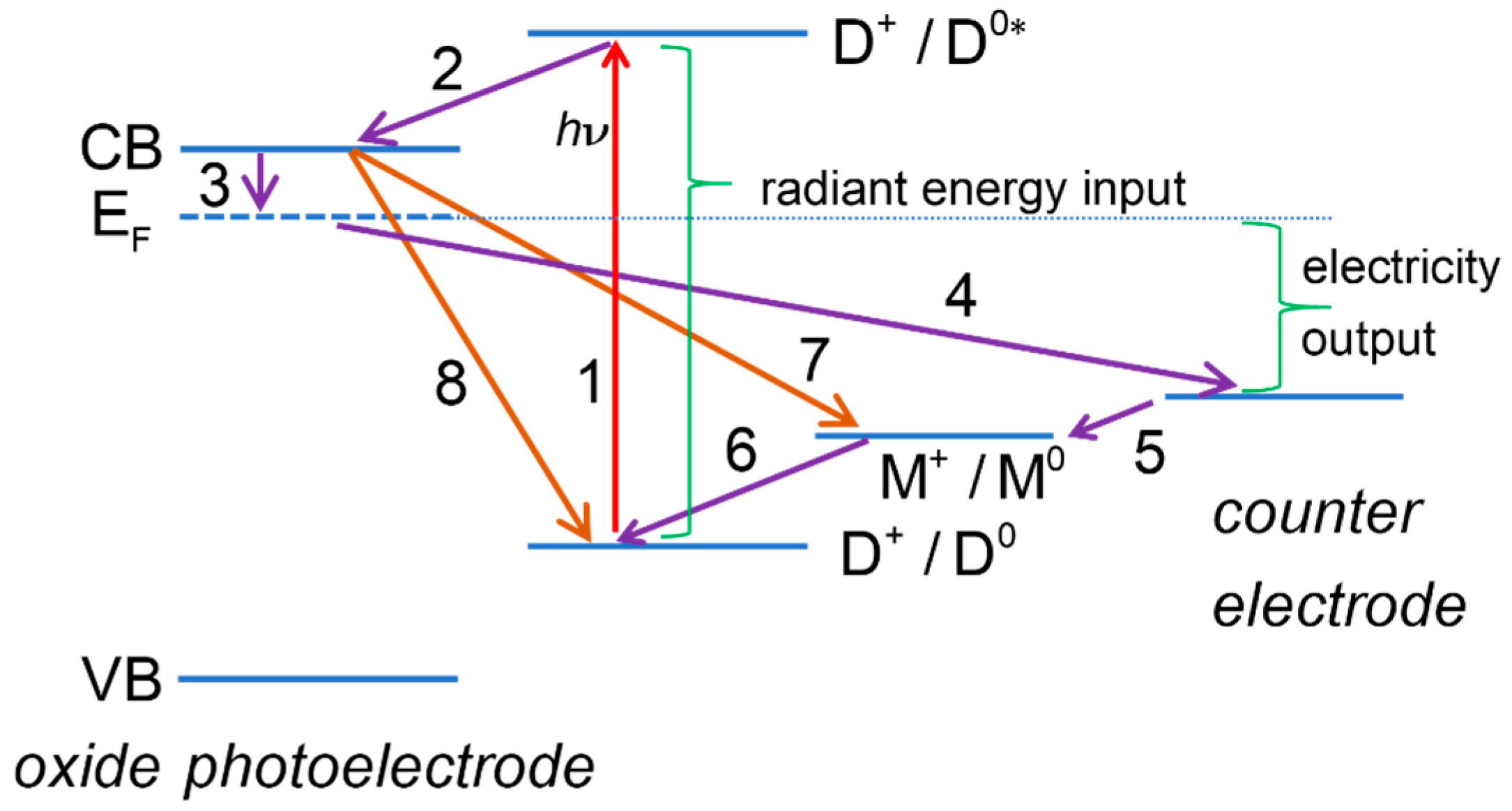
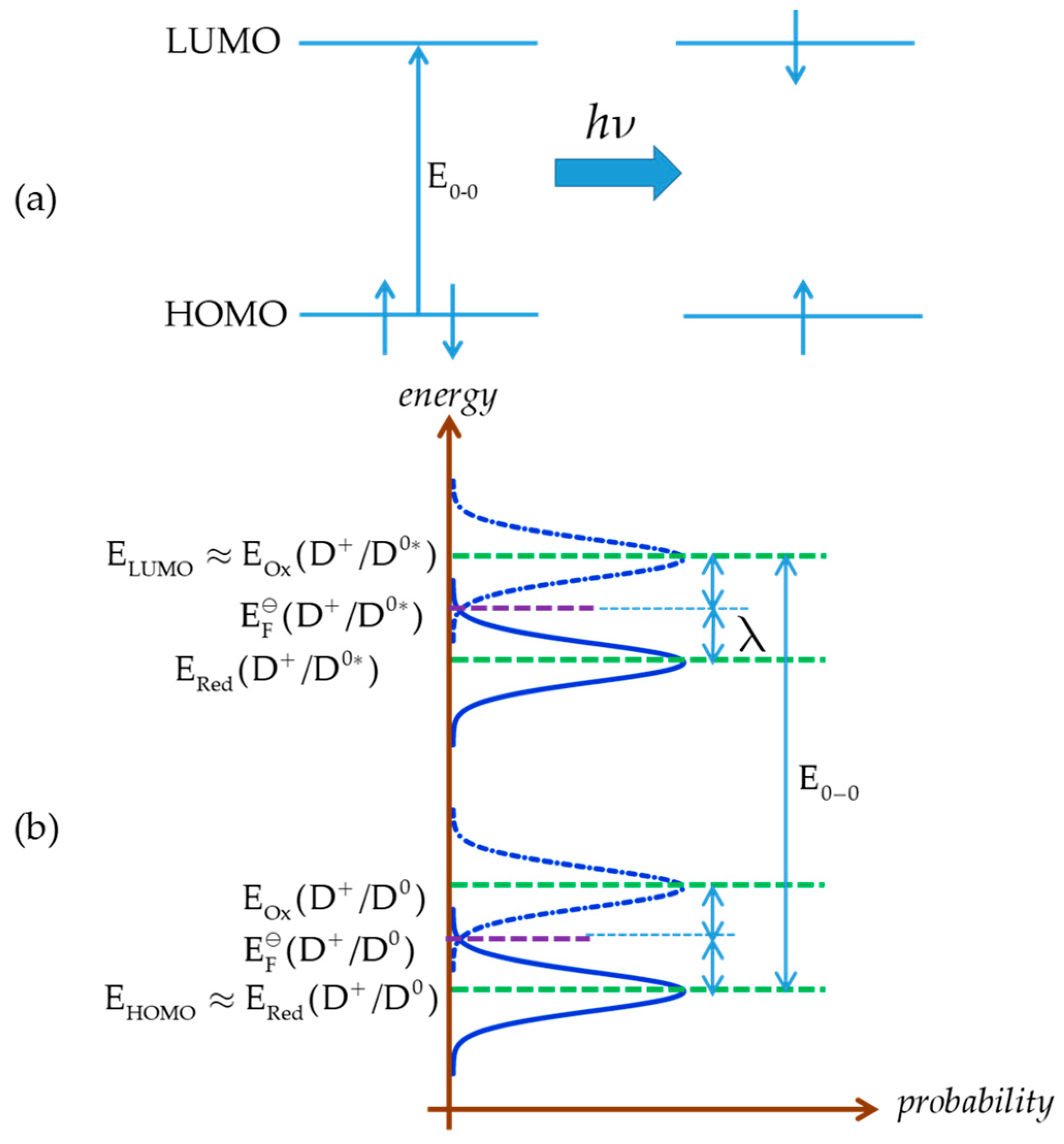
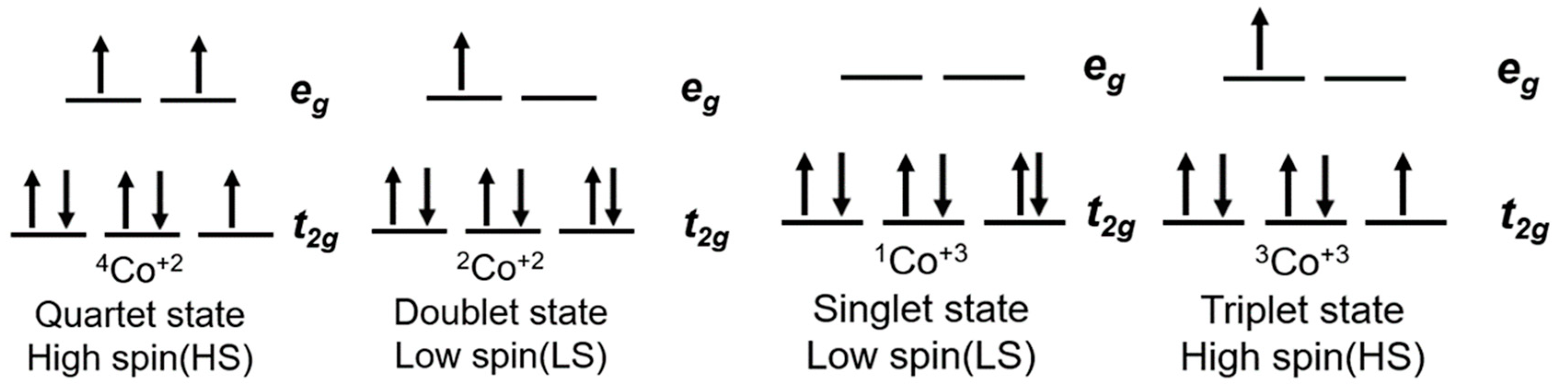
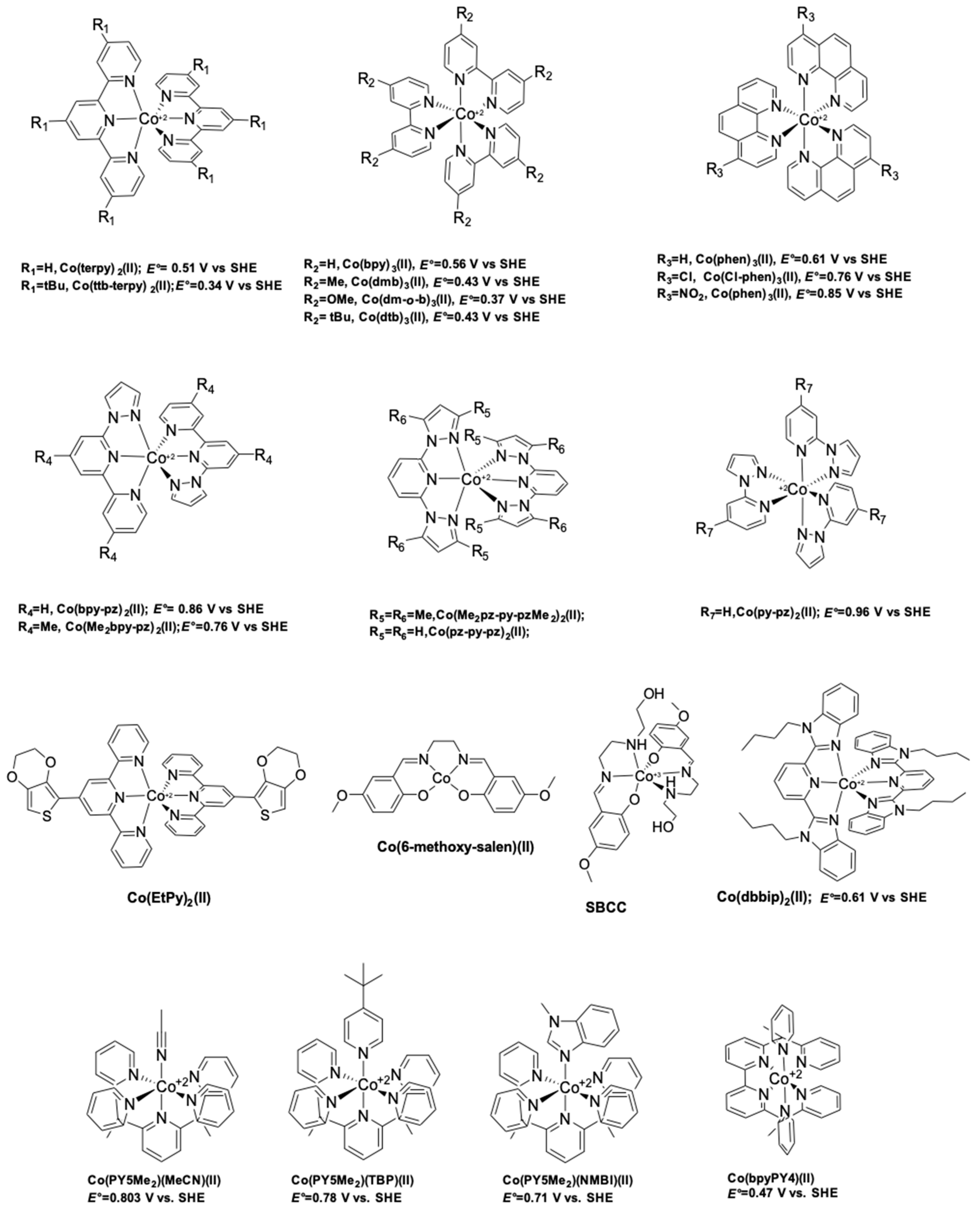
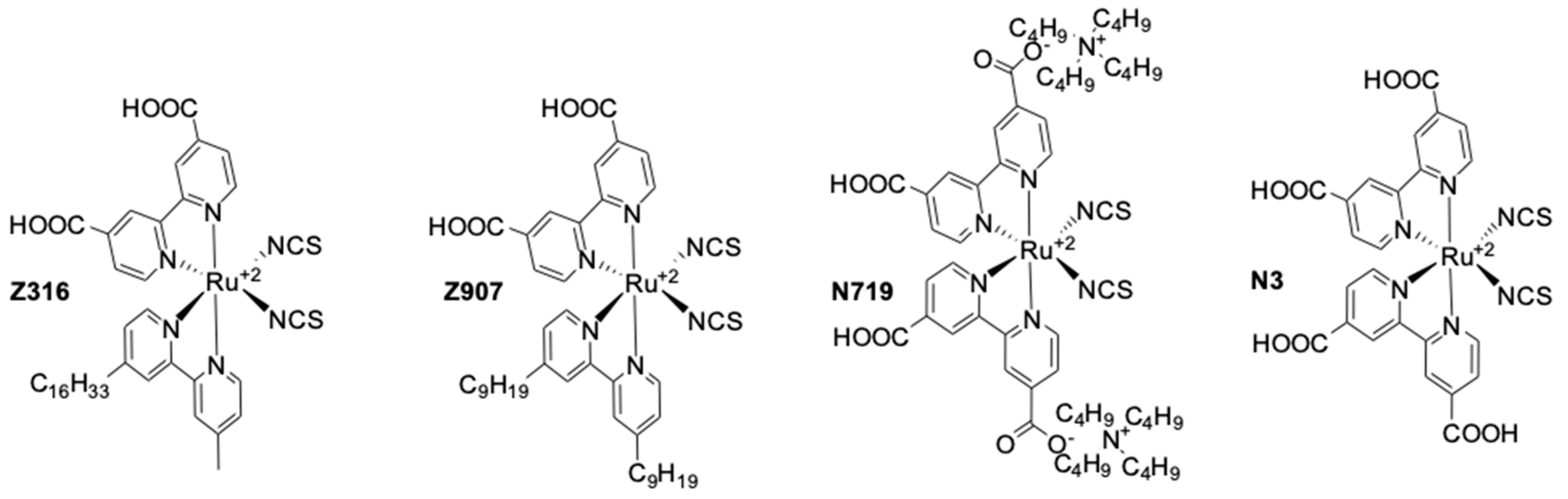
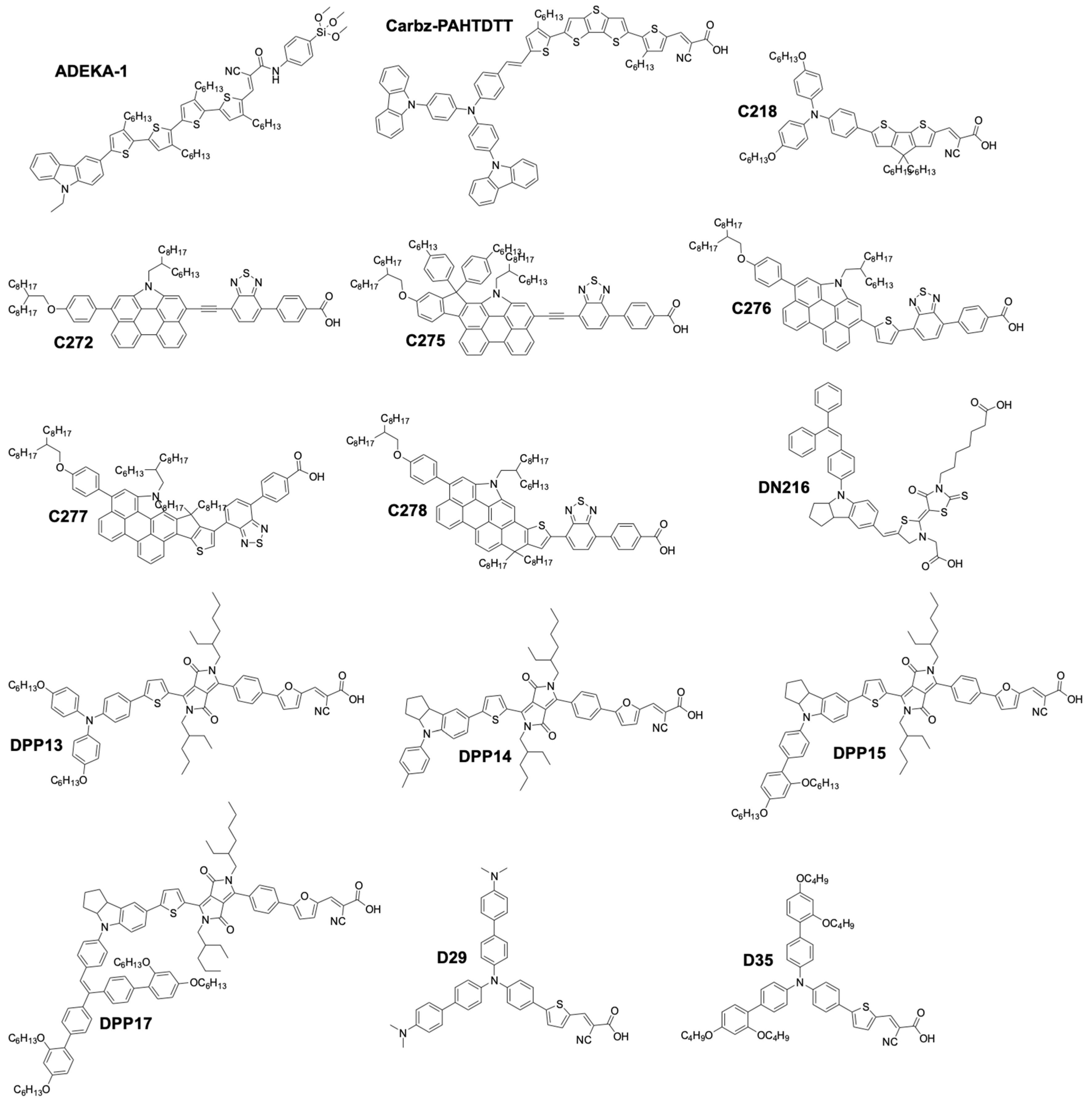
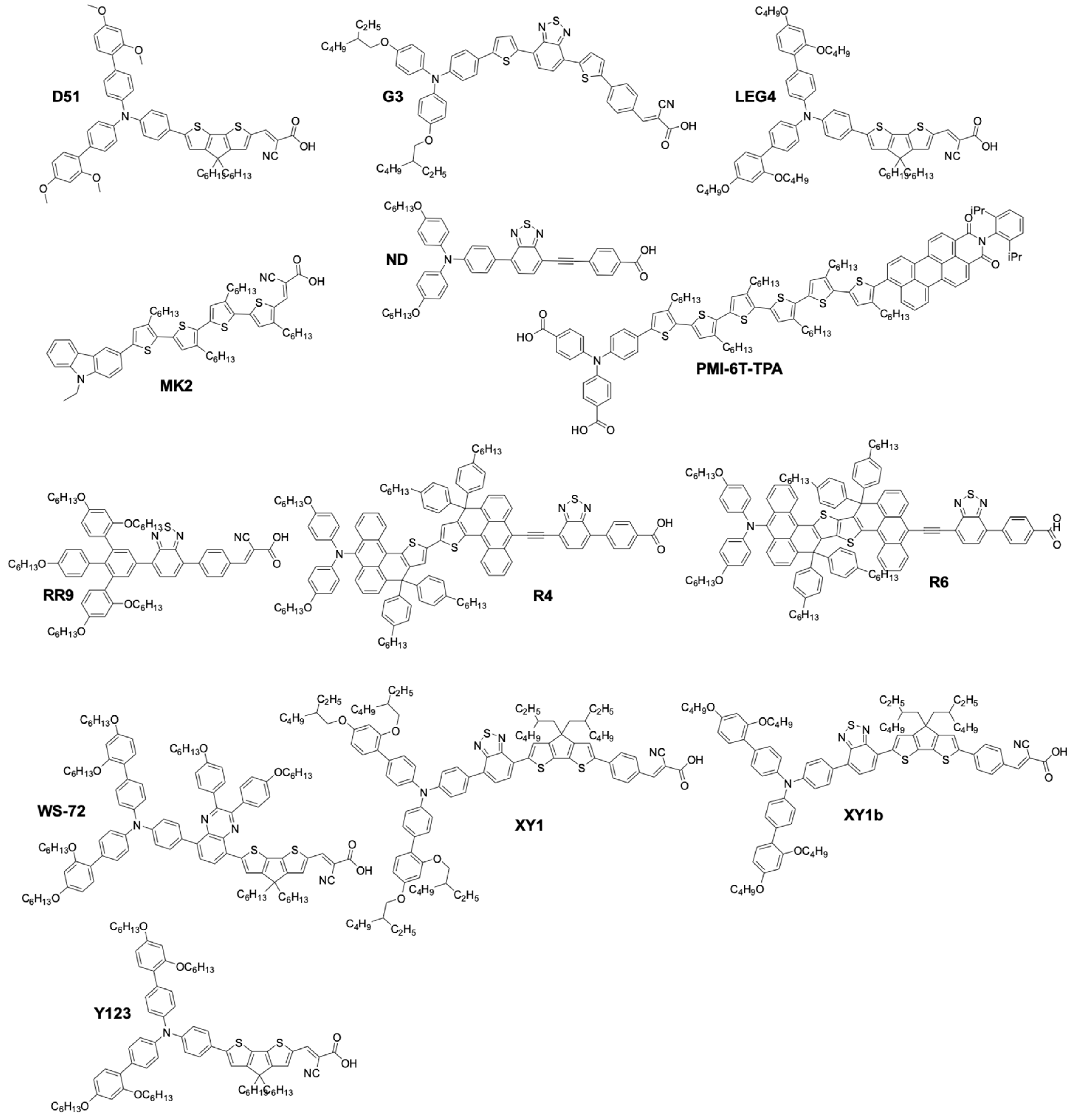
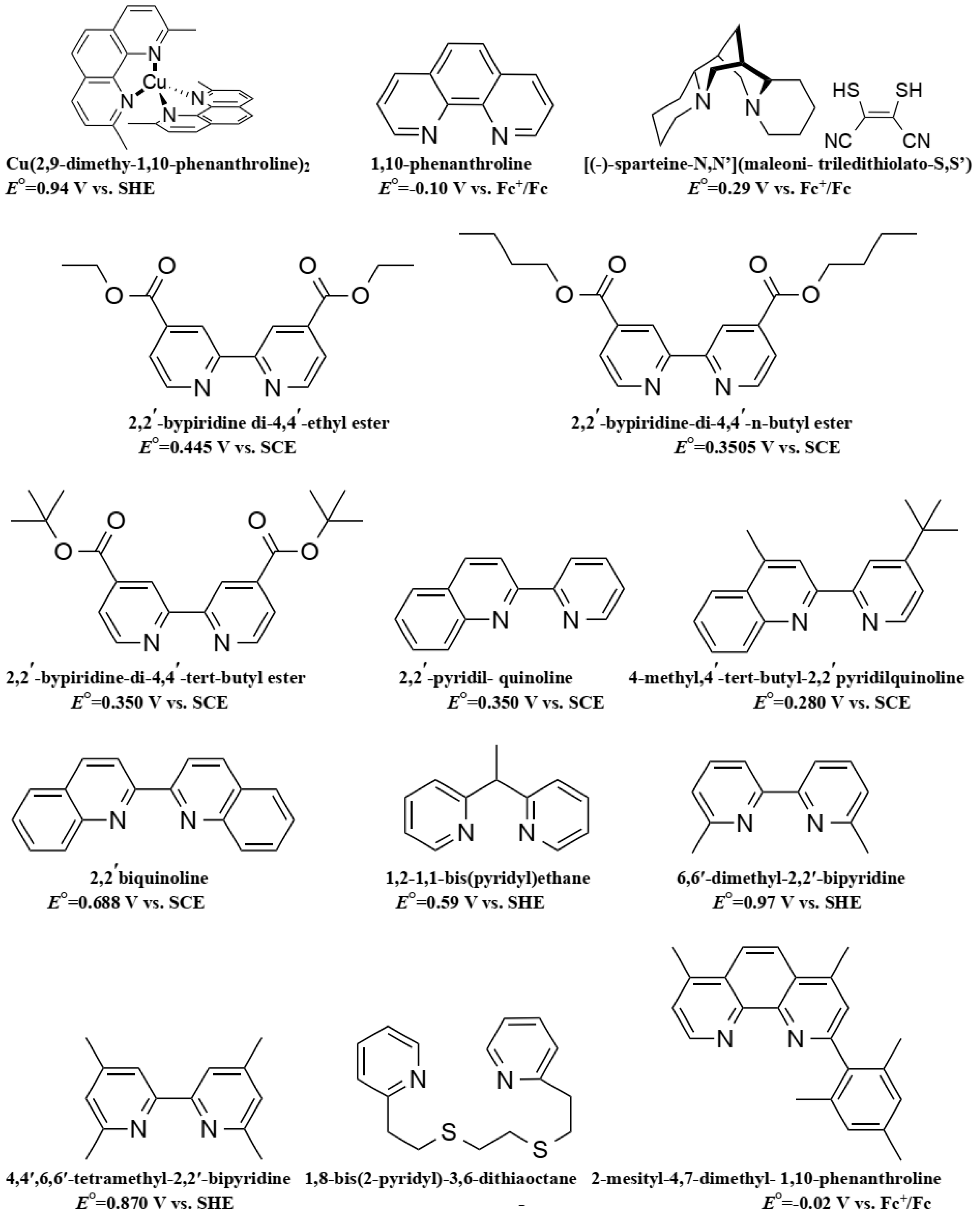
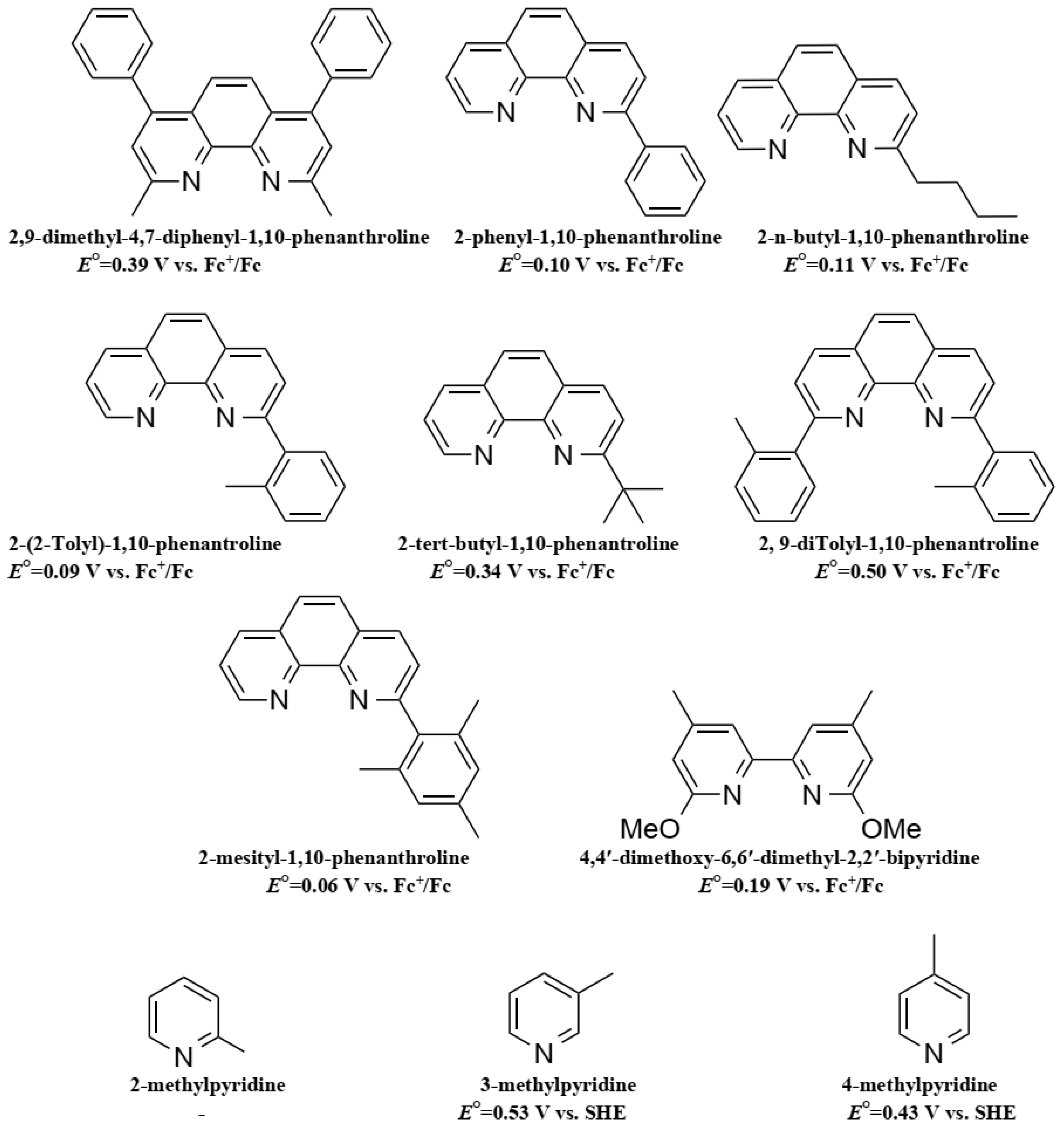
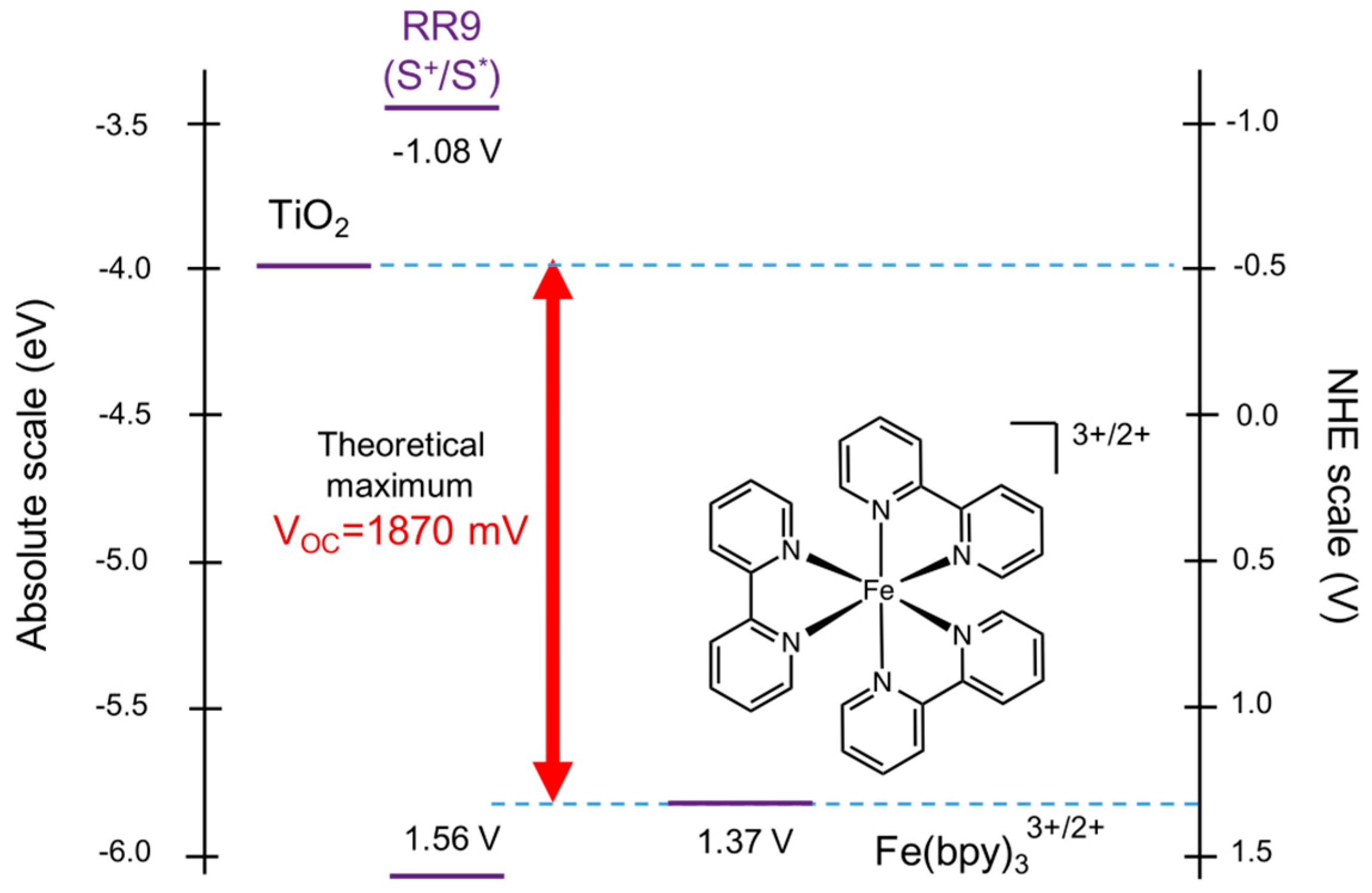
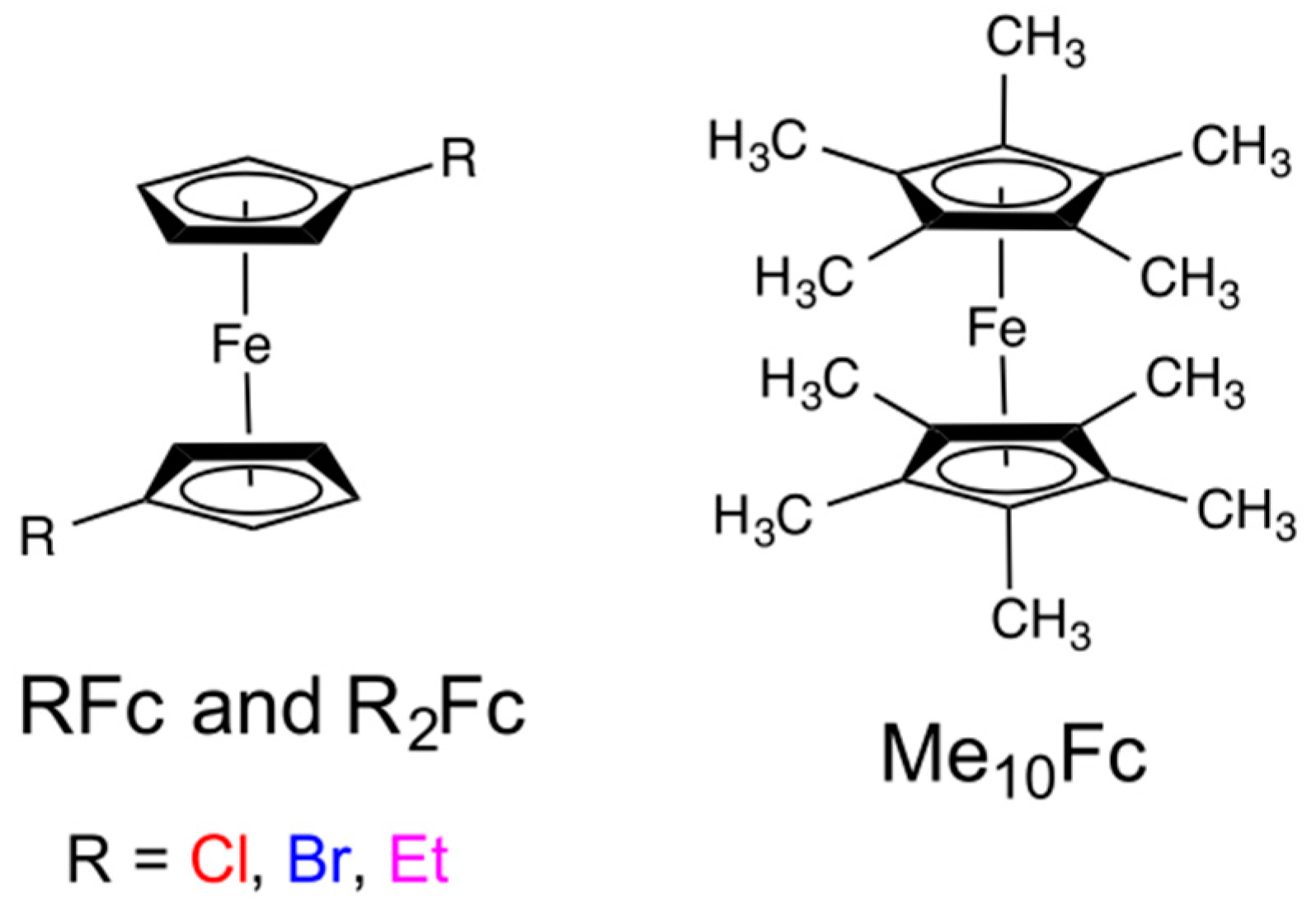
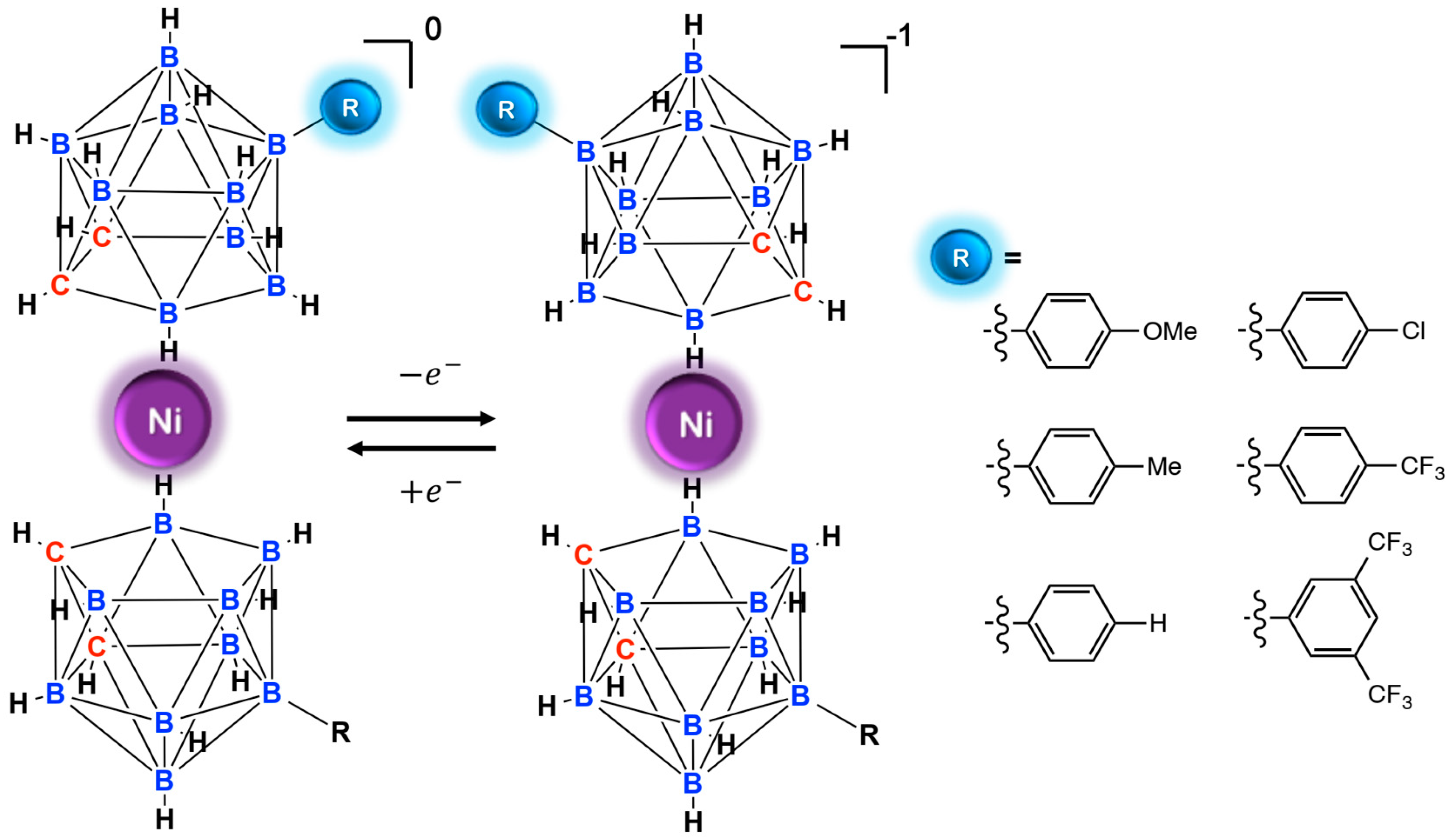
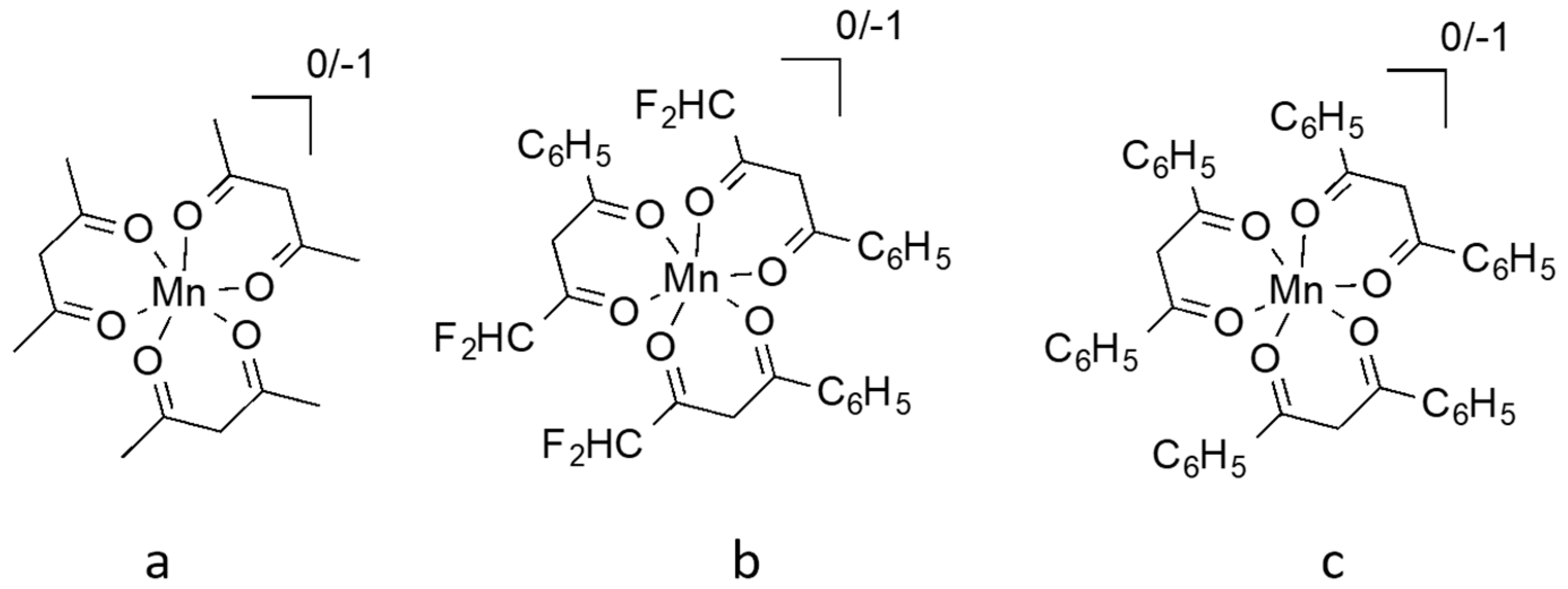
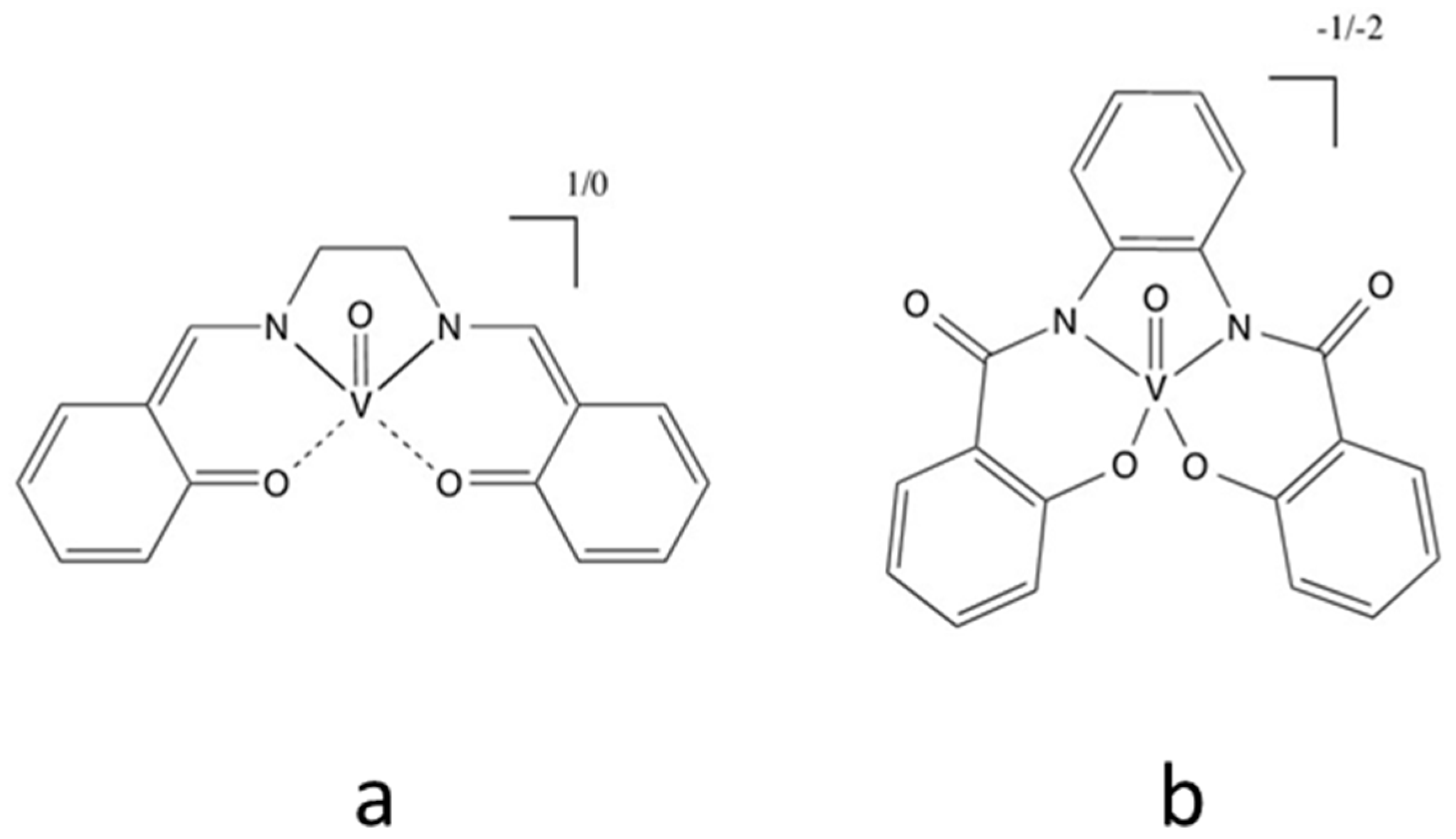
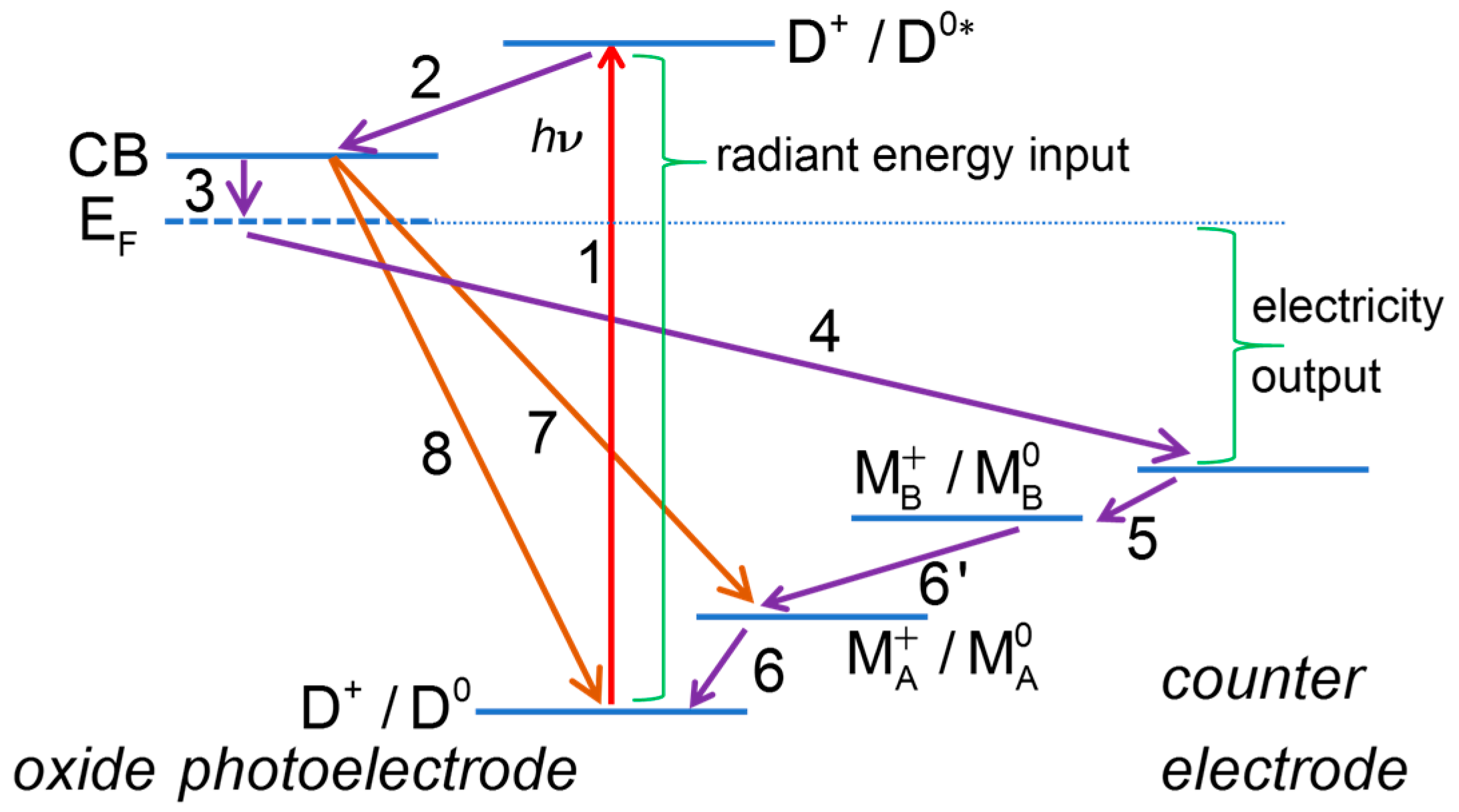
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).