Design of Molecular Water Oxidation Catalysts Stabilized by Ultrathin Inorganic Overlayers—Is Active Site Protection Necessary?


Abstract
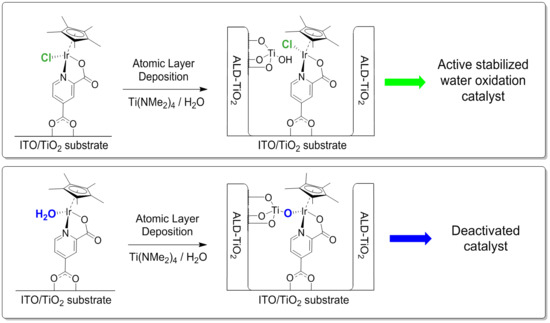
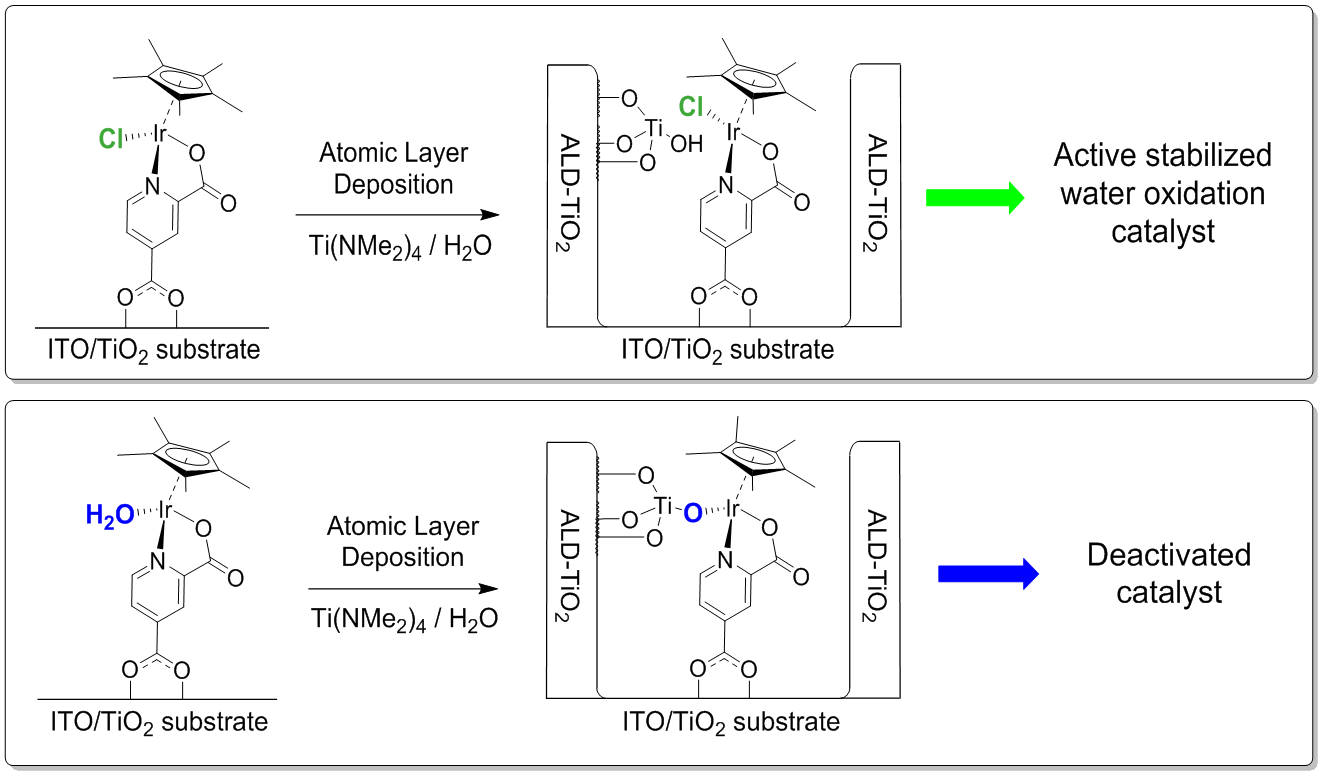{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
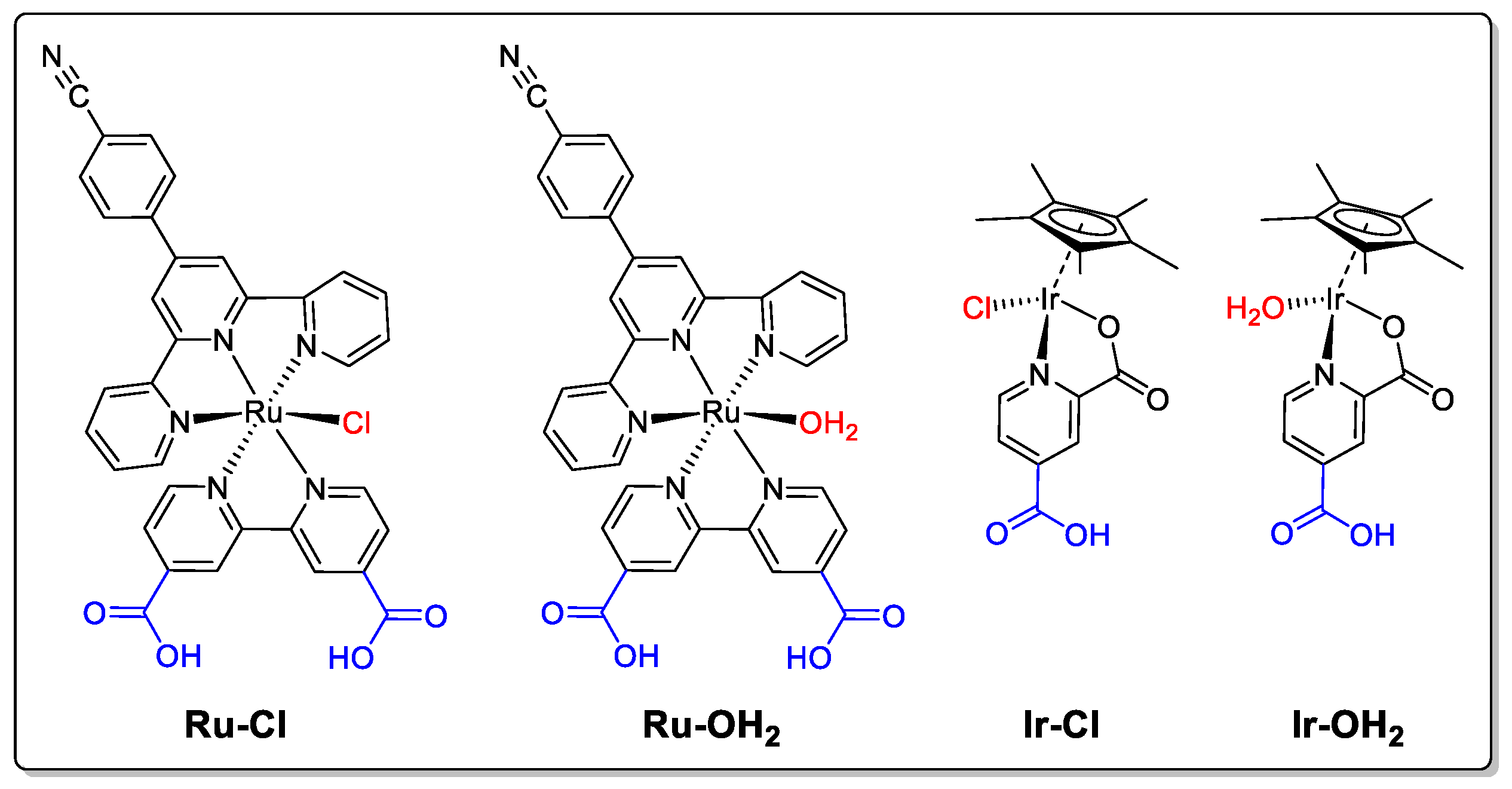
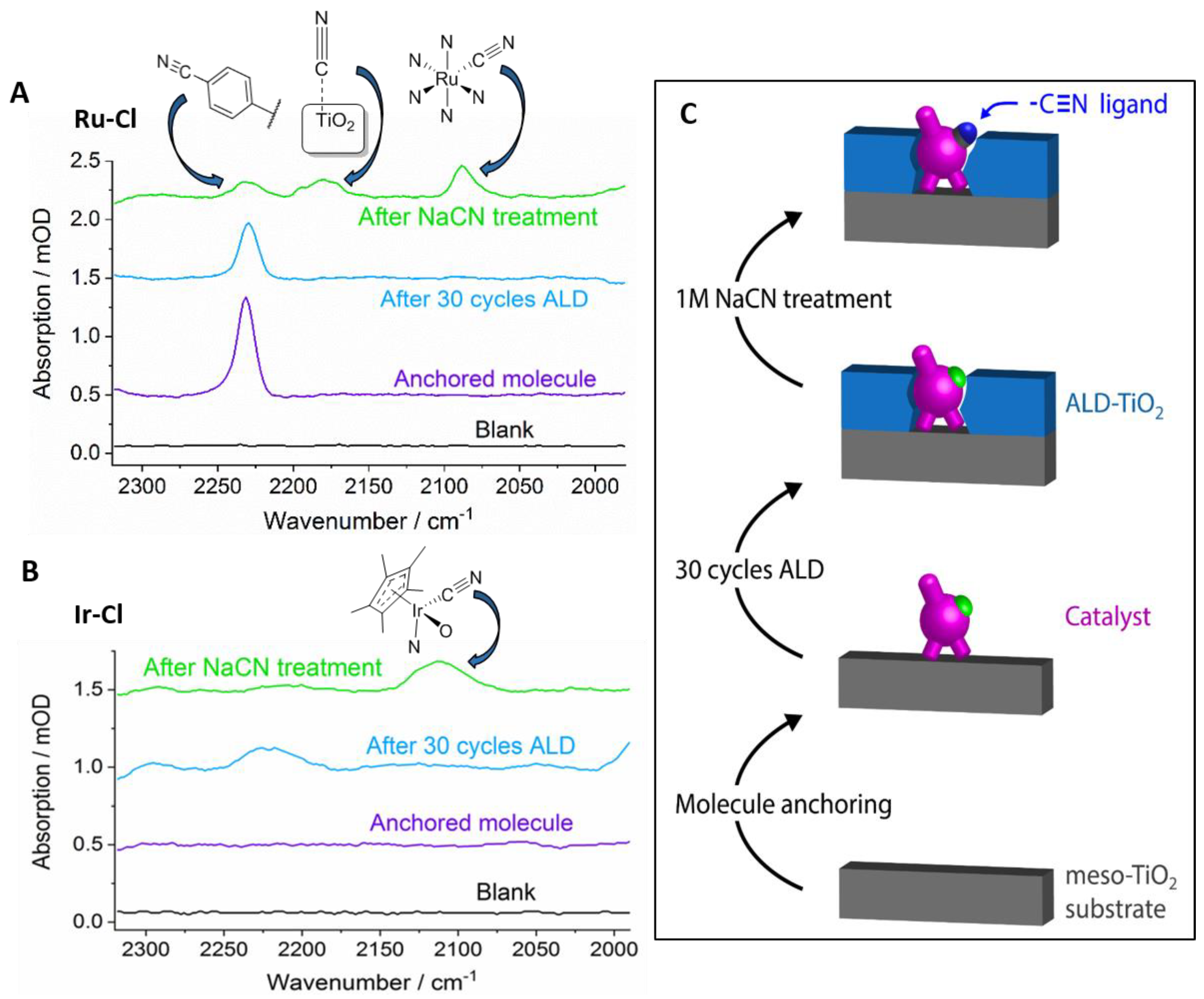
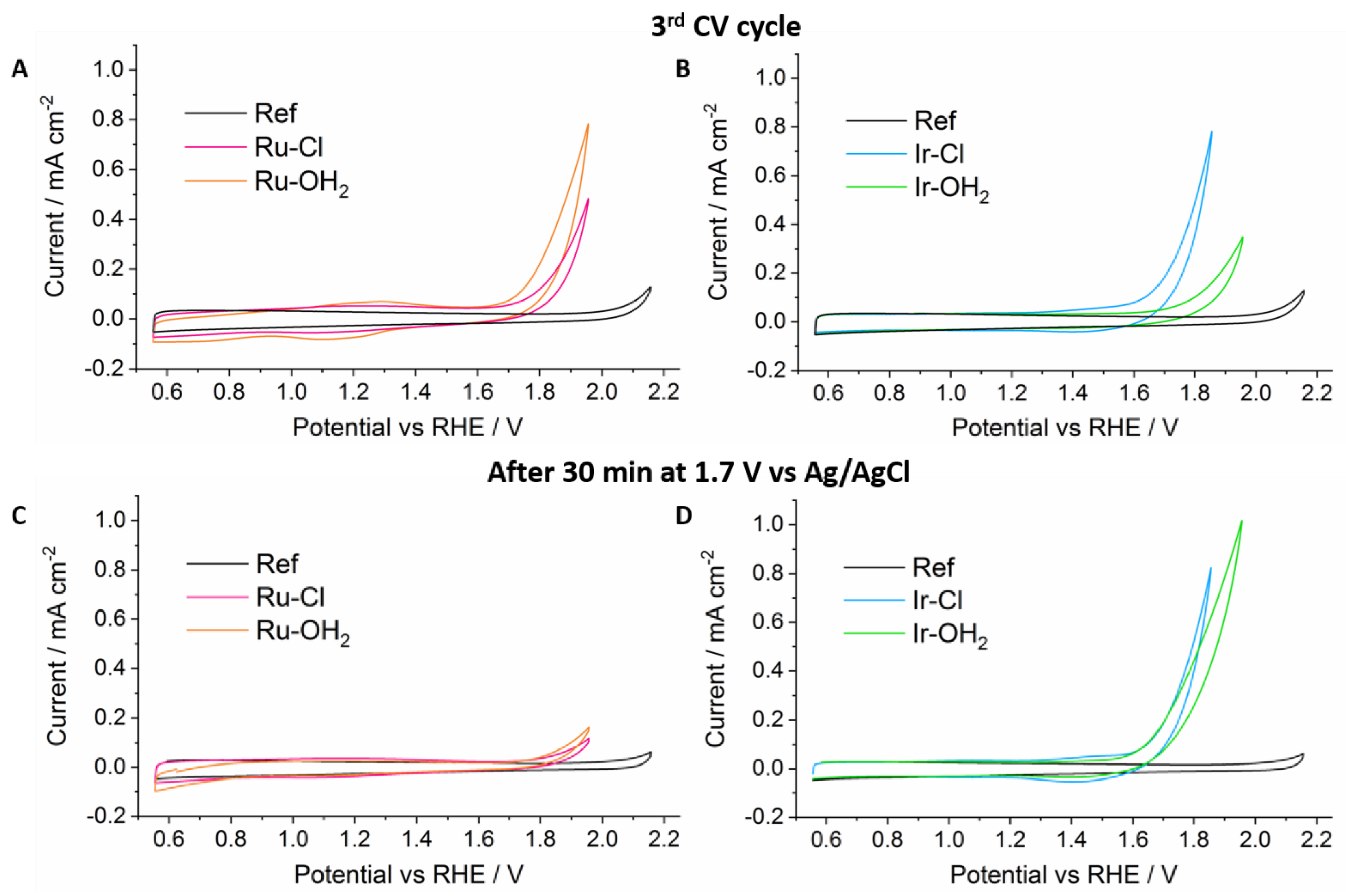
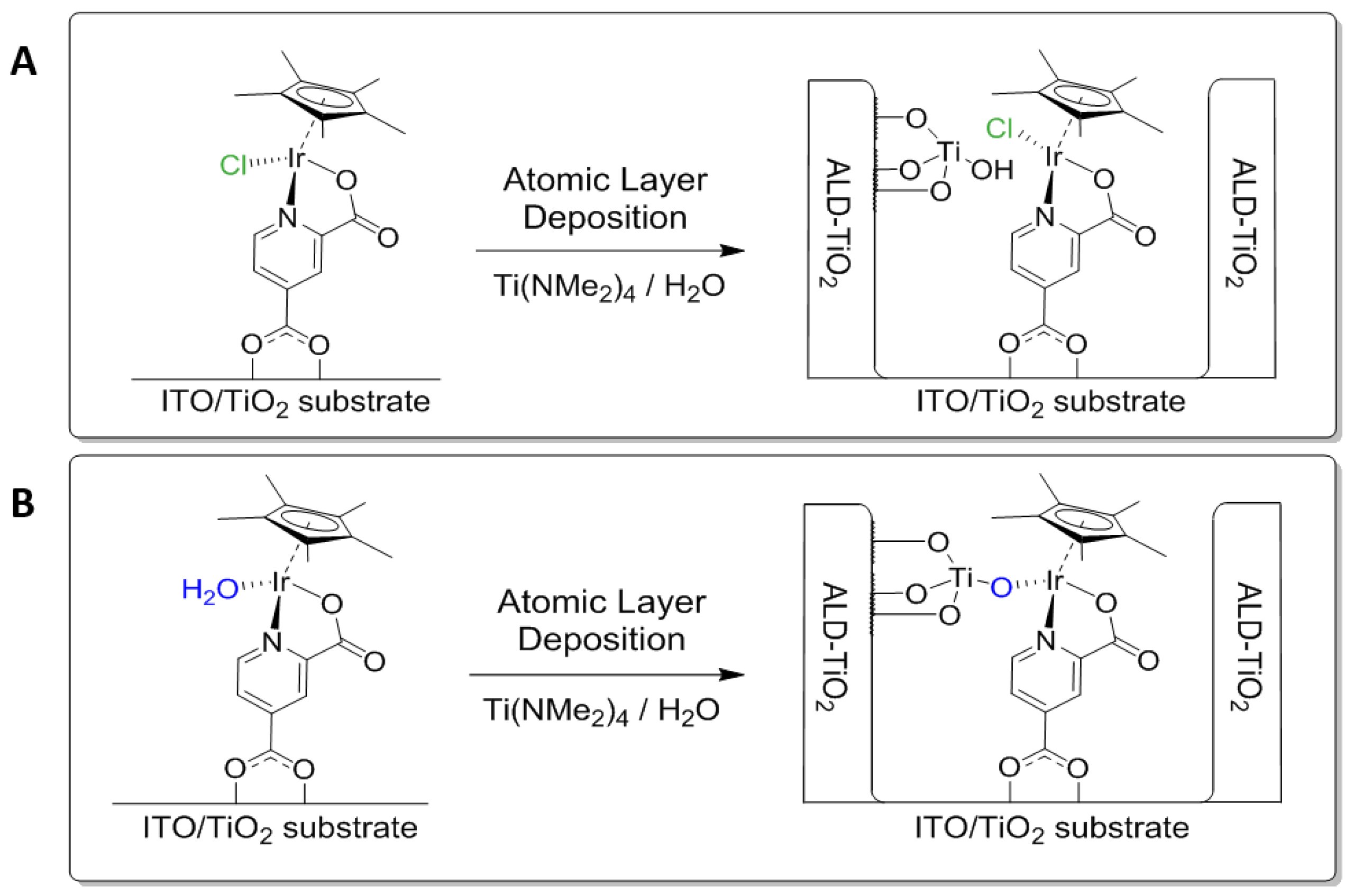
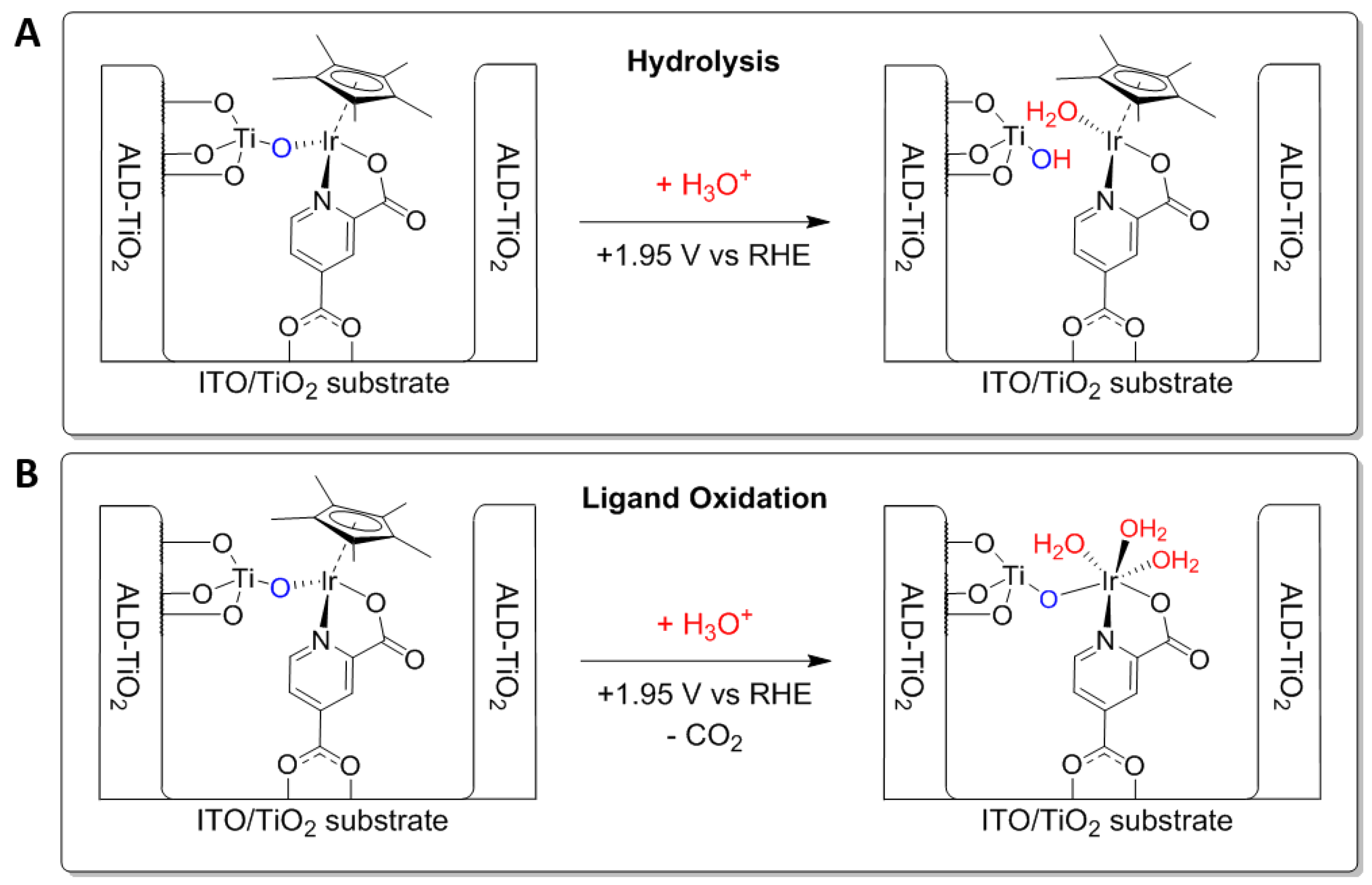
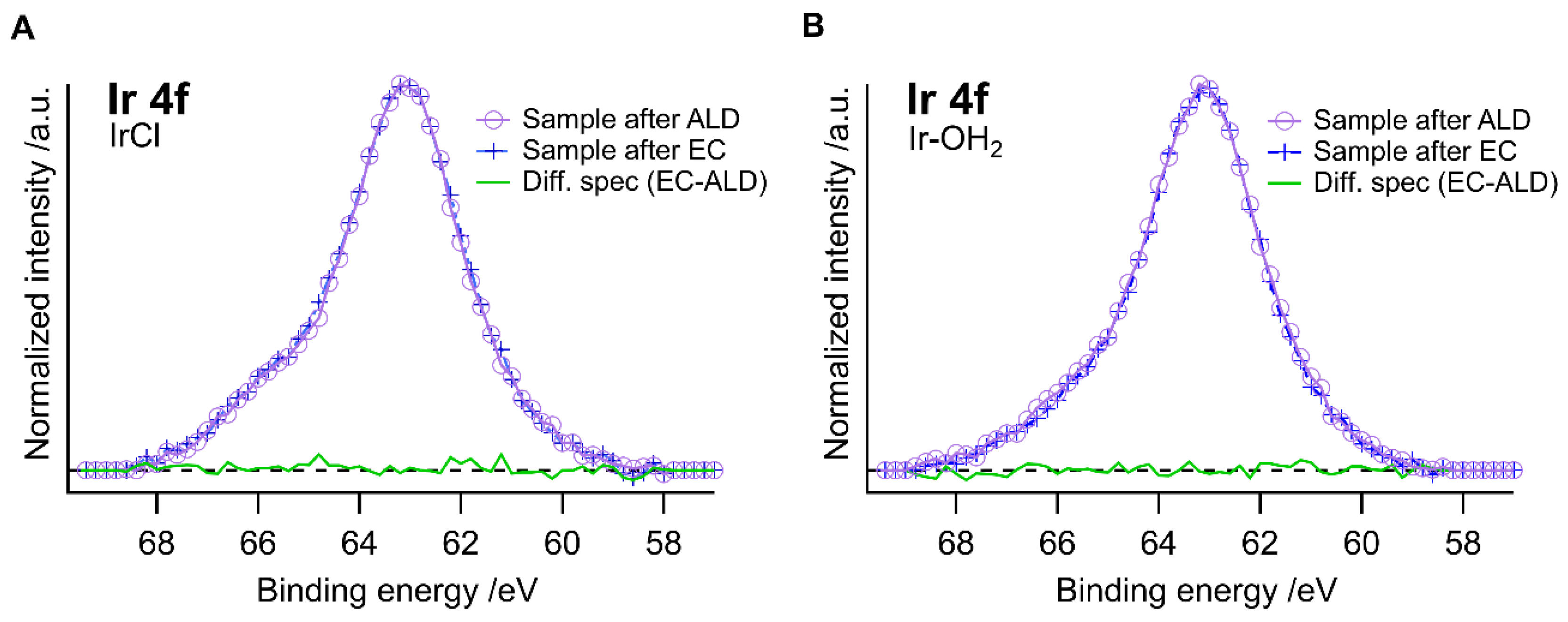
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Materials
3.2.1. 4′-(4-Cyanophenyl)-2,2′:6′,2″-terpyridine (tpy-PhCN)
3.2.2. Synthesis of [Ru(tpy-PhCN)Cl3]
3.2.3. Synthesis of [Ru(tpy-PhCN)(bda)Cl]Cl (Ru–Cl)
3.2.4. Synthesis of [Ru(tpy-PhCN)(bda)(OH2)](ClO4)2 (Ru–OH2)
3.2.5. Synthesis of [Cp*Ir(κ-N,O-lutidine)(OH2)] (ClO4) (Ir–OH2)
3.2.6. Preparation of ITO Spin Coating Solution
3.2.7. Preparation of TiO2 Spin Coating Solution
3.2.8. Preparation of Mesoporous Substrates
3.2.9. Anchoring of Molecular Catalysts on Substrates
3.2.10. Atomic Layer Deposition
3.3. Methods
3.3.1. Ligand Exchange Experiments and IR Spectroscopy
3.3.2. Electrochemical Analyses
3.3.3. X-ray Photoelectron Spectroscopy (XPS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar water splitting cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Moniz, S.J.A.; Wang, A.; Zhang, T.; Tang, J. Photoelectrochemical devices for solar water splitting—Materials and challenges. Chem. Soc. Rev. 2017, 46, 4645–4660. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, R.R.; Septina, W.; Siol, S.; Moehl, T.; Wick-Joliat, R.; Tilley, S.D. Photocorrosion-resistant Sb2Se3 photocathodes with earth abundant MoSx hydrogen evolution catalyst. J. Mater. Chem. A 2017, 5, 23139–23145. [Google Scholar] [CrossRef]
- Wick, R.; Tilley, S.D. Photovoltaic and Photoelectrochemical Solar Energy Conversion with Cu2O. J. Phys. Chem. C 2015, 119, 26243–26257. [Google Scholar] [CrossRef]
- Abdi, F.F.; Han, L.; Smets, A.H.M.; Zeman, M.; Dam, B.; Van De Krol, R. Efficient solar water splitting by enhanced charge separation in a bismuth vanadate-silicon tandem photoelectrode. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Guijarro, N.; Liu, Y.; Schouwink, P.; Wells, R.A.; Le Formal, F.; Sun, S.; Gao, C.; Sivula, K. Spinel Structural Disorder Influences Solar-Water-Splitting Performance of ZnFe2O4 Nanorod Photoanodes. Adv. Mater. 2018, 1801612. [Google Scholar] [CrossRef] [PubMed]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1. [Google Scholar] [CrossRef]
- Hou, Y.; Zhuang, X.; Feng, X. Recent Advances in Earth-Abundant Heterogeneous Electrocatalysts for Photoelectrochemical Water Splitting. Small Methods 2017, 1, 1700090. [Google Scholar] [CrossRef]
- Benck, J.D.; Chen, Z.; Kuritzky, L.Y.; Forman, A.J.; Jaramillo, T.F. Amorphous molybdenum sulfide catalysts for electrochemical hydrogen production: Insights into the origin of their catalytic activity. ACS Catal. 2012, 2, 1916–1923. [Google Scholar] [CrossRef]
- Qian, M.; Cui, S.; Jiang, D.; Zhang, L.; Du, P. Highly Efficient and Stable Water-Oxidation Electrocatalysis with a Very Low Overpotential using FeNiP Substitutional-Solid-Solution Nanoplate Arrays. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Joliat, E.; Schnidrig, S.; Probst, B.; Bachmann, C.; Spingler, B.; Baldridge, K.K.; Von Rohr, F.; Schilling, A.; Alberto, R. Cobalt complexes of tetradentate, bipyridine-based macrocycles: Their structures, properties and photocatalytic proton reduction. Dalton Trans. 2016, 45, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Matheu, R.; Ertem, M.Z.; Benet-Buchholz, J.; Coronado, E.; Batista, V.S.; Sala, X.; Llobet, A. Intramolecular Proton Transfer Boosts Water Oxidation Catalyzed by a Ru Complex. J. Am. Chem. Soc. 2015, 137, 10786–10795. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.D.; Newell, R.H.; McNevin, M.J.; Muckerman, J.T.; DuBois, M.R.; DuBois, D.L. Hydrogen oxidation and production using nickel-based molecular catalysts with positioned proton relays. J. Am. Chem. Soc. 2006, 128, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, U.J.; Roberts, J.A.S.; Pool, D.H.; Appel, A.M.; Stewart, M.P.; DuBois, M.R.; Dougherty, W.G.; Kassel, W.S.; Bullock, R.M.; DuBois, D.L. [Ni(PPh 2 N C6H4X2)2]2+ Complexes as Electrocatalysts for H2 Production: Effect of Substituents, Acids, and Water on Catalytic Rates. J. Am. Chem. Soc. 2011, 133, 5861–5872. [Google Scholar] [CrossRef] [PubMed]
- Junge, H.; Marquet, N.; Kammer, A.; Denurra, S.; Bauer, M.; Wohlrab, S.; Gärtner, F.; Pohl, M.M.; Spannenberg, A.; Gladiali, S.; et al. Water oxidation with molecularly defined iridium complexes: Insights into homogeneous versus heterogeneous catalysis. Chem. Eur. J. 2012, 18, 12749–12758. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.F.; Lebedev, D.; Waltar, K.; Povia, M.; Nachtegaal, M.; Fabbri, E.; Copéret, C.; Schmidt, T.J. Iridium Oxide for the Oxygen Evolution Reaction: Correlation between Particle Size, Morphology, and the Surface Hydroxo Layer from Operando XAS. Chem. Mater. 2016, 28, 6591–6604. [Google Scholar] [CrossRef]
- Codolà, Z.; Cardoso, J.M.S.; Royo, B.; Costas, M.; Lloret-Fillol, J. Highly Effective Water Oxidation Catalysis with Iridium Complexes through the Use of NaIO4. Chem. Eur. J. 2013, 19, 7203–7213. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Maegawa, Y.; Goto, Y.; Hara, K.; Inagaki, S. Heterogeneous Catalysis for Water Oxidation by an Iridium Complex Immobilized on Bipyridine-Periodic Mesoporous Organosilica. Angew. Chem Int. Ed. 2016, 55, 7943–7947. [Google Scholar] [CrossRef] [PubMed]
- Rodenberg, A.; Orazietti, M.; Mosberger, M.; Bachmann, C.; Probst, B.; Alberto, R.; Hamm, P. Quinones as Reversible Electron Relays in Artificial Photosynthesis. ChemPhysChem 2016, 17, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Windisch, J.; Orazietti, M.; Hamm, P.; Alberto, R.; Probst, B. General Scheme for Oxidative Quenching of a Copper Bis-Phenanthroline Photosensitizer for Light-Driven Hydrogen Production. ChemSusChem 2016, 9, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Güttinger, R.; Moré, R.; Song, F.; Wan, W.; Patzke, G.R. Frontiers of water oxidation: the quest for true catalysts. Chem. Soc. Rev. 2017, 46, 6124–6147. [Google Scholar] [CrossRef] [PubMed]
- Odrobina, J.; Scholz, J.; Pannwitz, A.; Francàs, L.; Dechert, S.; Llobet, A.; Jooss, C.; Meyer, F. Backbone Immobilization of the Bis(bipyridyl)pyrazolate Diruthenium Catalyst for Electrochemical Water Oxidation. ACS Catal. 2017, 7, 2116–2125. [Google Scholar] [CrossRef]
- Zhang, L.; Cole, J.M. Anchoring Groups for Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 3427–3455. [Google Scholar] [CrossRef] [PubMed]
- Galoppini, E. Linkers for anchoring sensitizers to semiconductor nanoparticles. Coord. Chem. Rev. 2004, 248, 1283–1297. [Google Scholar] [CrossRef]
- Boissezon, R.; Muller, J.; Beaugeard, V.; Monge, S.; Robin, J.-J. Organophosphonates as anchoring agents onto metal oxide-based materials: synthesis and applications. RSC Adv. 2014, 4, 35690. [Google Scholar] [CrossRef]
- Materna, K.L.; Rudshteyn, B.; Brennan, B.J.; Kane, M.H.; Bloomfield, A.J.; Huang, D.L.; Shopov, D.Y.; Batista, V.S.; Crabtree, R.H.; Brudvig, G.W. Heterogenized Iridium Water-Oxidation Catalyst from a Silatrane Precursor. ACS Catal. 2016, 6, 5371–5377. [Google Scholar] [CrossRef]
- Lauinger, S.M.; Piercy, B.D.; Li, W.; Yin, Q.; Collins-Wildman, D.L.; Glass, E.N.; Losego, M.D.; Wang, D.; Geletii, Y.V.; Hill, C.L. Stabilization of Polyoxometalate Water Oxidation Catalysts on Hematite by Atomic Layer Deposition. ACS Appl. Mater. Interfaces 2017, 9, 35048–35056. [Google Scholar] [CrossRef] [PubMed]
- Son, H.J.; Prasittichai, C.; Mondloch, J.E.; Luo, L.; Wu, J.; Kim, D.W.; Farha, O.K.; Hupp, J.T. Dye stabilization and enhanced photoelectrode wettability in water-based dye-sensitized solar cells through post-assembly atomic layer deposition of TiO2. J. Am. Chem. Soc. 2013, 135, 11529–11532. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, A.K.; Alibabaei, L.; Losego, M.D.; Concepcion, J.J.; Kalanyan, B.; Parsons, G.N.; Meyer, T.J. Crossing the divide between homogeneous and heterogeneous catalysis in water oxidation. Proc. Natl. Acad. Sci. USA 2013, 110, 20918–20922. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.; Losego, M.D.; Kalanyan, B.; Parsons, G.N.; Meyer, T.J. Stabilizing small molecules on metal oxide surfaces using atomic layer deposition. Nano Lett. 2013, 13, 4802–4809. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Jiang, Y.-L.; Detavernier, C.; Deduytsche, D.; Van Meirhaeghe, R.L.; Ru, G.-P.; Li, B.-Z.; Qu, X.-P. Atomic layer deposition of TiO2 from tetrakis-dimethyl-amido titanium or Ti isopropoxide precursors and H2O. J. Appl. Phys. 2007, 102, 083521. [Google Scholar] [CrossRef]
- Burke, M.; Blake, A.; Povey, I.M.; Schmidt, M.; Petkov, N.; Carolan, P.; Quinn, A.J. Low sheet resistance titanium nitride films by low-temperature plasma-enhanced atomic layer deposition using design of experiments methodology. J. Vac. Sci. Technol. A 2014, 32, 031506. [Google Scholar] [CrossRef]
- Takeuchi, K.J.; Thompson, M.S.; Pipes, D.W.; Meyer, T.J. Redox and spectral properties of monooxo polypyridyl complexes of ruthenium and osmium in aqueous media. Inorg. Chem. 1984, 23, 1845–1851. [Google Scholar] [CrossRef]
- Bucci, A.; Savini, A.; Rocchigiani, L.; Zuccaccia, C.; Rizzato, S.; Albinati, A.; Llobet, A.; MacChioni, A. Organometallic iridium catalysts based on pyridinecarboxylate ligands for the oxidative splitting of water. Organometallics 2012, 31, 8071–8074. [Google Scholar] [CrossRef]
- Bignozzi, C.A.; Argazzi, R.; Scandola, F.; Schoonover, J.R.; Gordon, K.C.; Dyer, R.B. Electronic Coupling in Cyano-Bridged Ruthenium Polypyridine Complexes and Role of Electronic Effects on Cyanide Stretching Frequencies. Inorg. Chem. 1992, 31, 5260–5267. [Google Scholar] [CrossRef]
- Ali, N.M.; Ward, M.D.; Hashim, N.; Daud, N. Synthesis and photophysical properties of bis(phenylpyridine) iridium(III) dicyanide complexes. Mater. Res. Innov. 2017, 1–6. [Google Scholar] [CrossRef]
- Wrzolek, P.; Schwalbe, M. Affecting the Catalytic Activity of the Known [Ru(tpy)(bpy)(OH2)] 2+ Complex in Water Oxidation by Utilization of a Hangman Ligand. Eur. J. Inorg. Chem. 2015, 2015, 4373–4378. [Google Scholar] [CrossRef]
- Cargill Thompson, A.M.W. The synthesis of 2,2′:6′,2″-terpyridine ligands—Versatile building blocks for supramolecular chemistry. Coord. Chem. Rev. 1997, 160, 1–52. [Google Scholar] [CrossRef]
- Zuccaccia, C.; Bellachioma, G.; Bortolini, O.; Bucci, A.; Savini, A.; Macchioni, A. Transformation of a Cp*-iridium(III) precatalyst for water oxidation when exposed to oxidative stress. Chem. Eur. J. 2014, 20, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, J.L.; Lin, W. Elucidating molecular iridium water oxidation catalysts using metal-organic frameworks: A comprehensive structural, catalytic, spectroscopic, and kinetic study. J. Am. Chem. Soc. 2012, 134, 19895–19908. [Google Scholar] [CrossRef] [PubMed]
- Hintermair, U.; Sheehan, S.W.; Parent, A.R.; Ess, D.H.; Richens, D.T.; Vaccaro, P.H.; Brudvig, G.W.; Crabtree, R.H. Precursor transformation during molecular oxidation catalysis with organometallic iridium complexes. J. Am. Chem. Soc. 2013, 135, 10837–10851. [Google Scholar] [CrossRef] [PubMed]
- Park-Gehrke, L.S.; Freudenthal, J.; Kaminsky, W.; DiPasquale, A.G.; Mayer, J.M. Synthesis and oxidation of Cp*IrIII compounds: functionalization of a Cp* methyl group. Dalt. Trans. 2009, 0, 1972. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.M.; Sheehan, S.W.; Hashmi, S.M.; Campos, J.; Hintermair, U.; Crabtree, R.H.; Brudvig, G.W. Electrochemical activation of Cp* iridium complexes for electrode-driven water-oxidation catalysis. J. Am. Chem. Soc. 2014, 136, 13826–13834. [Google Scholar] [CrossRef] [PubMed]
- Goodall, W.; Wild, K.; Arm, K.J.; Williams, J.A.G. The synthesis of 4′-aryl substituted terpyridines by Suzuki cross-coupling reactions: substituent effects on ligand fluorescence. J. Chem. Soc. Perkin Trans. 2 2002, 1669–1681. [Google Scholar] [CrossRef]
- Xi, Y.; Wei, W.; Xu, Y.; Huang, X.; Zhang, F.; Hu, C. Coordination Polymers Based on Substituted Terpyridine Ligands: Synthesis, Structural Diversity, and Highly Efficient and Selective Catalytic Oxidation of Benzylic C–H Bonds. Cryst. Growth Des. 2015, 15, 2695–2702. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sévery, L.; Siol, S.; Tilley, S.D. Design of Molecular Water Oxidation Catalysts Stabilized by Ultrathin Inorganic Overlayers—Is Active Site Protection Necessary? Inorganics 2018, 6, 105. https://doi.org/10.3390/inorganics6040105
Sévery L, Siol S, Tilley SD. Design of Molecular Water Oxidation Catalysts Stabilized by Ultrathin Inorganic Overlayers—Is Active Site Protection Necessary? Inorganics. 2018; 6(4):105. https://doi.org/10.3390/inorganics6040105
Chicago/Turabian StyleSévery, Laurent, Sebastian Siol, and S. David Tilley. 2018. "Design of Molecular Water Oxidation Catalysts Stabilized by Ultrathin Inorganic Overlayers—Is Active Site Protection Necessary?" Inorganics 6, no. 4: 105. https://doi.org/10.3390/inorganics6040105
APA StyleSévery, L., Siol, S., & Tilley, S. D. (2018). Design of Molecular Water Oxidation Catalysts Stabilized by Ultrathin Inorganic Overlayers—Is Active Site Protection Necessary? Inorganics, 6(4), 105. https://doi.org/10.3390/inorganics6040105