Abstract
Titanocene bis-arylthiolates [(C5H4X)(C5H4Y)Ti(SC6H4R)2] (X,Y = H, Cl; R = H, Me) can be prepared from the corresponding titanocene dichlorides by reacting with the thiols in the presence of DABCO as a base. They react with n-butyl lithium to give unstable Ti(III) radical anions. While the unsubstituted thiolates (X = Y = R = H) react with lithium Di-isopropylamide by decomposing to dimeric fulvalene-bridged and thiolate-bridged Ti(III) compounds, the ring-chlorinated compounds can be deprotonated with LDA and give appropriate electrophiles di-substituted and tri-substituted titanocene dithiolates.
1. Introduction
Titanocene compounds, i.e., compounds of the type “Cp2TiXn” where “Cp” stands for a substituted or unsubstituted cyclopentadienyl ligand and “X” for any anionic or neutral ligand and n = 0–2 are arguably the second-most studied metallocenes after ferrocene. Soon after the first synthesis of (C5H5)2TiCl2 in 1954 [1], its potential for acting as a polymerization catalyst when combined with certain aluminum compounds was discovered [2]. Further studies showed that a judicious choice of substituents on the cyclopentadienyl ring had a great influence on the stereochemistry of the produced polymers, which gave rise to an enormous number of publications and also many review articles [3,4,5,6,7,8]. However, it was also found that the titanium catalysts were rather quickly deactivated and the corresponding zirconium compounds showed much higher stability and catalytic activity. Thus, the mainstream research on metallocene catalysts stopped for titanium-based metallocenes nearly completely until recently when the concepts of “Green Chemistry” and “Sustainable Catalysis” came into play [9,10,11,12]. Another area of “applied research” opened up after the discovery that Cp2TiCl2 showed antitumor activity [13,14,15]. Here again, it turned out that the effectiveness of the titanocenes could be enhanced by introducing substituents on the cyclopentadienyl rings [16,17] as well as by modifying the “X”-ligands [17,18]. Lastly, titanocenes were the subject of purely “academic” studies, e.g. studies devoted to synthesis, isolation, and characterization of the “true” titanocenes “Cp2Ti” [19] or of “chiral at titanium” complexes “CpCp’TiXY” [20]. In addition, in these studies, the importance of cyclopentadienyl ring substituents was well established. All of the titanocenes with substituted cyclopentadienyl rings known so-far have been prepared by a reaction of the substituted cyclopentadiene with TiCln (n = 2–4). This method fails for very electronegative substituents or substituents with ligating properties. Since our group focuses on the synthesis of Cyclopentadienyl complexes with such substituents for a long time, using an approach of performing halogen-metal exchange reactions followed by electrophilic substitutions on already coordinated perhalogenated cyclopentadienyl ligands [21], we wondered if our approach was also suitable for the titanocene system. Since our synthetic protocol always involves the use of lithium organyls or lithium amides, there was also the need for a proper titanocene starting material. It has been known for a long time that titanocene chlorides react with alkyl lithiums to thermally unstable titanocene alkyls Cp2TiR2, which easily decompose to titanium(III) products [22,23] and with lithium amides to mono-cyclopentadienyl titanium tris-amides CpTi(NR2)3 [24]. In addition, with the often-used base KOtBu, a mixture of products including those of splitting off the cyclopentadienyl ligands was observed [25]. We, therefore, decided to look at the also long-known titanocene thiolates Cp2Ti(SR)2. These compounds are usually prepared either by a reaction of Cp2TiCl2 with thiols in the presence of the base, which were sometimes contaminated with the mixed chloride-thiolates Cp2Ti(SR)Cl [26,27,28] or by oxidative addition of disulfides to in-situ prepared “Cp2Ti” [29]. Some of these thiolates showed promising antitumor properties [30,31].
In this study, we report on the synthesis of several titanocene bis(aryl thiolates) (Cp)(Cp’)Ti(SAr)2 and their reactivity towards lithium alkyls and amides including functionalization of the cyclopentadienyl rings. It should be mentioned here that a related approach was used for deriving the so-called “Troticenes” CpTi(Cht) [32].
2. Results
2.1. Synthesis of Starting Materials
Treatment of the known titanocene dichlorides (C5H4X)(C5H4Y)TiCl2 (X = Y = H, 1a; X = H, Y = Cl, 1b; X = Y = Cl, 1c) [33,34] with HS–C6H4R (R = H, p-Me) in the presence of DABCO (1,4-diazabicyclo[2.2.2]octane) yields the corresponding titanocene arylthiolates (C5H4X)(C5H4Y)Ti(S–C6H4R)2 (R = H, 2a–c, Me: 3a–c), respectively, in good yields (64% to 89% except for 3b, 19%) (Scheme 1).
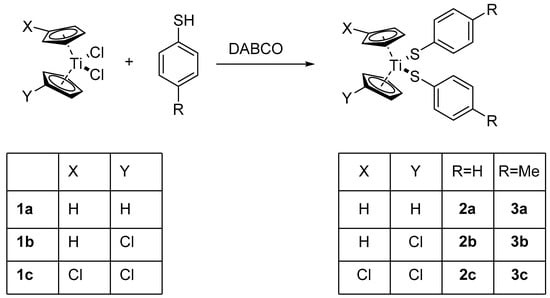
Scheme 1.
Synthesis of substituted titanocene thiolates.
Compounds 2a and 3a had been reported before [26,27]. All compounds were characterized by 1H-NMR and 13C-NMR and mass spectroscopy including HRMS. The NMR spectra of 3b/c show the presence of HS–C6H4CH3 or [S–C6H4–CH3]2 and very weak signals (<2%) derived from 1b/c and (C5H4X)(C5H4Y)Ti(S–C6H4R)Cl.
2.2. Reaction of Dithiolates 2a and 3a with Butyllithium
Treatment of a suspension of 2a or 3a with 1.1 equivalents of n-BuLi in THF at −78 °C results in the formation of yellow solutions. Warming to room temperature leads to a color change from yellow to brown within five minutes. The resulting solutions appear to be paramagnetic since it turns out impossible to obtain any good-quality 1H- or 13C-NMR spectra. However, the solutions resulting from 2a show a 7Li-NMR resonance at δ = 2.64 ppm while the solutions resulting from 3a exhibit a 7Li-NMR signal at δ = 2.74 ppm (Figures S5 and S6). For comparison, the 7Li-NMR signals of THF solutions of Li(C5H5), LiSC6H5, and LiSC6H4CH3 can be found at δ = −8.37, 1.98, and 1.90 ppm, respectively). Cooling these solutions down to −78 °C leads to a color change and it turns into a violet color. Warming up to room temperature reinstates the brown color and evaporation of the solvent in vacuum leaves a brown residue, 2a′ or 3a′, respectively. When a frozen solution made up from 3a and four equivalents of butyl lithium in THF is gradually warmed up within a NMR spectrometer, a 1H-NMR spectrum immediately taken at −80 °C shows no signals of the Li–CH2 protons. However, a signal due to LiC5H5 and several signals is presumably assigned to Tol-SBu. After warming to ambient, the signals due to 3a are re-installed and, while the LiCp signal has disappeared, the signals of Tol-SBu are still present (Figure S4).
Both solid compounds are extremely air-sensitive and yield within seconds in air violet solids, which can be identified as 2a or 3a by using 1H-NMR spectroscopy. When the yellow solutions, obtained from 2a or 3a and n-BuLi at −78 °C, are treated with dimethyldisulfide at this temperature and the mixture is warmed to ambient, the color changes to red-violet. Evaporation of the solvent leaves violet oils that contain a mixture of various compounds where Cp2Ti(S–C6H4R)(SCH3) (R = H, Me) and (C6H4R)2S2 can be identified as unreacted starting materials (Scheme 2).
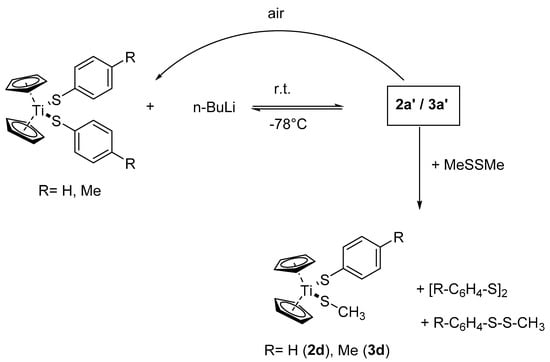
Scheme 2.
Reaction of 2a or 3a with n-BuLi and air or MeSSMe.
2.3. Ring Metalation of 2a–c with Lithium Diisopropylamide and Reaction with Electrophiles
When a THF solution of 2a is treated at −78 °C with a freshly prepared THF solution of LDA (1.2 equivalents), which is followed by the addition of an equimolar amount of SiMe3Cl and warming up to room temperature, the product isolated after work-up contains mostly apparently unreacted 2a together with small amounts of the silylated compounds (C5H4SiMe3)(C5H4Z)Ti(SC6H5)2 (Z = H, 4a; SiMe3, 4b) and a di-nuclear complex of formula C32H28S2Ti2 (5). When dimethyldisulfide was used as an electrophile, the only identifiable product was the dimer 5. Attempts to purify the compounds by column chromatography lead only to its decomposition. However, compounds 4 and 5 could be unambiguously characterized by HRMS.
Treatment of a THF solution of 2b with a solution of LDA at −78 °C followed by the addition of hexachloroethane leads, after a chromatographic workup, to a 72% yield of the desired 1,2-dichlorocyclopentadienyl complex (C5H3Cl2)(C5H5)Ti(SC6H5)2 (6a). Similar treatment of 2c with LDA and C2Cl6 led to an inseparable mixture of (C5H3Cl2)(C5H3ClY)Ti(SC6H5)2 (Y = H, 7a, Cl, 7b) in a combined yield of ca. 40% together with a 47% yield of the olefin C2(SC6H5)4. Similar treatment of 2b with LDA and chlorotrimethylsilane gave the chiral 1-chloro-2-trimethylsilyl-cyclopentadienyl complex 6b. While 6a,b could be characterized by NMR and mass spectrometry, compounds 7a,b could only be identified by HRMS. The NMR spectra of 6a show weak signals that might be assigned to the 1,3-regio-isomer of 6a (ca. 6%). The NMR spectra of 6b show weak signals that might be due to PhSH or PhSSPh. The mass spectra of both compounds show peaks due to (C5H3ClY)(C5H5)Ti(SC6H5)Cl (Y = Cl, 6a′, SiMe3, 6b′).
If the 1,2-dichlorocyclopentadienyl complex 6a is treated in THF solution with LDA, which is followed by hexachloroethane, chromatographic workup of the product mixture yields a 24% yield of the desired 1,2,3-trichlorocyclopentadienyl complex (C5H2Cl3)(C5H5)Ti(SC6H5)2 (8a) together with a 20% recovery of the starting material and 7% tetrakis(phenylthio)ethene. Starting from 6b, LDA and SiMe3Cl the 1-chloro-2,5-bis(trimethylsilyl)cyclopentadienyl complex 8b was obtained in a 75% yield (Scheme 3). 8a,b could be characterized by NMR and mass spectroscopy. The 1H-NMR spectrum of 8a shows a weak signal (ca. 4%) that might be assigned to the C5H5 group of the 1,2,4-regio-isomer of 8a. Signals of PhSH can be seen in NMR spectra of both compounds 8a/b. The mass spectrum of 8a shows the presence of [(C5H2Cl3)(C5H5)Ti(SC6H5)Cl] (8a′).
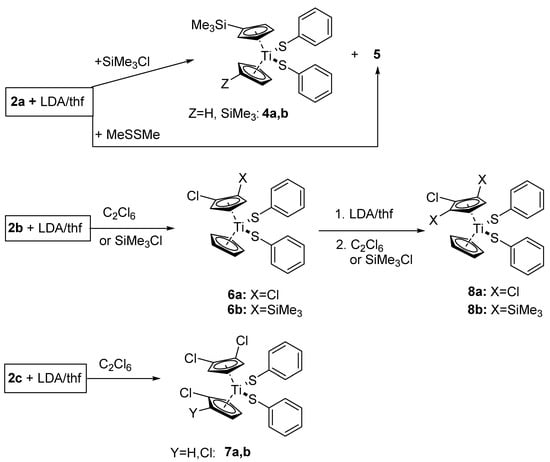
Scheme 3.
Reactions of 2a–c with LDA and electrophiles.
3. Discussion
Our previous approach towards functionalization of metal-coordinated cyclopentadienyl rings needs perhalogenated cyclopentadienyl complexes as starting materials, which are not known for the titanocene system. To obtain such titanocenes with perhalogenated Cp rings, stepwise introduction of halogens via alternate metalation-electrophilic halogenation sequences might be a useful strategy, which was successfully applied by us [35,36,37] and others [38,39] in the ferrocene system and also in the cymantrene system [40]. For the metalation step, we used lithium bases like butyl lithium or lithium amides. As outlined in the introduction, these reagents cannot be used with titanocene chlorides but may work with the corresponding aryl thiolates. Application of the known synthetic protocol “titanocene dichloride + aromatic thiol + base” with replacement of the usual NEt3 by DABCO gives not only slightly better yields for the already known bis-arylthiolates 2a and 3a but also allows the synthesis of the new chlorocyclopentadienyl thiolates 2b,c and 3b,c. All chlorocyclopentadienyl compounds are air-stable and are highly viscous violet oils that withstand all attempts of crystallization. Since the used starting materials [(C5H4Cl)(C5H4X)TiCl2] are always obtained as mixtures with the unsubstituted Cp2TiCl2, which are extremely difficult to separate, the corresponding dithiolates were also obtained as mixtures that were easily separated by using chromatography. The lower yields of 2b,c and 3b,c in comparison with their unsubstituted analogs 2a,3a are, therefore, probably due to losses in the purification step and not a consequence of an intrinsic instability. The particularly low yield of 3b is probably due to an adventitious presence of moisture in the reaction mixture.
The reaction of 2a/3a with one equivalent n-BuLi yields reversibly paramagnetic solutions, which are extremely air-sensitive and produce the starting materials quantitatively upon a deliberate addition of air. Treatment of the reaction solutions with dimethyldisulfide gives the mixed titanocene aryl-alkyl-thiolates 2d/3d together with the symmetric and asymmetric disulfides ArSSAr and ArSSMe. We think that this behavior is due to a temperature dependent redox-equilibrium with the room temperature solution containing a Ti(III) radical anion, according to Scheme 4.
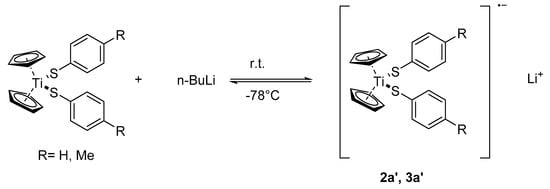
Scheme 4.
The redox equilibrium between 2a/3a and 2a’/3a’.
Such a redox equilibria are known from electrochemical studies of Cp2TiCl2 [41,42]. It was found that dependent on the cyclopentadienyl ring substituents, the intermediate radical anion might either reversibly split off a chloride anion yielding Cp2TiCl (the so-called “Nugent-Rajanbabu-reagent” [43]) or irreversibly one of the cyclopentadienyl ligands to give CpTiCl2. It was also reported that treatment of the dialkyltitanocenes Cp2TiR2 with organolithium compounds first produced unstable “back-onium complexes” [Cp2TiR3]−Li+, which decomposed at 20 °C to CpTiR2, RH, and LiCp [23]. At least in the observed time and temperature frame used by us, the presumed Ti(III) thiolates 2a′/3a′ are more stable since we could not observe any LiSAr or LiCp in the reaction solution. Quite interestingly, when a fourfold excess of butyl lithium was used in an NMR experiment, LiCp can be detected in the cold solution, but it disappears when warming-up. At the same time, Tol-SBu can be detected. Therefore, the first reactions in this case are outlined below.
Cp2Ti(SAr)2 + BuLi → “[Cp2Ti(SAr)2Bu]Li”→ “CpTi(SAr)” + ArSBu + LiCp
On warming, apparently the LiCp and the “CpTi(SAr)” fragment change back to the starting dithiolate and unidentified decomposition products. In the case of the 1:1 stoichiometric reaction, the first step might be the same, but a further reaction is different due to the absence of excessive butyl lithium.
Clearly, for the butyl lithium, the redox reaction is preferred over ring deprotonation. We, therefore, turned towards lithium diisopropylamide as a possible deprotonation agent. When 2a was used, with SiMe3Cl as quenching reagent, small amounts of the deprotonation products could be identified together with an unexpected dinuclear compound of formula C32H28S2Ti2 (5). 5 was the only identifiable product when MeSSMe was used as a quenching reagent. We believe that this compound is a di-titanium(III) compound triply bridged by a fulvalene-diide and two phenylthiolate ligands (Figure 1).
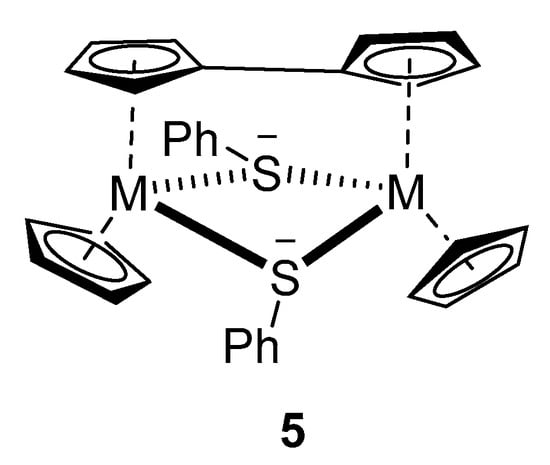
Figure 1.
Proposed structure of 5 (M = Ti).
A zirconium compound of an identical structure (5, M = Zr) was obtained by an oxidation reaction of a Zr(II) complex and characterized by NMR spectra and crystal structure determination [44]. Similar fulvalene-bridged Ti(III) complexes with chloride, hydride, or sulfide bridges instead of the aryl thiolate bridges were obtained by a sodium reduction of Cp2TiCl2 [45] or thermolysis of the Ti(II) complex Cp2Ti(Me3Si–C≡C–SiMe3) [46]. A related mono-cyclopentadienyl Ti(III) complex without a fulvalene bridge known as [CpTiCl]2[µ-SAr]2 was obtained from the corresponding mononuclear CpTiCl2(SAr) upon a reduction with sodium amalgam [47]. We assume that the ring-lithiated primary product of the 2a-LDA reaction is unstable under the reaction conditions and decomposes after splitting off LiSPh first to a Ti(III) radical centered on the Cp ring, which dimerizes to form the finally observed dinuclear compound 5.
However, when the chlorocyclopentadienyl dithiolates 2b,c were treated with LDA followed by quenching with hexachloroethane or chlorotrimethylsilane, the desired di-substituted complexes 6a,b and 7a,b could be isolated in moderate yields. Starting from 6b,b, repeating the treatment with LDA and C2Cl6 or SiMe3Cl gave the corresponding tri-substituted compounds 8a,b in yields of 24% and 75%, respectively. An attempt of a “one-pot” synthesis of pentachloro-titanocene thiolates using alternating additions of LDA and C2Cl6 gave a 52% yield of 6a (in the case of 2b as starting material) but no higher substituted products. Apart from the observations in the ferrocene system [35], either the reactivity towards deprotonation or the stability of the formed chlorinated products decreases with an increasing number of chlorine substituents. According to the 1H-NMR spectra, both 6a and 8a are formed as regio-isomers with one largely dominating. By comparing results from the ferrocene system, we conclude that the major isomers are the 1,2 and 1,2,3-substituted ones. There are also weak signals in the NMR and mass spectra of 3c, 6a, 6b and 8a that might be assigned to chloride-mono(phenylthiolate) complexes, which are always accompanied by signals attributable to PhSH. Since chloroform was used as a solvent both for NMR and mass spectra, it seems possible that the complexes are unstable towards this solvent according to the equation below.
CpCp’Ti(SPh)2 + CHCl3 → CpCp’Ti(SPh)Cl + PhSH
An alternative explanation would be that the starting materials were contaminated with the chloride-mono(thiolate) complexes. However, no signs of this can be seen in the spectra of 2b, 2c, 3a.
One interesting aspect in the mass spectra of nearly all compounds is the presence of peaks assignable to [C10H8Ti2(SAr)2]2+, which corresponds to the suggested structure of 5 (without the terminal C5H5-ligands).
4. Experimental Part
All solvents were of analytical grade and were distilled over the Na or Na/K alloy and stored over the Na wire. All reagents (n-BuLi: 1.6M in Hexane, thiophenol, thiocresol, Di-isopropylamine, hexachloroethane, dimethyldisulfide, and DABCO) and Cp2TiCl2 were obtained from commercial suppliers and were used as such. (C5H4Cl)(C5H4X)TiCl2 (X = H, Cl) were prepared according to literature procedures [33,34]. Fresh solutions of LDA were prepared from Di-isopropylamine and n-BuLi in THF. Chromatographic separations were performed in glass columns (30 × 7 cm2) filled with silica 60 (Merck, 0.063 to 0.2 mm). All reactions were run under N2 atmosphere using standard Schlenk equipment. Work-up and chromatographic purifications were performed in the air. All peaks found in the mass spectra, according to the fragmentation pattern, are included in Table S1 of the Supplementing Information.
4.1. Preparation of (C5H5)2Ti(SPh)2 (2a)
A suspension of Cp2TiCl2 (1.99 g, 8.0 mmol) in THF (50 mL) is treated with PhSH (1.64 mL, 16.0 mmol) and DABCO (1.79 g, 16.0 mmol) by stirring at r.t. After a few minutes, the color changes from red to violet. Stirring is continued for 90 min when the suspension is filtered through a glass frit. The residue on the frit is eluted with Et2O until the filtrate is colorless. The combined filtrates are evaporated to dryness to leave a red powder, which is transferred to the top of a silica gel column (L = 30 cm) and then eluted with toluene. The eluate is evaporated to give a red powder: 2a (2.75 g, 86%). 1H-NMR (400 MHz, CDCl3): δ = 7.57 (m, 4H), 7.30 (m, 4H), 7.15 (m, 2H), 6.04 (s, 10H); 13C-NMR (100 MHz, CDCl3): δ = 148.6, 132.4, 128.4, 125.6, 112.8 (CCp); MS (EI): m/z = 396 (M+, 15%), 287 (Cp2TiSPh, 100%), 221 (CpTiSPh–H, 14%), 218 (PhSSPh, 4%), 178 (Cp2Ti, 80%); UV (in CHCl3): λmax = 539 nm, 399 nm.
4.2. Preparation of (C5H5)2Ti(SC6H4CH3-p)2 (3a)
2b was prepared and purified in exactly the same manner as 2a by using thiocresol (1.99 g, 16.0 mmol). A violet powder was obtained: 2b (3.02 g, 89%). 1H-NMR (400 MHz, CDCl3): δ = 7.43 (d, 4H), 7.11 (d, 4H), 6.02 (s, 10H), 2.35 (s, 6H); 13C-NMR (100 MHz, CDCl3): δ = 145.1, 135.2, 132.2, 129.1, 112.7 (CCp), 21.1 (CMe); MS (EI): m/z = 424 (M+, 14%), 301 (Cp2TiSTol, 100%), 235 (CpTiSTol–H, 11%), 246 (TolSSTol, 7%), 178 (Cp2Ti, 66%); UV (in CHCl3): λmax = 543 nm, 401 nm.
4.3. Preparation of (C5H5)(C5H4Cl)Ti(SC6H5)2 (m/z)
A solution of (C5H5)(C5H4Cl)TiCl2 (0.50 g, 1.70 mmol) in THF (15 mL) is treated with PhSH (0.39 g, 3.5 mmol) and DABCO (0.40 g, 3.50 mmol). Within a few seconds, the color changes from orange-brown to violet. Stirring is continued for 60 min when the formed suspension is filtered through a glass frit. The residue on the frit is treated with several portions of Et2O until the extract becomes color-less. The combined filtrates are evaporated to dryness. The residue is placed on top of a silica gel column (30 × 7 cm2) and eluted with toluene. Three fractions can be eluted, which are evaporated to dryness. The first (violet) fraction yields 2c as a violet oil (3 mg). The second (also violet) gives 2b as a violet oil (0.53 g, 70%) and, from the third (red) fraction, 2a can be obtained as a red powder (32 mg). Analytical data for 2b: 1H-NMR (270 MHz, CDCl3): δ = 7.55 (m, 4H), 7.31 (m, 4H), 7.16 (m, 2H), 6.15 (t, 2H), 6.09 (s, 5H), 5.89 (t, 2H); 13C-NMR (68 MHz, CDCl3): δ = 148.1, 131.8, 128.3, 125.5, 116.8 (Ccp,i), 115.1 (CCp) 112.1 (Ccp,α), 111.6 (CCp,β); HRMS (EI, C10H935Cl48Ti): 211.9839 (calcd. 211.9873).
4.4. Preparation of (C5H5)(C5H4Cl)Ti(SC6H4CH3-p)2 (3b)
A solution of (C5H5)(C5H4Cl)TiCl2 (0.459 g, 1.60 mmol) in THF (20 mL) is treated with p-TolSH (0.397 g, 3.20 mmol) and DABCO (0.359 g, 3.20 mmol). Work-up was performed in exactly the same manner as for 3a. Chromatography yielded two fractions. 3b was obtained as a violet oil (0.135 g, 19%) from the first fraction while the second gave 3a as a red powder (13 mg). Analytical data for 3b include: 1H-NMR (400 MHz, CDCl3): δ = 7.42 (m, 4H), 7.11 (m, 4H), 6.16 (t, 2H), 6.07 (s, 5H), 5.87 (t, 2H), 2.36 (s, 6H). Additional weak peaks are assigned to 1b (at 6.63 and 6.45 ppm) and to 3b′ (6.34 and 6.29 ppm); 13C-NMR (68 MHz, CDCl3): δ = 144.7, 135.3, 131.7, 129.1, 116.7 (Ccp,i), 115.1 (CCp), 112.9 (Ccp,α), 111.5 (CCp,β), 21.1 (CMe); HRMS (EI, C24H2335Cl48Ti32S2): 458.0421 (calcd. 458.0409).
4.5. Preparation of (C5H4Cl)2Ti(SC6H5)2 (2c)
A suspension of (C5H4Cl)2TiCl2 (0.636 g, 2.00 mmol) in THF (30 mL) was treated with PhSH (0.41 mL, 4.0 mmol) and DABCO (0.449 g, 4.0 mmol). Within a few seconds, the color of the suspension changed from orange to violet. Stirring was continued for 60 min. Work-up was performed as described for 2b. (for a 30 × 7 cm2 column, a maximum of 0.25 crude product can be separated by using a total of 3.0 L of toluene). Three fractions can be collected. The first (violet) fraction yields 2c as a violet oil (0.205 g, 70%). The second (violet) fraction gives 2b as a violet oil (25 mg) and the third (red) fraction contains 2a obtained as a red powder (11 mg). Analytical data for 2c: 1H-NMR (400 MHz, CDCl3): δ = 7.55 (m, 4H), 7.33 (m, 4H), 7.17 (m, 2H), 6.10 (t, 4H), 6.02 (t, 4H); 13C-NMR (100 MHz, CDCl3): δ = 148.0, 131.8, 128.4, 125.8, 118.1 (Ccp,i), 115.7 (Ccp,α), 112.8 (CCp,β); HRMS (EI, C22H1835Cl248Ti32S2): 463.9710 (calcd. 463.9707).
4.6. Preparation of (C5H4Cl)2Ti(SC6H4–CH3-p)2 (3c)
A suspension of (C5H4Cl)2TiCl2 (0.30 g, 0.94 mmol) in THF (25 mL) was treated with TolSH (0.234 g, 1.88 mmol) and DABCO (0.211 g, 1.88 mmol). Within a few seconds, the color of the suspension changed from orange to violet. Stirring was continued for 60 min. A work-up was performed as described for 2b. Three fractions could be collected. The first yielded 3c as a violet oil (0.294 g, 64%), the second yielded 3b as a violet oil (20 mg), and the third yielded 3a as a red powder (14 mg). Analytical data for 3c: 1H-NMR (400 MHz, CDCl3): δ = 7.42 (d, 4H), 7.11 (d, 4H), 6.10 (t, 4H), 6.01 (t, 4H), 2.36 (s, 6H). An additional, very weak signal at 6.48 ppm could be assigned to 1c: 13C-NMR (100 MHz, CDCl3): δ = 144.6, 135.5, 131.6, 129.2, 117.8 (Ccp,i), 115.6 (Ccp,α), 112.7 (CCp,β), 21.1 (CMe); HRMS (EI, C17H1535Cl37Cl48Ti32S): 370.9727 (calcd. 370.9718).
4.7. Preparation of (C5H5)(C5H3Cl2)Ti(SC6H5)2 (6a)
A deep violet solution of 2b (0.413 g, 0.96 mmol) in THF (25 mL) is treated at −78 °C with a freshly prepared THF solution of LDA (from HN(i-Pr)2 (0.14 mL, 0.96 mmol) and n-BuLi (0.60 mL of a 1.6 M solution in hexane) in THF (15 mL) at 0 °C). After stirring for 10 min, hexachloroethane (0.341 g, 1.44 mmol) is added and the cooling bath is removed. When the solution has reached r.t., the solvent is evaporated in vacuo. The residue is placed on top of a silica gel column. Toluene elutes three fractions (the first one is yellow, the second one is violet, and the third one is reddish). The second fraction yields 6a as a violet oil (0.317 g, 72%). 1H-NMR (400 MHz, CDCl3): δ = 7.53 (m, 4H), 7.30 (m, 4H), 7.15 (m, 2H), 6.39 (t, 1H), 6.10 (s, 5H), 6.01 (d, 2H). Additional weak signals at 6.17 (s), 6.30 (d), 5.79 (t) with significantly smaller coupling constants can be assigned to the 1,3-regio-isomer. 13C-NMR (100 MHz, CDCl3): δ = 147.9, 131.8, 128.4, 125.8, 117.5 (Ccp), 116.6 (CCp,Cl), 115.6 (Ccp,α), 112.5 (CCp,β); HRMS (EI, C22H1835Cl248Ti32S2): 463.9727 (calcd. 463.9707).
4.8. Preparation of (C5H5)(C5H3ClSiMe3)Ti(SC6H5)2 (6b)
A deep violet solution of 2b (0.139 g, 0.32 mmol) in THF (20 mL) is treated at −78 °C with a freshly prepared THF solution of LDA (from HN(i-Pr)2 (0.06 mL, 0.40 mmol) and n-BuLi (0.25 mL of a 1.6 M solution in hexane) in THF (10 mL) at 0 °C). After stirring for 10 min, chlorotrimethylsilane (0.05 mL, 0.40 mmol) is added and the cooling bath is removed. When the solution has reached r.t., the solvent is evaporated in vacuo. The residue is placed on top of a silica gel column and eluted with toluene. The first two fractions (yellow and grey) are discarded. The third fraction yields 6b as a violet oil (0.068 g, 44%) and the fourth is apparently unreacted with 2b (0.011 g, 9%). 1H-NMR (270 MHz, CDCl3): δ = 7.53 (m, 4H), 7.29 (m, 4H), 7.14 (m, 2H), 6.54 (m, 2H), 6.03 (s, 5H), 5.65 (t, 1H), 0.33 (s, 9H). An additional very weak peak (<2%) at 6.30 ppm is assigned to 6b′. 13C-NMR (68 MHz, CDCl3): δ = 148.9, 148.2, 131.8, 131.7, 128.28, 128.24, 127.48, 127.12, 121.8, 118.9, 115.9 (C5H5), 108.5, −0.1 (SiCH). HRMS (EI, C25H2735Cl28Si48Ti32S2): 502.0493 (calcd. 502.0491).
4.9. Preparation of (C5H5)(C5H2Cl3)Ti(SC6H5)2 (8a)
A deep violet solution of 6a (0.25 g, 0.50 mmol) in THF (10 mL) is treated at −78 °C with a freshly prepared THF solution of LDA (from HN(i-Pr)2 (0.07 mL, 0.50 mmol) and n-BuLi (0.31 mL of a 1.6 M solution in hexane) in THF (10 mL) at 0 °C). After stirring for 10 min, hexachloroethane (0.176 g, 0.75 mmol) is added and the cooling bath is removed. When the solution has reached r.t., the solvent is evaporated in vacuo. The residue is placed on top of a silica gel column. Toluene elutes three fractions (the first one is yellow, the second one is violet, and the third one is reddish). The first fraction yields tetrakis(phenylthio)ethane (8 mg), the second gives 8a as a violet oil (59 mg, 24%), and unreacted 6a can be recovered from the third fraction (25 mg, 10% recovery). 1H-NMR (400 MHz, CDCl3): δ = 7.56 (m, 4H), 7.32 (m, 4H), 7.17 (m, 2H), 6.46 (s, 2H), 6.11 (s, 5H). An additional weak singlet at 6.13 ppm is assigned to the 1,2,4-regio-isomer. 13C-NMR (100 MHz, CDCl3): δ = 147.9, 131.8, 128.5, 125.9, 118.6 (CCp), 116.0 (2Ccp,Cl), 113.5 (Ccp,H), 111.2 (CCp,Cl); HRMS (EI, C10H735Cl348Ti32): 279.9076 (calcd. 279.9093).
4.10. Preparation of (C5H5)[C5H2Cl(SiMe3)2]Ti(SC6H5)2 (8b)
A deep violet solution of 6b (0.040 g, 0.08 mmol) in THF (10 mL) is treated at −78 °C with a freshly prepared THF solution of LDA (from HN(i-Pr)2 (0.02 mL, 0.13 mmol) and n-BuLi (0.08 mL of a 1.6 M solution in hexane) in THF (10 mL) at 0 °C). After stirring for 10 min, trimethylchlorosilane (0.02 mL, 0.16 mmol) is added and the cooling bath is removed. When the solution has reached r.t., the solvent is evaporated in vacuo. The residue is placed on top of a silica gel column. tertBuOMe elutes two fractions of which only the first one is collected. 8b is obtained as a violet oil (37 mg, 75%), 1H-NMR (400 MHz, CDCl3): δ = 7.55 (m, 4H), 7.29 (m, 4H), 7.12 (m, 2H), 6.75 (s, 2H), 6.03 (s, 5H), 0.30 (s, 18H); 13C-NMR (100 MHz, CDCl3): δ = 148.9, 131.7, 128.2, 125.2, 123.9(CCp,Cl), 121.7 (Ccp,H), 120.7(CCp,Si), 116.2 (CCp), 0.03 (SiCH); both 1H- and 13C-NMR spectra also show signals of the starting compound 8b. HRMS (EI, C28H3535Cl48Ti32S228Si2): 574.0891 (calcd. 574.0887).
4.11. Reaction of 2a with LDA and SiMe3Cl
A violet solution of 2a (0.199 g, 0.50 mmol) in THF (15 mL) is treated at −78 °C with a freshly prepared THF solution of LDA (from HN(i-Pr)2 (0.085 mL, 0.60 mmol) and n-BuLi (0.38 mL of a 1.6 M solution in hexane) in THF (15 mL) at 0 °C). After stirring for 10 min, SiMe3Cl (0.063 mL, 0.50 mmol) is added and is continually stirred at this temperature for 10 min. Then the cooling bath is removed. After the solution has reached r.t., the solvent is evaporated in vacuo. The residue is extracted with benzene (10 mL). Evaporation of the violet extract in vacuo yields a violet oil (0.217 g). 1H-NMR analysis shows that it mainly consists of the starting material 2a. MS-analysis shows the presence of small amounts of (C5H5)(C5H4SiMe3)Ti(SPh)2 (4a) together with (C5H4SiMe3)Ti(SPh)2 (4b) and a compound 5 analyzed as “C32H28S2Ti2”. Compounds 4b and 5 can be separated and isolated by chromatography in trace amounts. EI-MS-data: for 4a: m/z = 359 (M+-SPh), 250 (M+-2 SPh), for 4b: m/z = 431 (M+-SPh, 53%), 357 (M+-SPh-SiMe3H, 20%), 322 (M+-2 SPh, 100%), for 5: m/z = 572 (M+, 4%), 463 (M+-SPh, 5%), 396 (Cp2Ti(SPh)2, 7%), 333 (C10H8Ti2SPh, 38%), 287 (Cp2TiSPh, 58%), 224 (C10H8Ti2, 39%), 178 (Cp2Ti, 100%). HRMS: 4a: C19H2332S28Si48Ti: 359.0744 (calcd: 359.0746); 4b: C22H3132S28Si248Ti: 431.1162 (calcd. 431.1164); 5: C32H2832S248Ti2: 572.0591 (calcd. 572.0591).
4.12. Reaction of 2a with LDA and MeSSMe
A violet solution of 2a (0.199 g, 0.50 mmol) in THF (15 mL) is treated at −78 °C with a freshly prepared THF solution of LDA (from HN(i-Pr)2 (0.085 mL, 0.60 mmol) and n-BuLi (0.38 mL of a 1.6 M solution in hexane) in THF (15 mL) at 0 °C). After stirring for 10 min, MeSSMe (5 drops) is added and continuously stirred at this temperature for 15 min. Then the cooling bath is removed. After the solution has reached r.t., the solvent is evaporated in vacuo. The residue is extracted with benzene (20 mL). Evaporation of the violet extract in vacuo yields a violet oil. MS analysis of this oil shows the presence of 5 as the only identifiable product. Attempts of chromatographic purification lead only to complete decomposition.
4.13. Reaction of 2c with LDA and Hexachloroethane
A deep violet solution of 2c (0.200 g, 0.43 mmol) in THF (20 mL) is treated at −78 °C with a freshly prepared THF solution of LDA (from HN(i-Pr)2 (0.13 mL, 0.90 mmol) and n-BuLi (0.56 mL of a 1.6 M solution in hexane) in THF (20 mL) at 0 °C). After stirring for 5 min, hexachloroethane (0.407 g, 1.72 mmol) is added and the cooling bath is removed. When the solution has reached r.t., the solvent is evaporated in vacuo. The residue is placed on top of a silica gel column. Toluene elutes two fractions. Evaporation of the first brown fraction leaves tetrakis(phenylthio)ethene as a brown oil (47 mg). The second violet fraction yields an inseparable mixture of (C5H4Cl)(C5H3Cl2)Ti(SPh)2 (7a) and (C5H3Cl2)2Ti(SPh)2 (7b) as a violet oil (0.114 g). Due to strongly overlapping signals of the two compounds, no NMR data can be attributed to the single components. However, identification is possible via HRMS: 7a: C22H1735Cl332S248Ti: 497.9349 (calcd. 497.9319); 7b: C22H1635Cl432S248Ti: 531.8946 (calcd. 531.8927).
5. Conclusions
The unsubstituted titanocene arylthiolates Cp2Ti(SAr)2 cannot be ring-functionalized either by a n-BuLi/electrophile or an LDA/electrophile sequence. In both cases, unstable Ti(III) radicals are formed, which either form the starting materials, dimerize to fulvalene bridged dititanocenes, or decompose completely. With chloro-substituted (ClCp)(XCp)Ti(SPh)2 (X = H, Cl), α-deprotonation can be achieved with LDA and Di-substituted and tri-substituted-cyclopentadienyl complexes can be obtained. However, the stability of the chloro-substituted titanocene thiolates decreases with an increasing degree of ring-substitution and, thus, the desired perchlorotitanocenes could not be obtained.
Supplementary Materials
The following are available online at http://www.mdpi.com/2304-6740/6/3/85/s1, Figure S1: VT-1H-NMR of compound 3a. Figure S2: 1H-NMR of a THF/hexane-solution of n-butyl lithium at −90 °C; Figure S3: 1H-NMR of a freshly prepared THF solution of LiC5H5 at −80 °C, Figure S4: VT-1H-NMR of the reaction of 3a with 4 eqiv. BuLi in THF, mixed at −120 °C. Figure S5: 7Li-NMR of the reaction mixture of 2a with 1.1 equiv BuLi, measured at r.t. Figure S6: 7Li-NMR of the reaction mixture of 3a with 1.1 equiv BuLi, measured at r.t. Figures S7, 9, 11, 13, 15, 17, 19, 21, 23: 1H-NMR spectra in CDCl3 of 2b, 2c, 3b, 3c, 6a, 6b, 7ab, 8a, 8b. Figures S8, 10, 12, 14, 16, 18, 20, 22, 24: 13C-NMR spectra in CDCl3 of 2b, 2c, 3b, 3c, 6a, 6b, 7ab, 8a, 8b. Figures S25–33: EI-mass spectra of 2b, 2c, 3b, 3c, 6a, 6b, 7ab, 8a, 8b. Table S1 contains the EI-mass spectral data of 2, 3, 6, 8.
Author Contributions
T.G.K. performed the preparative and analytical work in the course of his Ph.D. thesis. K.S. supervised the work and wrote the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wilkinson, G.; Birmingham, J.M. Bis-cyclopentadienyl Compounds of Ti, Zr, V, Nb and Ta. J. Am. Chem. Soc. 1954, 76, 4281–4284. [Google Scholar] [CrossRef]
- Natta, G.; Pino, P.; Mazzanti, G.; Giannini, U. A Crystallizable Organometallic Complex Containing Titanium and Aluminum. J. Am. Chem. Soc. 1957, 79, 2975–2976. [Google Scholar] [CrossRef]
- Coates, G.W. Precise Control of Polyolefin Stereochemistry Using Single-Site Metal Catalysts. Chem. Rev. 2000, 100, 1223–1252. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, U.; Burlakov, V.V.; Bach, M.A.; Beweries, T. Five-membered metallacycles of titanium and zirconium—Attractive compounds for organometallic chemistry and catalysis. Chem. Soc. Rev. 2007, 36, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C. Metallocene and related catalysts for olefin, alkyne and silane dimerization and oligomerization. Coord. Chem. Rev. 2006, 250, 66–94. [Google Scholar] [CrossRef]
- Vassylyev, O.; Panarello, A.; Khinast, J.G. Enantioselective Hydrogenations with Chiral Titanocenes. Molecules 2005, 10, 587–619. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, W. Polymerization catalysis. Catal. Today 2000, 62, 23–34. [Google Scholar] [CrossRef]
- Morcillo, S.P.; Miguel, D.; Campana, A.G.; Alvarez de Cienfuegos, L.; Justicia, J.; Cuerva, J.M. Recent applications of Cp2TiCl in natural product synthesis. Org. Chem. Front. 2014, 1, 15–33. [Google Scholar] [CrossRef]
- Richrath, R.B.; Olyschläger, T.; Hildebrandt, S.; Enny, D.G.; Fianu, G.D.; Flowers, R.A., II; Gansäuer, A. Cp2TiX Complexes for Sustainable Catalysis in Single-Electron Steps. Chem. Eur. J. 2018, 24, 6371–6379. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.C.; Garcia, I.R.; Maecker, R.N.R.; Morales, L.P.; Oltra, J.E.; Martinez, A.R. Cp2TiCl: An Ideal Reagent for Green Chemistry? Org. Process Res. Dev. 2017, 21, 911–923. [Google Scholar] [CrossRef]
- Machat, M.R.; Jandl, C.; Rieger, B. Titanocenes in Olefin Polymerization: Sustainable Catalyst System or an Extinct Species? Organometallics 2017, 36, 1408–1418. [Google Scholar] [CrossRef]
- Martinez, A.R.; Rodriguez, M.C.; Rodriguez-Garcia, I.; Morales, L.P.; Rodriguez Maecker, R.N. Titanocene dichloride: A new green reagent in organic chemistry. Chin. J. Catal. 2017, 38, 1659–1663. [Google Scholar] [CrossRef]
- Köpf, H.; Köpf-Mayer, P. Titanocen-dichlorid-das erste Metallocen mit cancerostatischer Wirksamkeit. Angew. Chem. Int. Ed. Engl. 1979, 18, 477–478. [Google Scholar]
- Gomez-Ruis, S.; Maksimovic-Ivanic, D.; Mijatovic, S.; Kaluderovic, G.N. On the Discovery, Biological Effects and Use of Cisplatin and Metallocenes in Anticancer Chemotherapy. Bioinorg. Chem. Appl. 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.; Tacke, M. Titanocenes: Cytotoxic and Anti-angiogenic Chemotherapy Against Advanced Renal-Cell Cancer. In Medicinal Organometallic Chemistry; Jaouen, G., Metzler-Nolte, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 32, pp. 119–140. [Google Scholar]
- Skoupilova, H.; Hrstka, R.; Bartosik, M. Titanocenes as Anticancer Agents: Recent Insights. Med. Chem. 2017, 13, 334–344. [Google Scholar] [CrossRef]
- Ellahioui, Y.; Prashar, S.; Gomez-Ruiz, S. Anticancer Applications and Recent Investigations of Metallodrugs Based on Gallium, Tin and Titanium. Inorganics 2017, 5, 4. [Google Scholar] [CrossRef]
- Immel, T.A.; Martin, J.T.; Dürr, C.J.; Groth, U.; Huhn, T. Dimethyl titanocene Y: A valuable precursor for libraries of cytotoxic titanocene derivatives. J. Inorg. Biochem. 2010, 104, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Chirik, P.J. Group 4 Transition Metal Sandwich Complexes: Still Fresh after Almost 60 Years. Organometallics 2010, 29, 1500–1517. [Google Scholar] [CrossRef]
- Alcock, N.W.; Clase, H.J.; Duncalf, D.J.; Hart, S.L.; McCamley, A.; McCormack, P.J.; Taylor, P.C. Synthesis of racemic chiral-at-metal complexes of the Group 4 metals. J. Organomet. Chem. 2000, 605, 45–54. [Google Scholar] [CrossRef]
- Sünkel, K. Coordination Chemistry of Pentahalocyclopentadienyls. Chem. Berichte 1997, 130, 1721–1730. [Google Scholar] [CrossRef]
- Chang, B.H.; Tung, H.S.; Brubaker, C.H., Jr. The Thermal Decomposition of Dibutylbis(η5-cyclopentadienyl)titanium, other Dialkyl Metallocenes, and Di(η5-pentamethylcyclopentadienyl) itanium. Inorg. Chim. Acta 1981, 51, 143–148. [Google Scholar] [CrossRef]
- Vyshinskaya, L.I.; Korneva, S.P.; Kulikova, G.P. Reactions of ansa-Titanocenes with Organolithium Compounds. Russ. J. Gen. Chem. 2002, 72, 68–70. [Google Scholar] [CrossRef]
- Morillo, A.; Chirinos, J.; Rajmankina, T.; Ibarra, D.; Arevalo, J.; Parada, A. Ethene polymerization with titanium half-sandwich amido-complexes. J. Chem. Res. 2003, 238–239. [Google Scholar] [CrossRef]
- Boyde, N.C.; Rightmire, N.R.; Bierschenk, E.J.; Steelman, G.W.; Hanusa, T.P.; Brennessel, W.W. Reaction environment and ligand lability in group 4 Cp2MXY (X,Y = Cl, OtBu) complexes. Dalton Trans. 2016, 45, 18635–18642. [Google Scholar] [CrossRef] [PubMed]
- Köpf, H.; Schmidt, H. Bis-π-cyclopentadienyl-titan-mercaptide. Z. Anorg. Allg. Chem. 1965, 340, 139–145. [Google Scholar] [CrossRef]
- Giddings, S.A. Bis(cyclopentadienyl)titanium(IV) Compounds with Sulfur-Containing Groups. Inorg. Chem. 1967, 6, 849–851. [Google Scholar] [CrossRef]
- Horacek, M. Titanocene sulfide chemistry. Coord. Chem. Rev. 2016, 314, 83–102. [Google Scholar] [CrossRef]
- Song, L.C.; Liu, P.C.; Han, C.; Hu, Q.M. Synthesis and characterization of Cp2Ti-containing organometallics via in situ oxidative-addition of ‘Cp2Ti’ intermediate. Crystal Structures of (1-C10H7S)2TiCp2, [(η5-C5H5)Fe(η5-C5H4CH2S)]2TiCp2 and [η2-OC(Ph)=C(Ph)O]TiCp2. J. Organomet. Chem. 2002, 648, 119–125. [Google Scholar] [CrossRef]
- Meyer, R.; Brink, S.; vanRensburg, C.E.J.; Joone, G.K.; Görls, H.; Lotz, S. Synthesis, characterization and antitumor properties of titanocene derivatives with thiophene containing ligands. J. Organomet. Chem. 2005, 690, 117–125. [Google Scholar] [CrossRef]
- Gomez-Ruiz, S.; Stanojkovic, T.P.; Kaluderovic, G.N. Synthesis, characterization, biological studies and in vitro cytotoxicity on human cancer cell lines of titanium(IV) and tin(IV) derivatives with the α,α’-dimercapto-o-xylene ligand. Appl. Organomet. Chem. 2012, 26, 383–389. [Google Scholar] [CrossRef]
- Pinkas, J.; Lamac, M. Transformation of functional groups attached to cyclopentadienyl or related ligands in group 4 metal complexes. Coord. Chem. Rev. 2015, 296, 45–90. [Google Scholar] [CrossRef]
- Conway, B.G.; Rausch, M.D. Formation and Reactivity of Halogen Derivatives of (η5-Cyclopentadienyl)thallium. Organometallics 1985, 4, 688–693. [Google Scholar] [CrossRef]
- Anslyn, E.A.; Grubbs, R.H.; Felten, C.; Rehder, D. Dichlorobis(η5-Chlorocyclopentadienyl)-Titanium, (ClC5H4)2TiCl2. Inorg. Synth. 1992, 29, 198–201. [Google Scholar]
- Bernhartzeder, S.; Sünkel, K. Coordination Chemistry of Perhalogenated Cyclopentadienes and Alkynes. Part 30. New High-Yield Syntheses of Monochloroferrocene and 1,2,3,4,5-Pentachloroferrocene. Molecular Structures of 1,2-Dichloroferrocene and 1,2,3-Trichloroferrocene. J. Organomet. Chem. 2012, 716, 146–149. [Google Scholar] [CrossRef]
- Sünkel, K.; Bernhartzeder, S. Coordination Chemistry of Perhalogenated Cyclopentadienes and Alkynes. XXVIII New High-yield Synthesis of Monobromoferrocene and Simplified Procedure for the Synthesis of Pentabromoferrocene. Molecular structures of 1,2,3-tribromoferrocene and 1,2,3,4,5-pentabromoferrocene. J. Organomet. Chem. 2011, 696, 1536–1540. [Google Scholar]
- Sünkel, K.; Weigand, S.; Hoffmann, A.; Blomeyer, S.; Reuter, C.G.; Vishnevskiy, Y.V.; Mitzel, N.W. Synthesis and Characterization of 1,2,3,4,5-Pentafluoroferrocene. J. Amer. Chem. Soc. 2015, 137, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, F.L.; Rosenberg, H. Preparation and reactions of decachloroferrocene ad decachlororuthenocene. J. Am. Chem. Soc. 1973, 95, 870–875. [Google Scholar] [CrossRef]
- Butler, I.R. 1,2,3,4,5-Pentabromoferrocene and related compounds: A simple synthesis of useful precursors. Inorg. Chem. Commun. 2008, 11, 484–486. [Google Scholar] [CrossRef]
- Klein-Heßling, C.; Stramm, C.; Sünkel, K. Metalation Studies on Carbonyl-Halocyclopentadienyl Manganese Complexes. Manuscript in preparation. 2018. [Google Scholar]
- Gansäuer, A.; Kube, C.; Daasbjerg, K.; Sure, R.; Grimme, S.; Fianu, G.D.; Sadasivam, D.V.; Flowers, R.A., II. Substituent Effects and Supramolecular Interactions of Titanocene(II) Chloride: Implications for Catalysis in Single Electron Steps. J. Am. Chem. Soc. 2014, 136, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.F.; Borjas, R.E.; Furilla, J.L. Investigation of the electrochemical properties of substituted titanocene dichlorides. Electrochim. Acta 1995, 40, 473–477. [Google Scholar] [CrossRef]
- Rosales, A.; Rodriguez-Garcia, I.; Munoz-Bascon, J.; Roldan-Molina, E.; Padial, N.M.; Morales, L.P.; Garcia-Ocana, M.; Oltra, J.E. The Nugent Reagent: A Formidable Tool in Contemporary Radical and Organometallic Chemistry. Eur. J. Org. Chem. 2015, 4567–4591. [Google Scholar]
- Wielstra, Y.; Gambarotta, S.; Spek, A.L.; Smeets, W.J.J. Binuclear Zirconium(III) and Zirconium(IV) Complexes: Mild Oxidation of Low-Valent Zirconium as a Synthetic Pathway to a Class of Tri- and Tetravalent Zirconium Fulvalene Complexes. Molecular Structures of [η5:η5-C10H8][CpZrSPh]2, [η5:η5-C10H8][CpZr(CH3)2]2 and [η5:η5-C10H8][CpZr]2[µ-S]2. Organometallics 1990, 9, 2142–2148. [Google Scholar]
- Cuenca, T.; Herrmann, W.A.; Ashworth, T.V. Chemistry of Oxophilic Transition Metals. 2. Novel Derivatives of Titanocene and Zirconocene. Organometallics 1986, 5, 2514–2516. [Google Scholar] [CrossRef]
- Pinkas, J.; Gyepes, R.; Cisarova, I.; Kubista, J.; Horacek, M.; Zilkova, N.; Mach, K. Hydrogenation of titanocene and zirconocene bis(trimethylsilyl)acetylene complexes. Dalton Trans. 2018, 47, 8921–8932. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, A.E.; Fanwick, P.E.; Rothwell, I.P. Synthesis, Characterization, and Reduction Chemistry of Mixed Cyclopentadienyl/Arylsulfide Titanium Dichlorides. Organometallics 2003, 22, 535–540. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).