Gd3Li3Te2O12:U6+,Eu3+: A Tunable Red Emitting Garnet Showing Efficient U6+ to Eu3+ Energy Transfer at Room Temperature



Abstract

1. Introduction
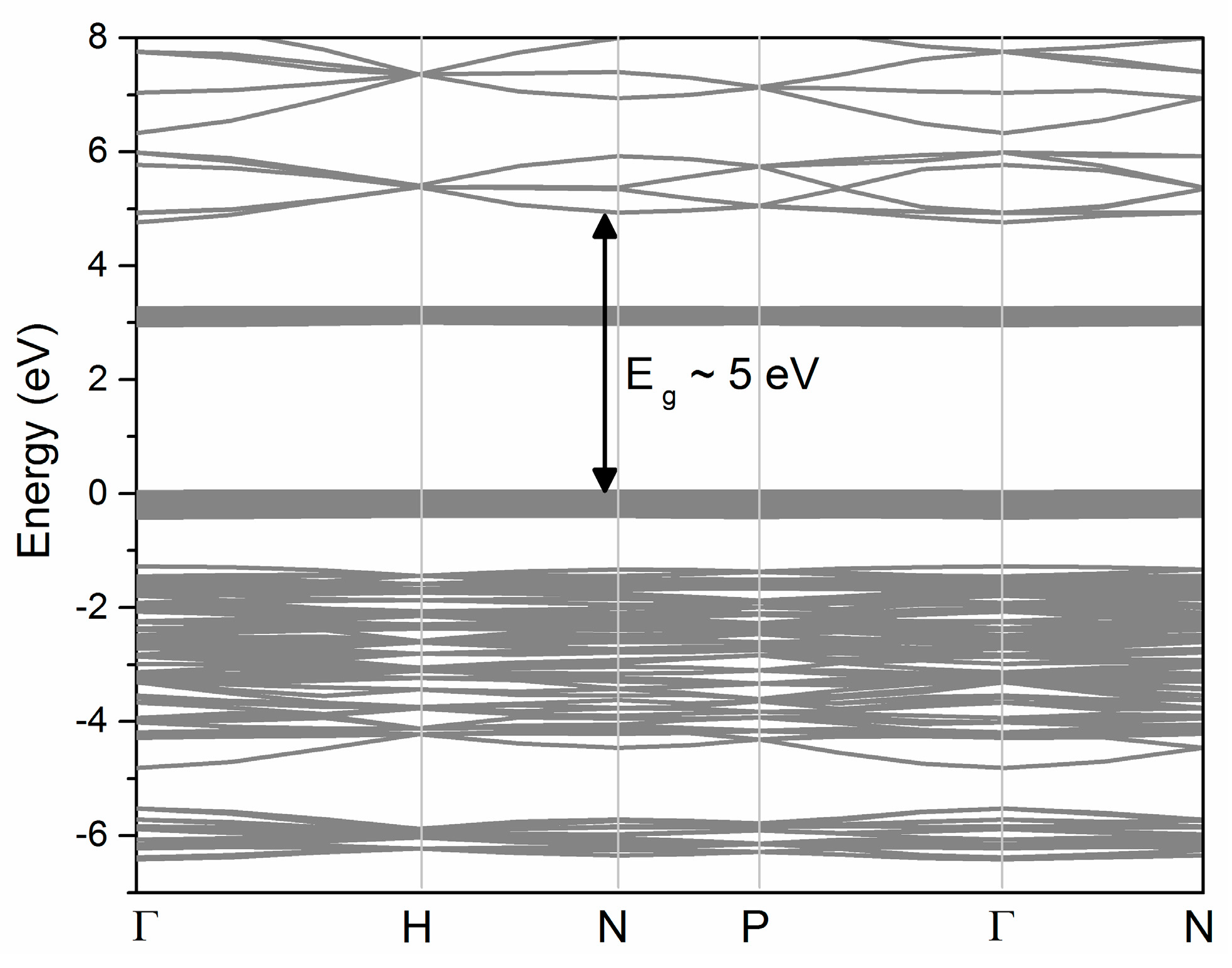
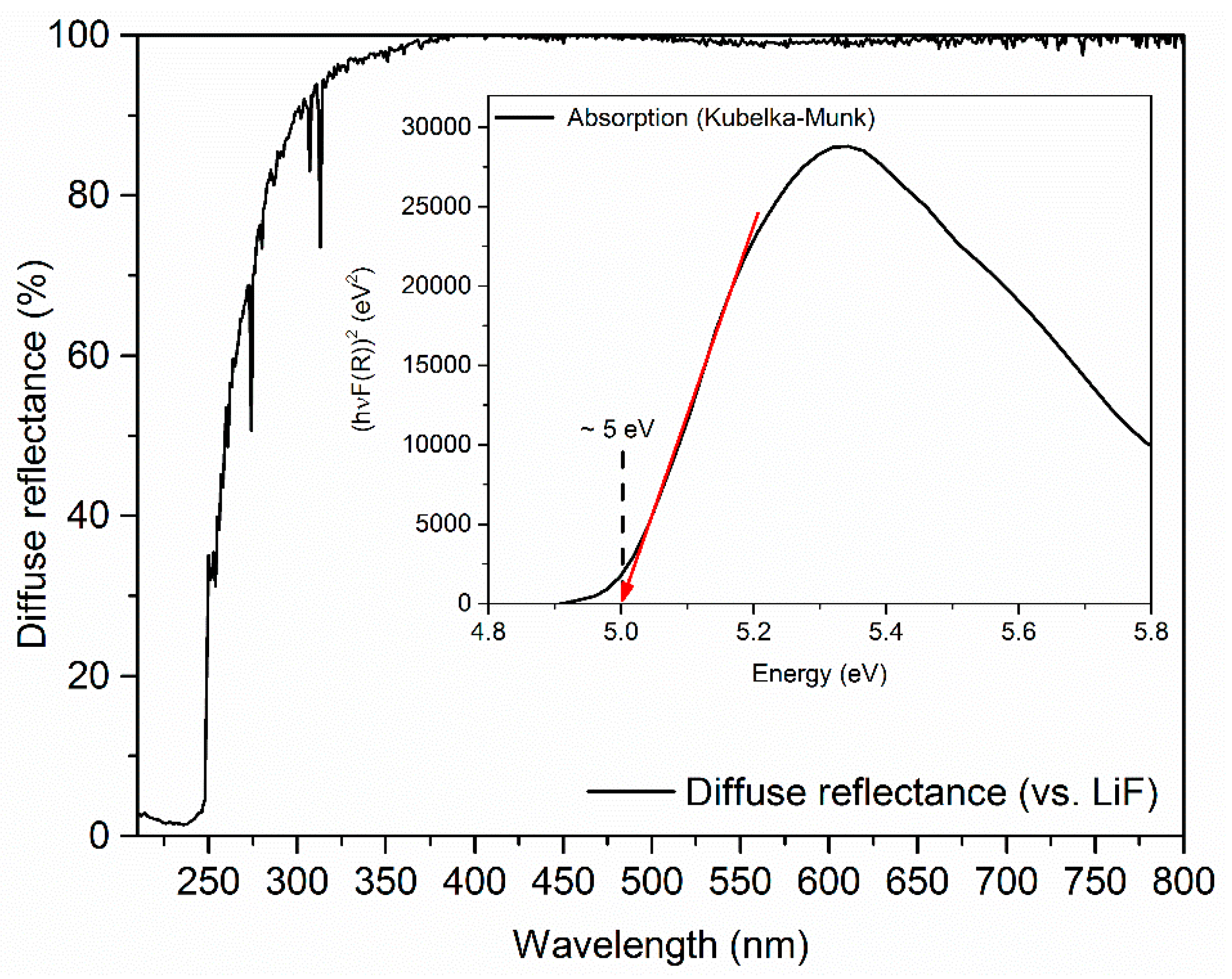
2. Method of Calculation and Electronic Properties
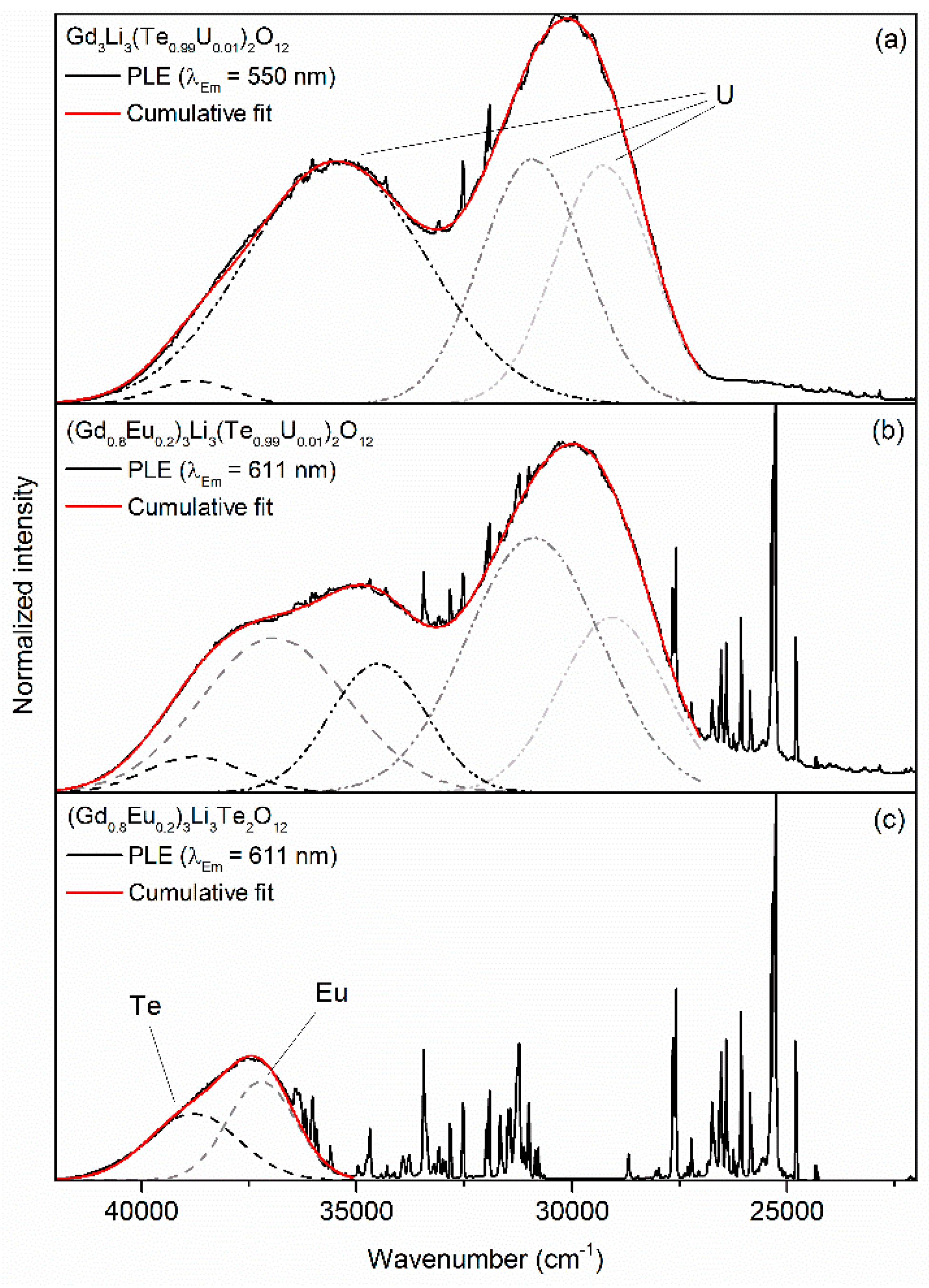
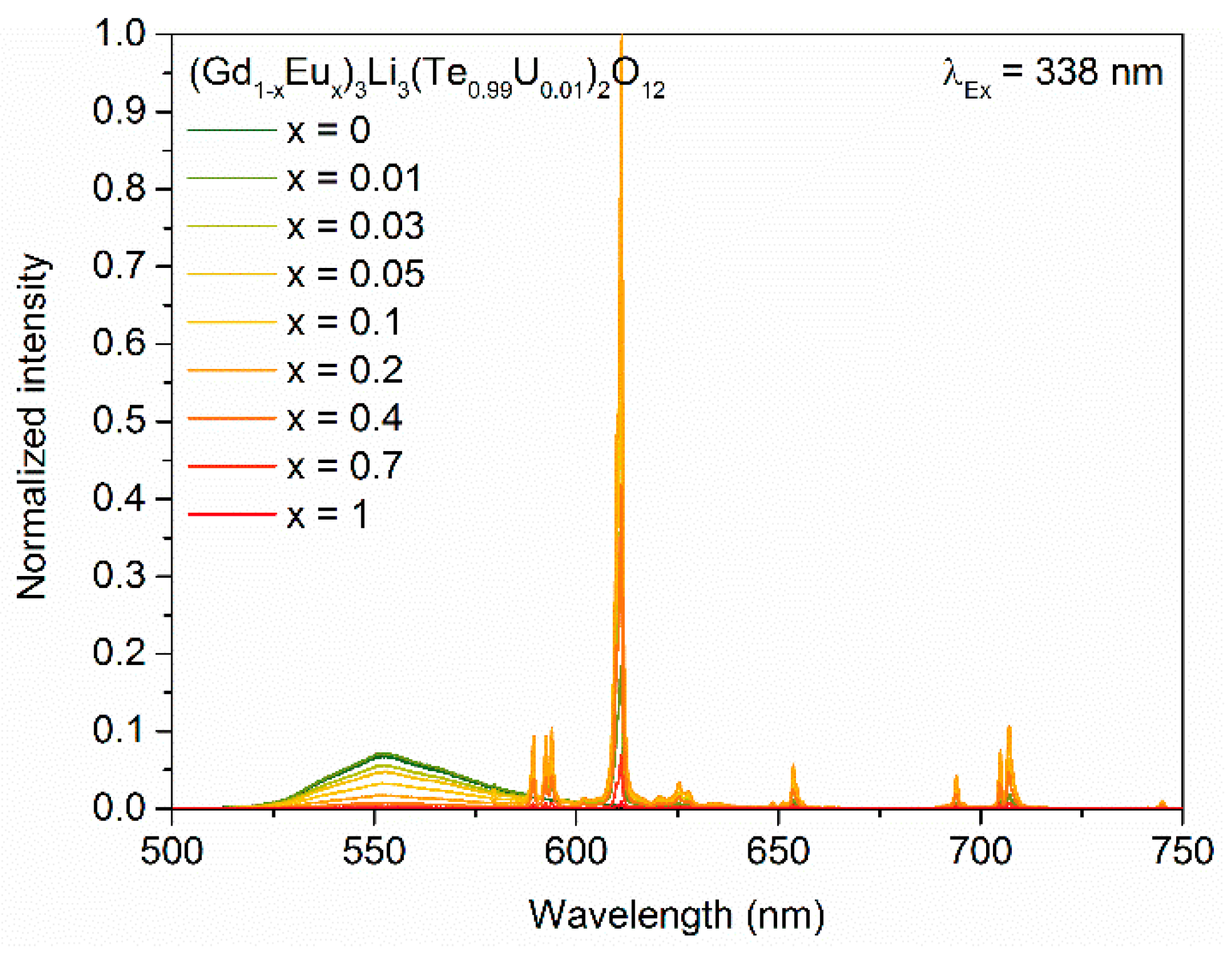
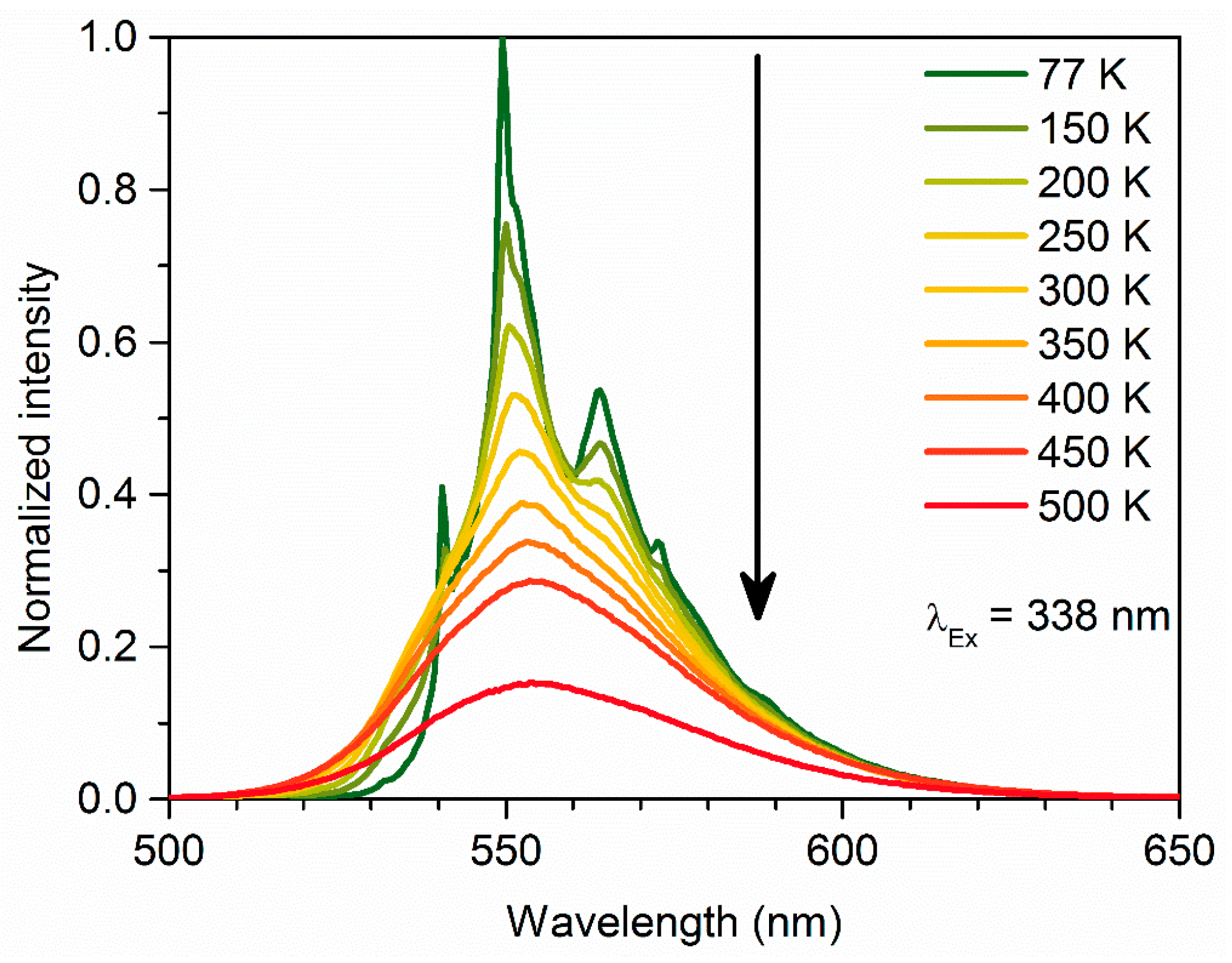
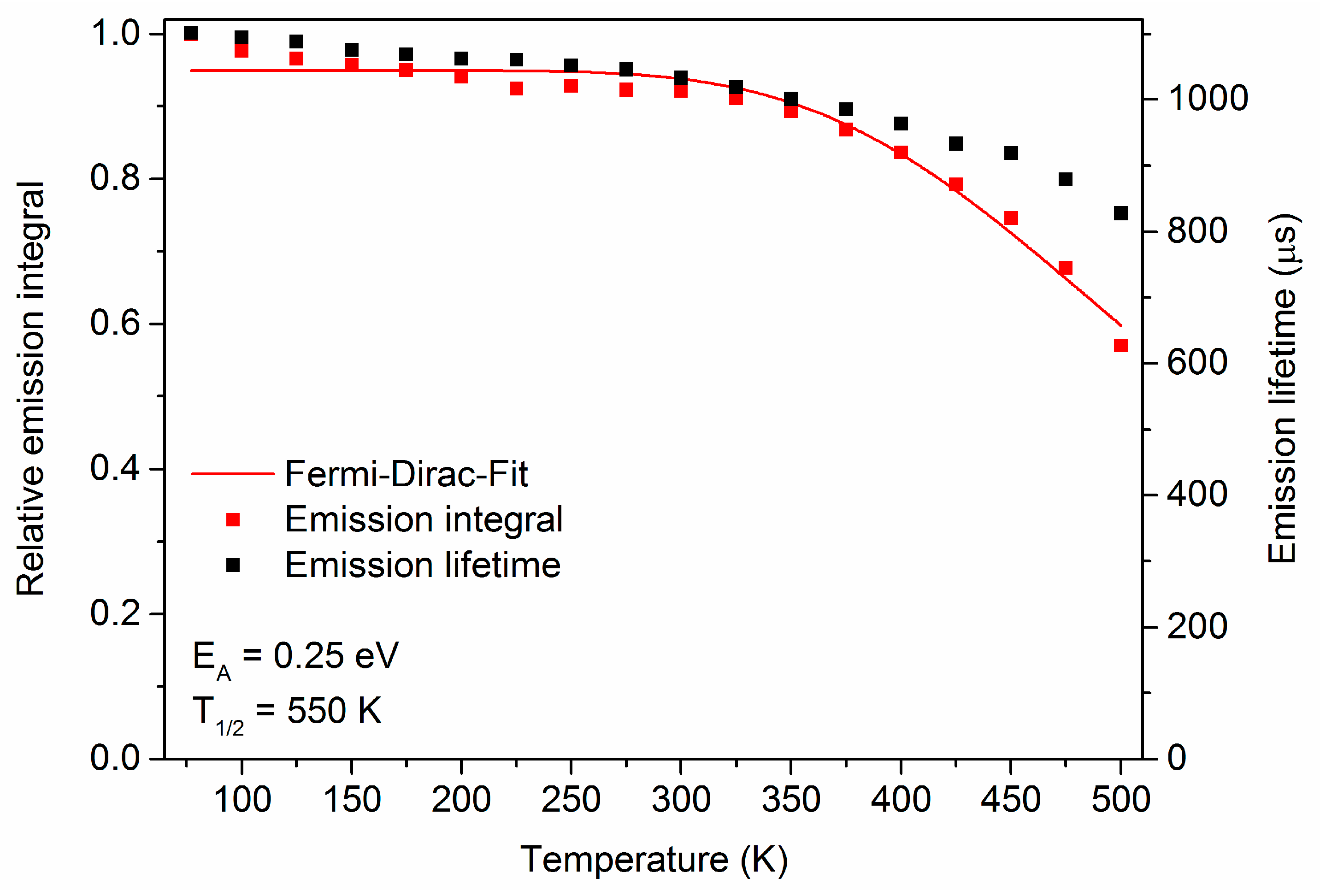
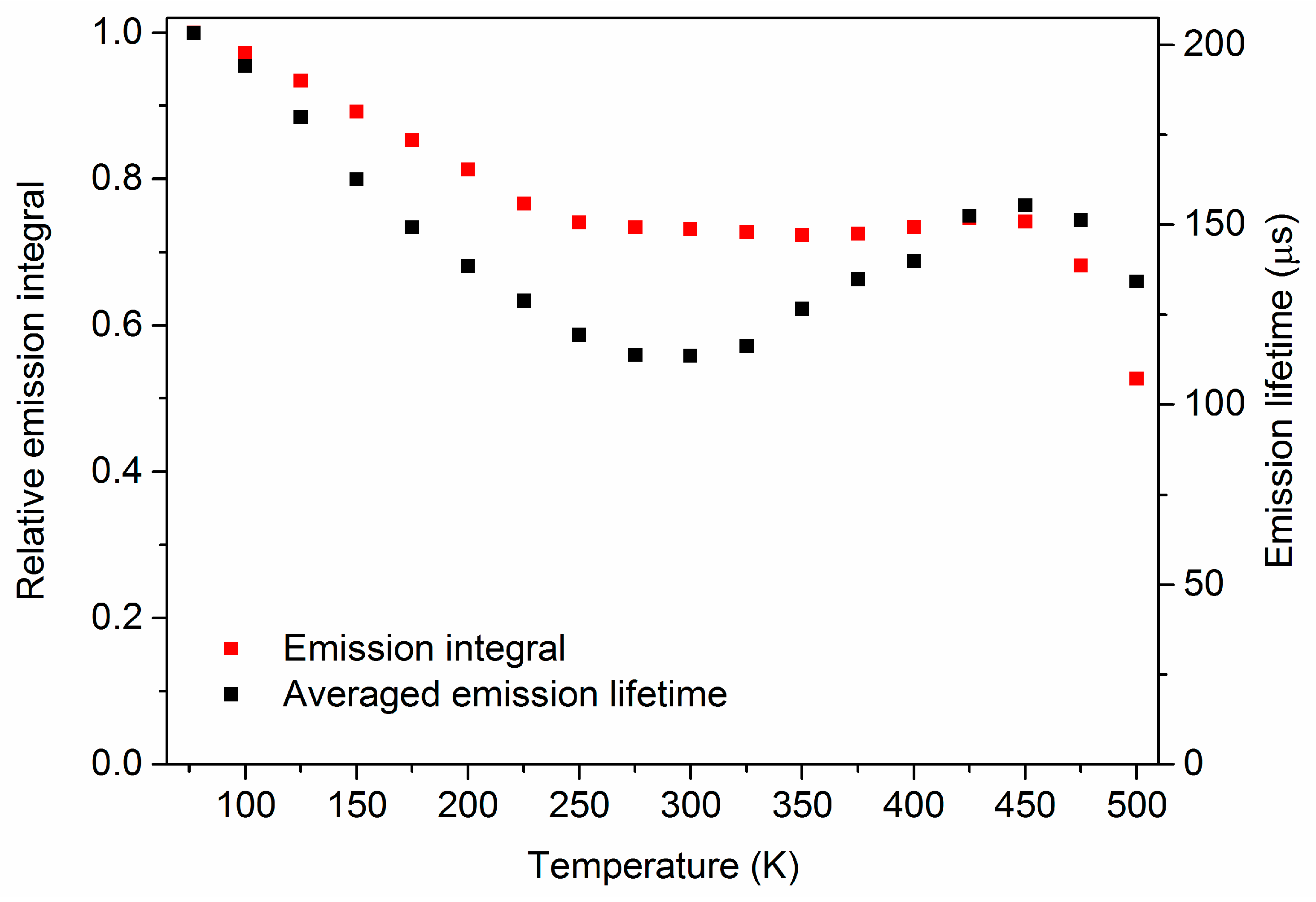
3. Results and Discussion
4. Experimental Section
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Welker, T. Recent developments on phosphors for fluorescent lamps and cathode-ray tubes. J. Lumin. 1991, 48–49, 49–56. [Google Scholar] [CrossRef]
- Feldmann, C.; Jüstel, T.; Ronda, C.R.; Schmidt, P.J. Inorganic Luminescent Materials: 100 Years of Research and Application. Adv. Funct. Mater. 2003, 13, 511–516. [Google Scholar] [CrossRef]
- Baur, F.; Jüstel, T. New Red-Emitting Phosphor La2Zr3(MoO4)9:Eu3+ and the Influence of Host Absorption on its Luminescence Efficiency. Aust. J. Chem. 2015, 68, 1727. [Google Scholar] [CrossRef]
- Katelnikovas, A.; Plewa, J.; Sakirzanovas, S.; Dutczak, D.; Enseling, D.; Baur, F.; Winkler, H.; Kareiva, A.; Jüstel, T. Synthesis and optical properties of Li3Ba2La3(MoO4)8:Eu3+ powders and ceramics for pcLEDs. J. Mater. Chem. 2012, 22, 22126. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, K.H.; Unithrattil, S.; Yoon, H.S.; Jang, H.G.; Im, W.B. Melilite-Structure CaYAl3O7:Eu3+ Phosphor: Structural and Optical Characteristics for Near-UV LED-Based White Light. J. Phys. Chem. C 2012, 116, 26850–26856. [Google Scholar] [CrossRef]
- Baur, F.; Glocker, F.; Jüstel, T. Photoluminescence and energy transfer rates and efficiencies in Eu3+ activated Tb2Mo3O12. J. Mater. Chem. C 2015, 3, 2054–2064. [Google Scholar] [CrossRef]
- Žukauskas, A.; Vaicekauskas, R.; Ivanauskas, F.; Vaitkevičius, H.; Shur, M.S. Spectral optimization of phosphor-conversion light-emitting diodes for ultimate color rendering. Appl. Phys. Lett. 2008, 93, 51115. [Google Scholar] [CrossRef]
- Park, W.J.; Jung, M.K.; Yoon, D.H. Influence of Eu3+, Bi3+ co-doping content on photoluminescence of YVO4 red phosphors induced by ultraviolet excitation. Sens. Actuator B Chem. 2007, 126, 324–327. [Google Scholar] [CrossRef]
- Böhnisch, D.; Baur, F.; Jüstel, T. Photoluminescence and energy transfer behavior of narrow band red light emitting Li3Ba2Tb3(MoO4)8:Eu3+. Dalton Trans. 2018, 47, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Blasse, G.; Krol, D.M. Energy transfer phenomena in ordered perovskites of the type Sr2Na0.5Ln3+0.5X6+O6. J. Lumin. 1981, 22, 389–396. [Google Scholar] [CrossRef]
- Krol, D.M.; Ros, J.P.M.; Roos, A. The influence of crystal structure and chemical composition on the energy transfer processes in uranates. J. Chem. Phys. 1980, 73, 1521–1526. [Google Scholar] [CrossRef]
- Smit, W.M.A.; Blasse, G. Luminescence and energy migration in the garnet Gd3Li3Te2O12 doped with several rare earths and uranium. J. Solid State Chem. 1986, 63, 308–315. [Google Scholar] [CrossRef]
- Köngeter, B.; Kemmler-Sack, S. Photolumineszenz und Energeitransfer in Seltenerd-aktivierten Granat Gd3Te2Li3O12. Z. Naturforsch. A 1984, 39a, 490–494. [Google Scholar] [CrossRef]
- Alberda, R.H.; Blasse, G. Luminescence in a new garnet phase with hexavalent metal ions. J. Lumin. 1976, 12–13, 687–692. [Google Scholar] [CrossRef]
- Zhang, W.; Seo, H.J. Luminescence and structure of a novel red-emitting phosphor Eu3+-doped tellurate garnet Li3Y3Te2O12. J. Alloys Compd. 2013, 553, 183–187. [Google Scholar] [CrossRef]
- Sztajnkrycer, M.D.; Otten, E.J. Chemical and Radiological Toxicity of Depleted Uranium. Mil. Med. 2004, 169, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Baur, F.; Jüstel, T. Warm-white LED with ultra high luminous efficacy due to sensitisation of Eu 3+ photoluminescence by the uranyl moiety in K4(UO2)Eu2(Ge2O7)2. J. Mater. Chem. C 2018, 7, 1600826. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Perdew, J.P. Density functional theory and the band gap problem. Int. J. Quantum Chem. 1986, 30, 451. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef]
- Enseling, D.; Herden, B.; Katelnikovas, A.; Möller, S.; Winkler, H.; Petry, R.; Meyer, H.-J.; Jüstel, T. Powder Reflection Spectroscopy in the Vacuum UV range. J. Appl. Spectrosc. 2014, 81, 341–346. [Google Scholar] [CrossRef]
- Kubelka, P.; Munk, F. Ein Beitrag zur Optik der Farbanstriche. Z. Phys. 1931, 12, 593–601. [Google Scholar]
- Tauc, J. Optical properties and electronic structure of amorphous Ge and Si. Mater. Res. Bull. 1968, 3, 37–46. [Google Scholar] [CrossRef]
- Tauc, J.; Grigorovici, R.; Vancu, A. Optical Properties and Electronic Structure of Amorphous Germanium. Phys. Stat. Sol. 1966, 15, 627–637. [Google Scholar] [CrossRef]
- O’Callaghan, M.P.; Lynham, D.R.; Cussen, E.J.; Chen, G.Z. Structure and Ionic-Transport Properties of Lithium-Containing Garnets Li3Ln3Te2O12 (Ln = Y, Pr, Nd, Sm–Lu). Chem. Mater. 2006, 18, 4681–4689. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Blasse, G.; Bril, A. Luminescence of tungsten-activated tellurates. J. Solid State Chem. 1970, 2, 291–294. [Google Scholar] [CrossRef]
- Jørgensen, C.K.; Lundström, T.; Patoharju, O.; Noer, B.; Reio, L. Complexes of the 4d- and 5d-Groups. IV. Electron Transfer Bands with Special Application to M. Delépine’s Complexes and a Transition from Iridium(III) to Pyridine, with some Remarks about Intermediate Coupling in Halide Complexes and the Uranyl Ion. Acta Chem. Scand. 1957, 11, 166–178. [Google Scholar] [CrossRef]
- De Hair, J.T.W.; Blasse, G. Luminescence of the octahedral uranate group. J. Lumin. 1976, 14, 307–323. [Google Scholar] [CrossRef]
- Blasse, G. The Structure Sensitivity of the U6+ Ion Luminescence in Oxides. J. Electrochem. Soc. 1977, 124, 1280. [Google Scholar] [CrossRef]
- Blasse, G.; Bleijenberg, K.C.; Krol, D.M. The luminescence of hexavalent uranium in solids. J. Lumin. 1979, 18-19, 57–62. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Liang, C.-H.; Yan, S.-A.; Chang, Y.-S. Synthesis and Photoluminescence Characteristics of High Color Purity and Brightness Li3Ba2Gd3(MoO4)8:Eu3+ Red Phosphors. J. Phys. Chem. C 2010, 114, 3645–3652. [Google Scholar] [CrossRef]
- Blasse, G.; Grabmaier, B.C. Luminescent Materials; Springer Verlag: Utrecht, The Netherlands, 1994; pp. 91–106. [Google Scholar]
- Bleijenberg, K.C.; Breddels, P.A. On the vibrational structure in the luminescence spectra of uranium-activated sodium fluoride crystals. J. Chem. Phys. 1980, 72, 5390–5398. [Google Scholar] [CrossRef]
- Bleijenberg, K.C. Luminescence properties of uranate centres in solids. In Luminescence and Energy Transfer. Structure and Bonding; Springer Verlag: Utrecht, The Netherlands, 1980; pp. 97–217. [Google Scholar]
- Kawamura, Y.; Sasabe, H.; Adachi, C. Simple Accurate System for Measuring Absolute Photoluminescence Quantum Efficiency in Organic Solid-State Thin Films. Jpn. J. Appl. Phys. 2004, 43, 7729–7730. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eu3+ Conc. | EQE | CIE1931 Color Coordinate | Luminous Efficacy | |
|---|---|---|---|---|
| % | (λEx = 338 nm) | x | y | [lm/Wopt] |
| 0 | 32 | 0.3812 | 0.6119 | 632 |
| 1 | 39 | 0.4194 | 0.5748 | 576 |
| 3 | 39 | 0.4655 | 0.5297 | 511 |
| 5 | 41 | 0.5031 | 0.4931 | 464 |
| 10 | 42 | 0.5514 | 0.4459 | 405 |
| 20 | 38 | 0.5921 | 0.4061 | 357 |
| 40 | 16 | 0.5933 | 0.4048 | 352 |
| 70 | 2 | 0.5747 | 0.4224 | 364 |
| 100 | <1 | 0.6079 | 0.3855 | 271 |
| Sample (x) | Fraction 1 (%) | τ1 (μs) | Fraction 2 (%) | τ2 (μs) | τ (μs) |
|---|---|---|---|---|---|
| 0 | 100 | 127.5 | |||
| 1 | 78 | 107.8 | 22 | 186.8 | 125.2 |
| 3 | 58 | 88.8 | 42 | 163.7 | 120.2 |
| 5 | 43 | 71.0 | 57 | 146.1 | 113.7 |
| 10 | 40 | 61.6 | 60 | 144.0 | 111.0 |
| 20 | 36 | 52.4 | 64 | 138.7 | 108.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böhnisch, D.; Rosenboom, J.; Jansen, T.; Jüstel, T. Gd3Li3Te2O12:U6+,Eu3+: A Tunable Red Emitting Garnet Showing Efficient U6+ to Eu3+ Energy Transfer at Room Temperature. Inorganics 2018, 6, 84. https://doi.org/10.3390/inorganics6030084
Böhnisch D, Rosenboom J, Jansen T, Jüstel T. Gd3Li3Te2O12:U6+,Eu3+: A Tunable Red Emitting Garnet Showing Efficient U6+ to Eu3+ Energy Transfer at Room Temperature. Inorganics. 2018; 6(3):84. https://doi.org/10.3390/inorganics6030084
Chicago/Turabian StyleBöhnisch, David, Juri Rosenboom, Thomas Jansen, and Thomas Jüstel. 2018. "Gd3Li3Te2O12:U6+,Eu3+: A Tunable Red Emitting Garnet Showing Efficient U6+ to Eu3+ Energy Transfer at Room Temperature" Inorganics 6, no. 3: 84. https://doi.org/10.3390/inorganics6030084
APA StyleBöhnisch, D., Rosenboom, J., Jansen, T., & Jüstel, T. (2018). Gd3Li3Te2O12:U6+,Eu3+: A Tunable Red Emitting Garnet Showing Efficient U6+ to Eu3+ Energy Transfer at Room Temperature. Inorganics, 6(3), 84. https://doi.org/10.3390/inorganics6030084