
Chiral Amine Covalent Organic Cage Lingated with Copper for Asymmetric Decarboxylative Mannich Reaction

Abstract
1. Introduction
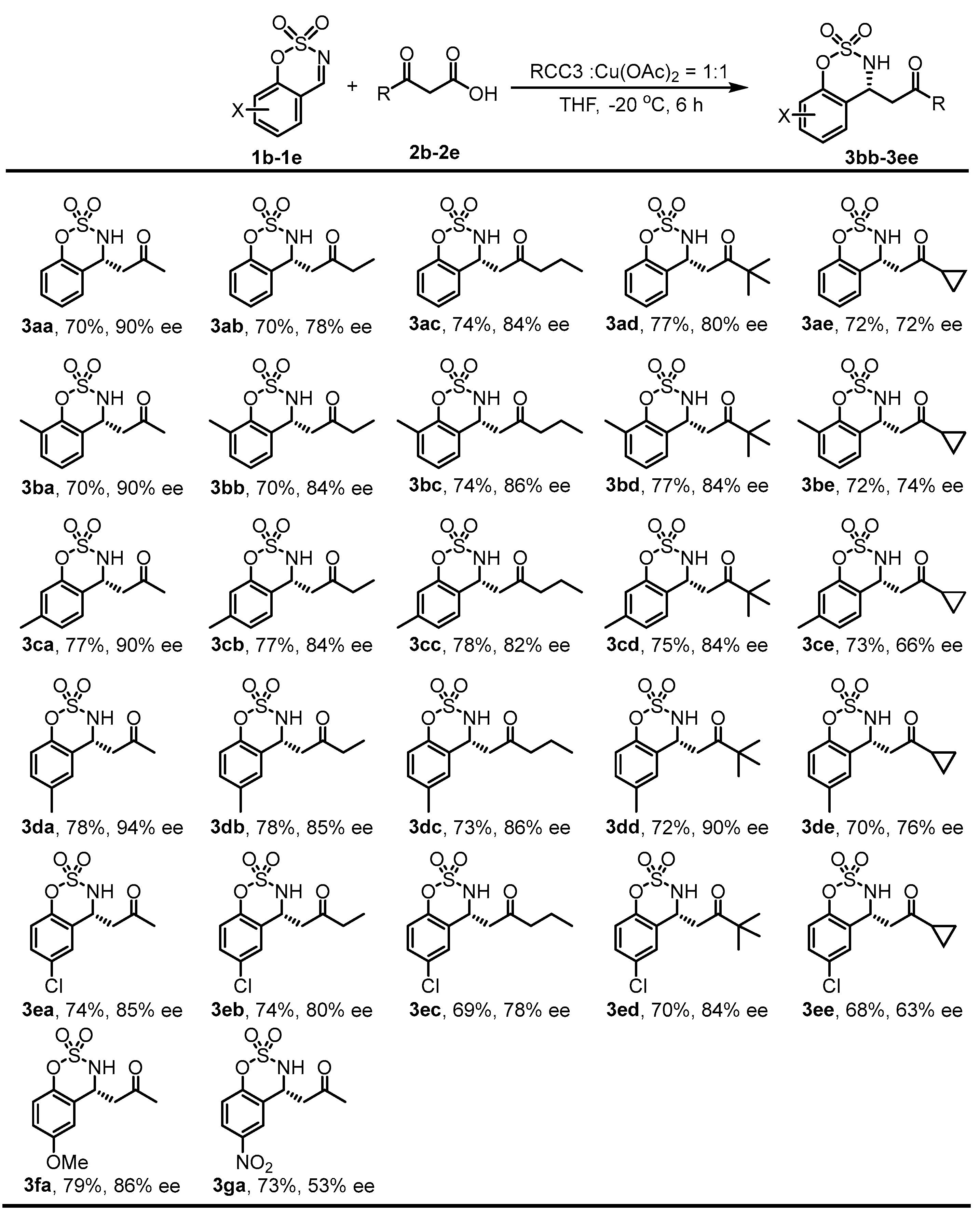
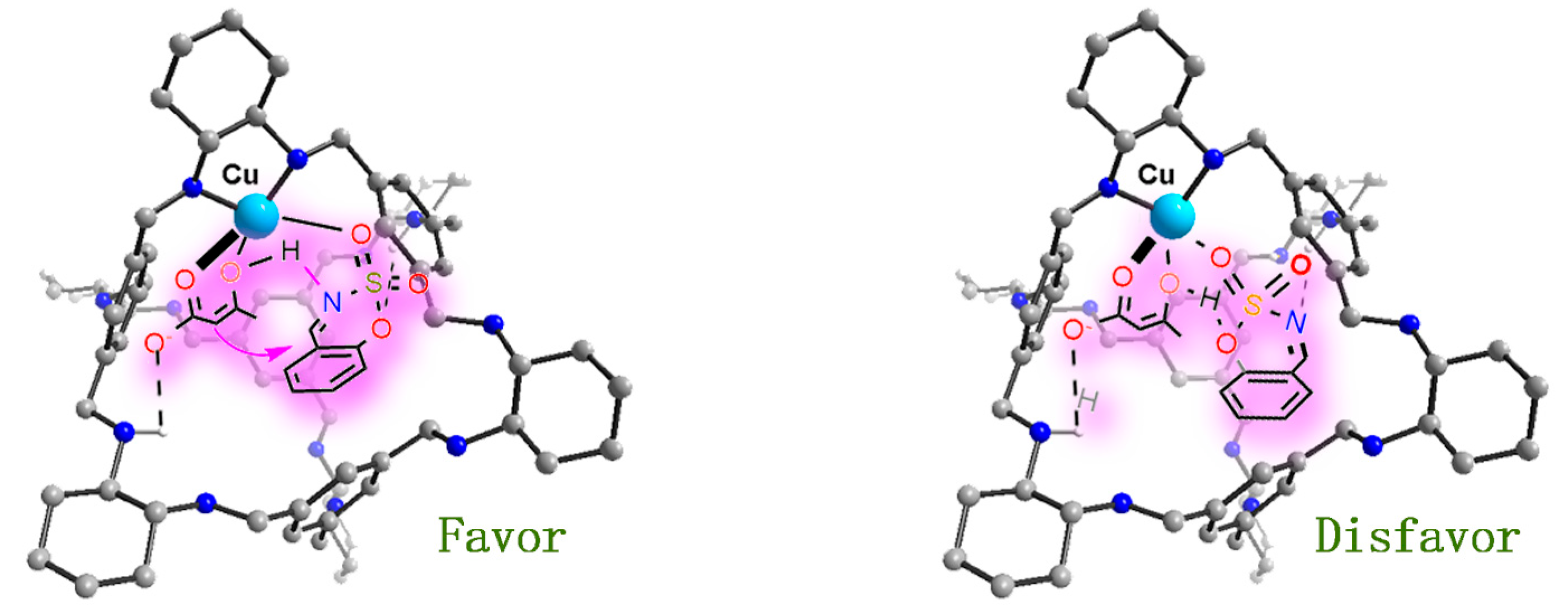
2. Results and Discussion
3. Materials and Methods
3.1. Materials and General Procedures
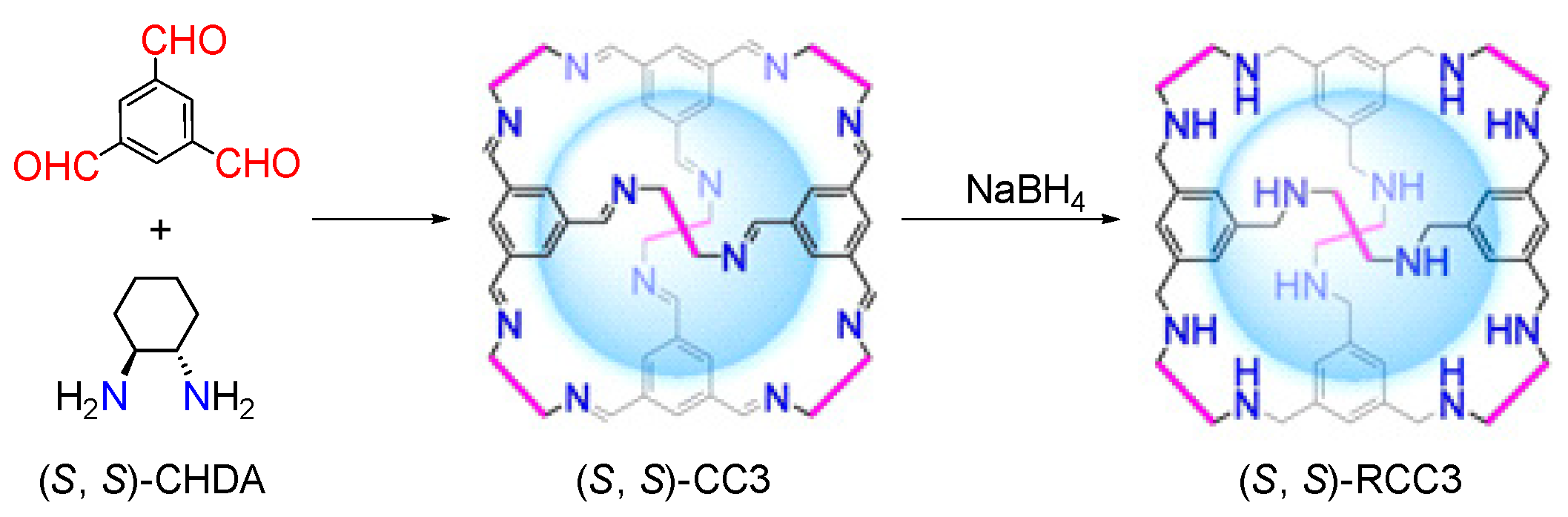
3.2. Synthesis of CC3
3.3. Synthesis of Corresponding Reduced RCC3
3.4. General Procedure for DMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, X.; Ullah, Z.; Stoddart, J.F.; Yavuz, C.T. Porous Organic Cages. Chem. Rev. 2023, 123, 4602–4634. [Google Scholar] [CrossRef]
- Hasell, T.; Little, M.A.; Chong, S.Y.; Schmidtmann, M.; Briggs, M.E.; Santolini, V.; Jelfs, K.E.; Cooper, A.I. Chirality as a tool for function in porous organic cages. Nanoscale 2017, 9, 6783–6790. [Google Scholar] [CrossRef]
- Gao, W.B.; Li, Z.H.; Tong, T.Y.; Dong, X.; Qu, H.; Yang, L.L.; Sue, A.C.H.; Tian, Z.Q.; Cao, X.Y. Chiral Molecular Cage with Tunable Stereoinversion Barriers. J. Am. Chem. Soc. 2023, 145, 17795–17804. [Google Scholar] [CrossRef]
- Zhou, H.; Ao, Y.F.; Wang, D.X.; Wang, Q.Q. Inherently Chiral Cages via Hierarchical Desymmetrization. J. Am. Chem. Soc. 2022, 144, 16767–16772. [Google Scholar] [CrossRef]
- Liu, C.; Jin, Y.; Qi, D.; Ding, X.; Ren, H.; Wang, H.; Jiang, J. Enantioselective assembly and recognition of heterochiral porous organic cages deduced from binary chiral components. Chem. Sci. 2022, 13, 7014–7020. [Google Scholar] [CrossRef]
- Monta-González, G.; Sancenón, F.; Martínez-Mán, R.; MartíCentelles, V. Purely Covalent Molecular Cages and Containers for Guest Encapsulation. Chem. Rev. 2022, 122, 13636–13708. [Google Scholar] [CrossRef]
- Tozawa, T.; Jones, J.T.A.; Swamy, S.I.; Jiang, S.; Adams, D.J.; Shakespeare, S.; Clowes, R.; Bradshaw, D.; Hasell, T.; Chong, S.Y.; et al. Porous organic cages. Nat. Mater. 2009, 8, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, P.; Mukherjee, P.S. Covalent Organic Cages in Catalysis. ACS Catal. 2023, 13, 6126–6143. [Google Scholar] [CrossRef]
- Du, Y.-J.; Zhou, J.-H.; Tan, L.-X.; Liu, S.-H.; Zhao, K.; Gao, Z.-M.; Sun, J.-K. Porous Organic Cage Nanostructures for Construction of Complex Sequential Reaction Networks. ACS Appl. Nano Mater. 2022, 5, 7974–7982. [Google Scholar] [CrossRef]
- Yang, X.-D.; Tan, L.; Sun, J.-K. Encapsulation of Metal Clusters within Porous Organic Materials: From Synthesis to Catalysis Applications. Chem. Asian J. 2022, 17, e202101289. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Y.B.; Shi, P.C.; Sartin, M.M.; Lin, X.J.; Zhang, P.; Fang, H.X.; Peng, P.X.; Tian, Z.Q.; Cao, X.Y. Chaperone-like chiral cages for catalyzing enantio-selective supramolecular polymerization. Chem. Sci. 2019, 10, 8076–8082. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Su, K.Z.; El-Sayed, E.S.M.; Ju, Z.F.; Yuan, D.Q. Chiral proline-substituted porous organic cages in asymmetric organocatalysis. Chem. Sci. 2022, 13, 3582–3588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Dutasta, J.P.; Dufaud, V.; Guy, L.; Martinez, A. Sulfoxidation inside a C3-Vanadium(V) Bowl-Shaped Catalyst. ACS Catal. 2017, 7, 7340–7345. [Google Scholar] [CrossRef]
- Wang, K.; Tang, X.; Anjali, B.A.; Dong, J.; Jiang, J.; Liu, Y.; Cui, Y. Chiral Covalent Organic Cages: Structural Isomerism and Enantioselective Catalysis. J. Am. Chem. Soc. 2024, 146, 6638–6651. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.Q.; Liu, Y.T.; Wang, L.H.; Wang, Z.H.; Liu, P.B.; Gao, J.; Jiang, Y.J. Incorporation of Metals and Enzymes with Porous Imine Molecule Cages for Highly Efficient Semiheterogeneous Chemoenzymatic Catalysis. ACS Catal. 2021, 11, 5544–5553. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Kochovski, Z.; Lee, H.-C.; Lu, Y.; Zhang, H.; Zhang, J.; Sun, J.-K.; Yuan, J. Ionic organic cage-encapsulating phase-transferable metal clusters. Chem. Sci. 2019, 10, 1450–1456. [Google Scholar] [CrossRef]
- Lu, Y.-L.; Qin, Y.-H.; Zheng, S.-P.; Ruan, J.; Huang, Y.-H.; Zhang, X.-D.; Liu, C.-H.; Hu, P.; Xu, H.-S.; Su, C.-Y. Anion-Mediated Allosteric Catalysis of [2 + 2] Photocycloaddition Based on a Flexible Metallo-Amine Cage for High Diastereoselectivity. ACS Catal. 2024, 14, 94–103. [Google Scholar] [CrossRef]
- Saha, R.; Mondal, B.; Mukherjee, P.S. Molecular Cavity for Catalysis and Formation of Metal Nanoparticles for Use in Catalysis. Chem. Rev. 2022, 122, 12244–12307. [Google Scholar]
- Wang, Z.-L. Recent Advances in Catalytic Asymmetric Decarboxylative Addition Reactions. Adv. Synth. Catal. 2013, 355, 2745–2755. [Google Scholar] [CrossRef]
- Nakamura, S. Catalytic enantioselective decarboxylative reactions using organocatalysts. Org. Biomol. Chem. 2014, 12, 394–405. [Google Scholar] [CrossRef]
- Evans, D.A.; Mito, S.; Seidel, D. Scope and Mechanism of Enantioselective Michael Additions of 1,3-Dicarbonyl Compounds to Nitroalkenes Catalyzed by Nickel(II)–Diamine Complexes. J. Am. Chem. Soc. 2007, 129, 11583–11592. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Xiong, H.-Y.; Nie, J.; Hua, M.-Q.; Ma, J.-A. Biomimetic catalytic enantioselective decarboxylative aldol reaction of β-ketoacids with trifluoromethyl ketones. Chem. Commun. 2012, 48, 4308–4310. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-F.; Shen, C.; Wang, J.-R.; Tian, S.-K. A Highly Diastereoselective Decarboxylative Mannich Reaction of β-Keto Acids with Optically Active N-Sulfinyl α-Imino Esters. Org. Lett. 2012, 14, 3092–3095. [Google Scholar] [CrossRef]
- Zhong, F.; Yao, W.; Dou, X.; Lu, Y. Enantioselective Construction of 3-Hydroxy Oxindoles via Decarboxylative Addition of β-Ketoacids to Isatins. Org. Lett. 2012, 14, 4018–4021. [Google Scholar] [CrossRef]
- Zuo, J.; Liao, Y.-H.; Zhang, X.-M.; Yuan, W.-C. Organocatalyzed Enantioselective Decarboxylative Stereoablation Reaction for the Construction of 3,3′-Disubstituted Oxindoles Using β-Ketoacids and 3-Halooxindoles. J. Org. Chem. 2012, 77, 11325–11332. [Google Scholar] [CrossRef]
- Moon, H.W.; Kim, D.Y. Enantioselective decarboxylative Michael addition of β-ketoacids to nitroalkenes catalyzed by binaphthyl-derived organocatalysts. Tetrahedron Lett. 2012, 53, 6569–6572. [Google Scholar] [CrossRef]
- Yuan, H.-N.; Wang, S.; Nie, J.; Meng, W.; Yao, Q.; Ma, J.-A. Hydrogen-Bond-Directed Enantioselective Decarboxylative Mannich Reaction of β-Ketoacids with Ketimines: Application to the Synthesis of Anti-HIV Drug DPC 083. Angew. Chem. Int. Ed. 2013, 52, 3869–3873. [Google Scholar] [CrossRef]
- Kang, Y.K.; Lee, H.J.; Moon, H.W.; Kim, D.Y. Organocatalytic enantioselective decarboxylative Michael addition of b-ketoacids to a,b-unsaturated ketones. RSC Adv. 2013, 3, 1332–1335. [Google Scholar] [CrossRef]
- Suh, C.W.; Chang, C.W.; Choi, K.W.; Lim, Y.J.; Kim, D.Y. Enantioselective decarboxylative aldol addition of β-ketoacids to isatins catalyzed by binaphthyl-modified organocatalyst. Tetrahedron Lett. 2013, 54, 3651–3654. [Google Scholar] [CrossRef]
- Zhu, B.-H.; Zheng, J.-C.; Yu, C.-B.; Sun, X.-L.; Zhou, Y.-G.; Shen, Q.; Tang, Y. One-Pot Highly Diastereoselective Synthesis of cis-Vinylaziridines via the Sulfur Ylide-Mediated Aziridination and Palladium(0)-Catalyzed Isomerization. Org. Lett. 2010, 12, 504–507. [Google Scholar] [CrossRef]
- Luo, Y.; Carnell, A.J.; Lam, H.W. Enantioselective Rhodium-Catalyzed Addition of Potassium Alkenyltrifluoroborates to Cyclic Imines. Angew. Chem. Int. Ed. 2012, 51, 6762–6766. [Google Scholar] [CrossRef]
- Luo, Y.; Hepburn, H.B.; Chotsaeng, N.; Lam, H.W. Enantioselective Rhodium-Catalyzed Nucleophilic Allylation of Cyclic Imines with Allylboron Reagents. Angew. Chem. Int. Ed. 2012, 51, 8309–8313. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, C.; Tan, J.-P.; Hu, H.-L.; Chen, Y.; Ren, X.; Zhang, H.-S.; Wang, T. Highly Enantioselective Construction of Fully Substituted Stereocenters Enabled by In Situ Phosphonium-Containing Organocatalysis. ACS Catal. 2020, 10, 5698–5706. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Nie, J.; Cai, H.; Ma, J.-A. Cyclic Aldimines as Superior Electrophiles for Cu-Catalyzed Decarboxylative Mannich Reaction of β-Ketoacids with a Broad Scope and High Enantioselectivity. Org. Lett. 2014, 16, 2542–2545. [Google Scholar] [CrossRef] [PubMed]
- Qian, P.; Dai, Y.; Mei, H.; Soloshonok, V.A.; Han, J.; Pan, Y. Ni-catalyzed asymmetric decarboxylative Mannich reaction for the synthesis of β-trifluoromethyl-β-amino ketones. RSC Adv. 2015, 5, 26811–26814. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Xie, Y.-C.; Yin, L. Copper(I)-catalyzed asymmetric decarboxylative Mannich reaction enabled by acidic activation of 2H-azirines. Nat. Commun. 2019, 10, 1699. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Li, J.-S.; Nie, J.; Ma, J.-A. Organocatalytic Asymmetric Decarboxylative Mannich Reaction of β-Keto Acids with Cyclic α-Ketiminophosphonates: Access to Quaternary α-Aminophosphonates. Org. Lett. 2018, 20, 3643–3646. [Google Scholar] [CrossRef]
- Xavier, T.; Condon, S.; Pichon, C.; Le Gall, E.; Presset, M. Decarboxylative Mannich Reactions with Substituted Malonic Acid Half-Oxyesters. J. Org. Chem. 2021, 86, 5452–5462. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, L.-W.; Zhou, H.; Meng, W.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Substrate-Induced Dimerization Assembly of Chiral Macrocycle Catalysts toward Cooperative Asymmetric Catalysis. Angew. Chem. Int. Ed. 2020, 59, 2623–2627. [Google Scholar] [CrossRef]
- Tang, X.; Hou, Y.D.; Tan, X.F.; Nie, J.; Cheung, C.W.; Ma, J.A. Synergistic Catalytic Asymmetric Decarboxylative Mannich Reaction of β-Ketoacids with Acyclic α-Arylenamides. ACS Catal. 2024, 14, 9701–9707. [Google Scholar] [CrossRef]
- Luo, N.; Ao, Y.F.; Wang, D.X.; Wang, Q.Q. Exploiting Anion–π Interactions for Efficient and Selective Catalysis with Chiral Molecular Cages. Angew. Chem. Int. Ed. 2021, 60, 20650–20655. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, H.; Liu, R.; Liu, K.; Hu, X.; Huang, J.; Wang, C.; Wang, L.; Liu, Y.; Liu, G.; et al. Copper(II)-catalyzed enantioselective decarboxylative Mannich reaction coordinated by supramolecular organic amine cages. Chem. Sci. 2025, 16, 4374–4382. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Cat. | Loading (mol%) | Time | Yield b/% | ee c/% |
| 1 | RCC3: Cu(OAc)2 (1:1) | 2.5 | 6 | 77 | 86 |
| 2 | RCC3: Cu(OAc)2 (1:1) | 5 | 6 | 79 | 91 |
| 3 | RCC3: Cu(OAc)2 (1:1) | 10 | 6 | 77 | 90 |
| 4 | RCC3: Cu(OAc)2 (1:1) | 5 | 6 | 86 d (68) e | 78 d (57) e |
| 5 | RCC3: Cu(OTf)2 (1:1) | 5 | 6 | 79 | 83 |
| 6 | RCC3: CuBr (1:1) | 5 | 6 | 78 | 81 |
| 7 | RCC3: CuCl2 (1:1) | 5 | 6 | 79 | 81 |
| 8 | RCC3: CuI2 (1:1) | 5 | 6 | 77 | 89 |
| 9 | RCC3: Cu(OAc)2 (1:2) | 5 | 6 | 78 | 55 |
| 10 | RCC3: Cu(OAc)2 (1:3) | 5 | 6 | 73 | 60 |
| 11 | RCC3: Cu(OAc)2 (1:4) | 5 | 6 | 80 | 55 |
| 12 | RCC3: Cu(OAc)2 (1:5) | 5 | 6 | 78 | 55 |
| 13 | RCC3: Cu(OAc)2 (1:6) | 5 | 6 | 79 | 57 |
| 14 | RCC3: Cu(OAc)2 (1:1) | 5 | 1 | 45 | 91 |
| 15 | RCC3: Cu(OAc)2 (1:1) | 5 | 2 | 62 | 88 |
| 16 | RCC3: Cu(OAc)2 (1:1) | 5 | 4.5 | 76 | 90 |
 | ||||
|---|---|---|---|---|
| Entry | Cat. | Time | Yield b/% | ee c/% |
| 1 | RCC3: Cu(OAc)2 (1:1) | 6 | 79 | 91 |
| 2 | CC3: Cu(OAc)2 (1:1) | 12 | 77 | 25 |
| 3 | RCC3: Cu(OAc)2 (1:2) | 6 | 75 | 90 |
| 4 | RCC3: Cu(OAc)2 (1:4) | 6 | 73 | 86 |
| 5 | CC3 | 12 | 63 | 5 |
| 6 | RCC3 | 12 | 66 | 9 |
| 7 | L2 | 12 | 61 | 6 |
| 8 | L2: Cu(OAc)2 (1:1) | 12 | 86 | 47 |
| 9 | Cu(OAc)2 | 12 | 40 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Tan, C.; Yuan, L. Chiral Amine Covalent Organic Cage Lingated with Copper for Asymmetric Decarboxylative Mannich Reaction. Inorganics 2025, 13, 245. https://doi.org/10.3390/inorganics13070245
Liu K, Tan C, Yuan L. Chiral Amine Covalent Organic Cage Lingated with Copper for Asymmetric Decarboxylative Mannich Reaction. Inorganics. 2025; 13(7):245. https://doi.org/10.3390/inorganics13070245
Chicago/Turabian StyleLiu, Kaihong, Chunxia Tan, and Lingli Yuan. 2025. "Chiral Amine Covalent Organic Cage Lingated with Copper for Asymmetric Decarboxylative Mannich Reaction" Inorganics 13, no. 7: 245. https://doi.org/10.3390/inorganics13070245
APA StyleLiu, K., Tan, C., & Yuan, L. (2025). Chiral Amine Covalent Organic Cage Lingated with Copper for Asymmetric Decarboxylative Mannich Reaction. Inorganics, 13(7), 245. https://doi.org/10.3390/inorganics13070245