
Employing Molecular Dynamics Simulations to Explore the Behavior of Diphenylalanine Dipeptides in Graphene-Based Nanocomposite Systems



Abstract

1. Introduction
2. Results and Discussion
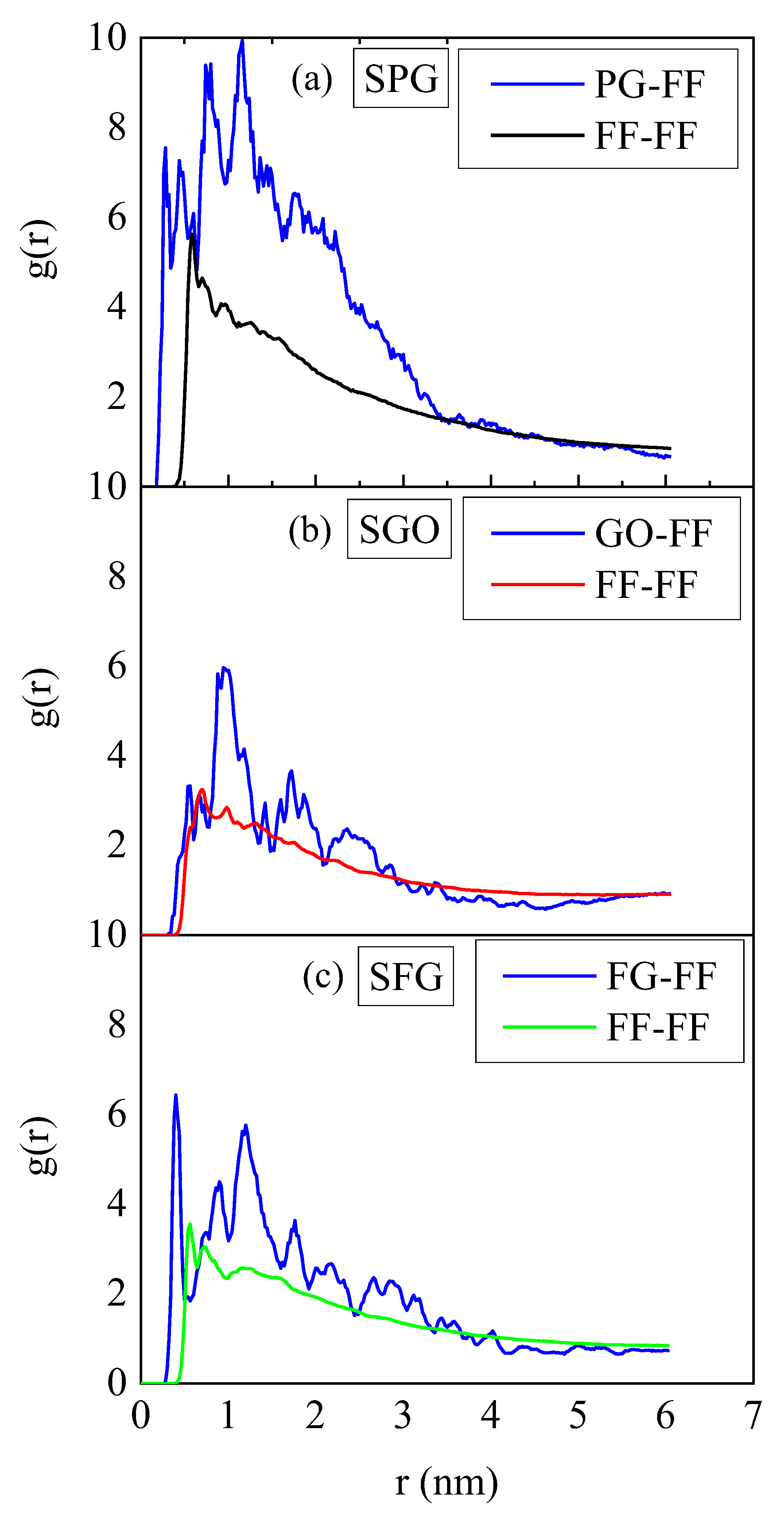
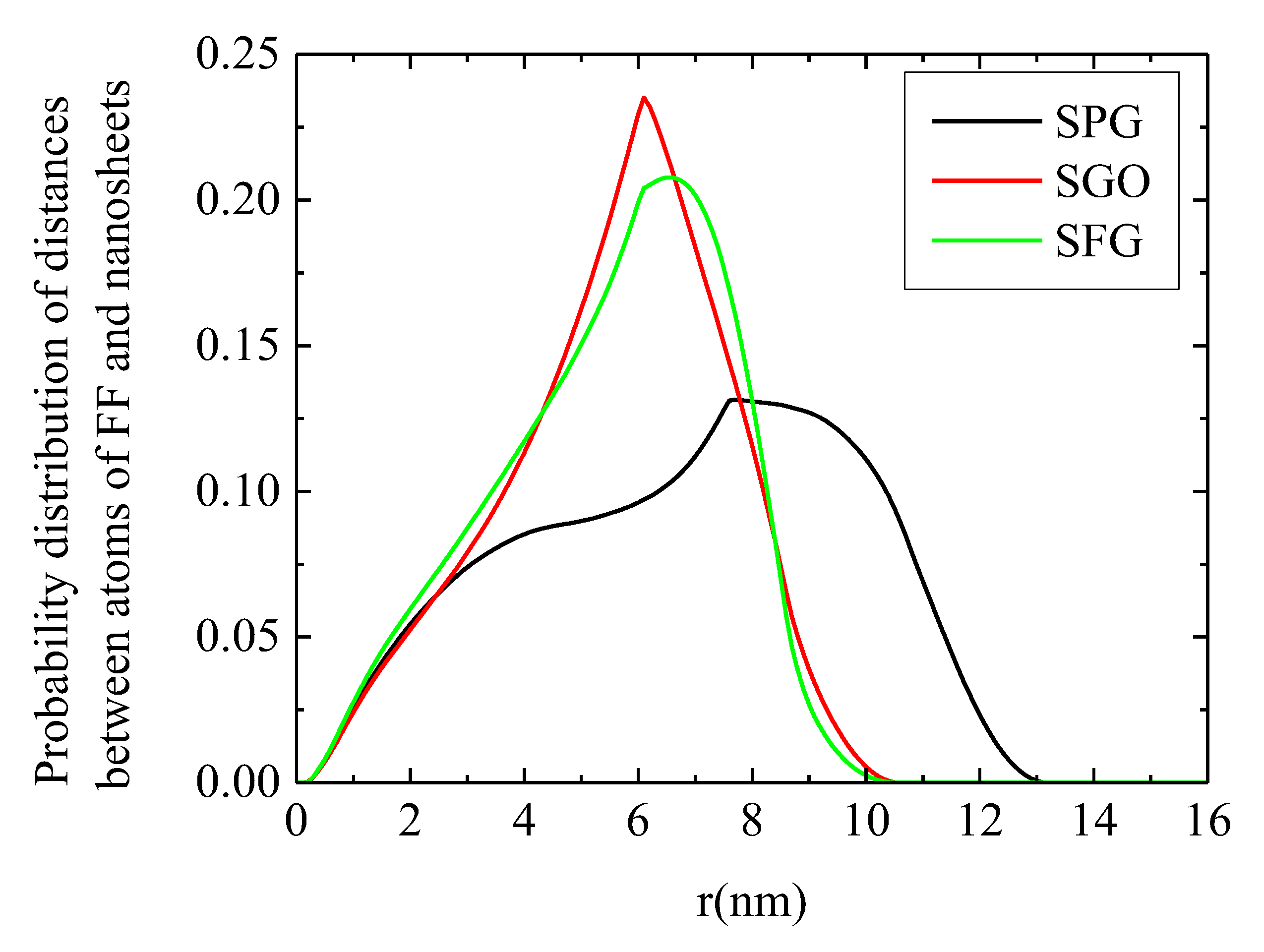
2.1. Conformational Properties
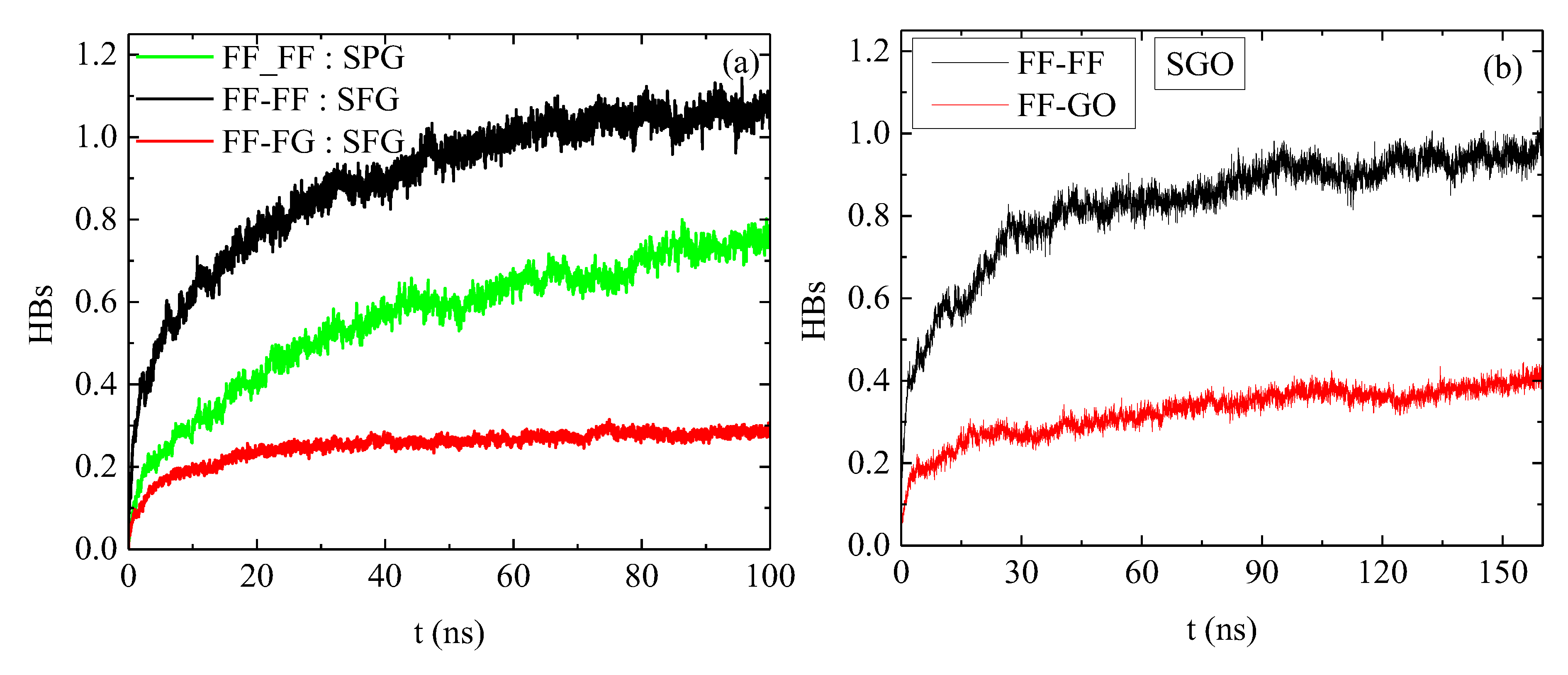
2.2. Energetics
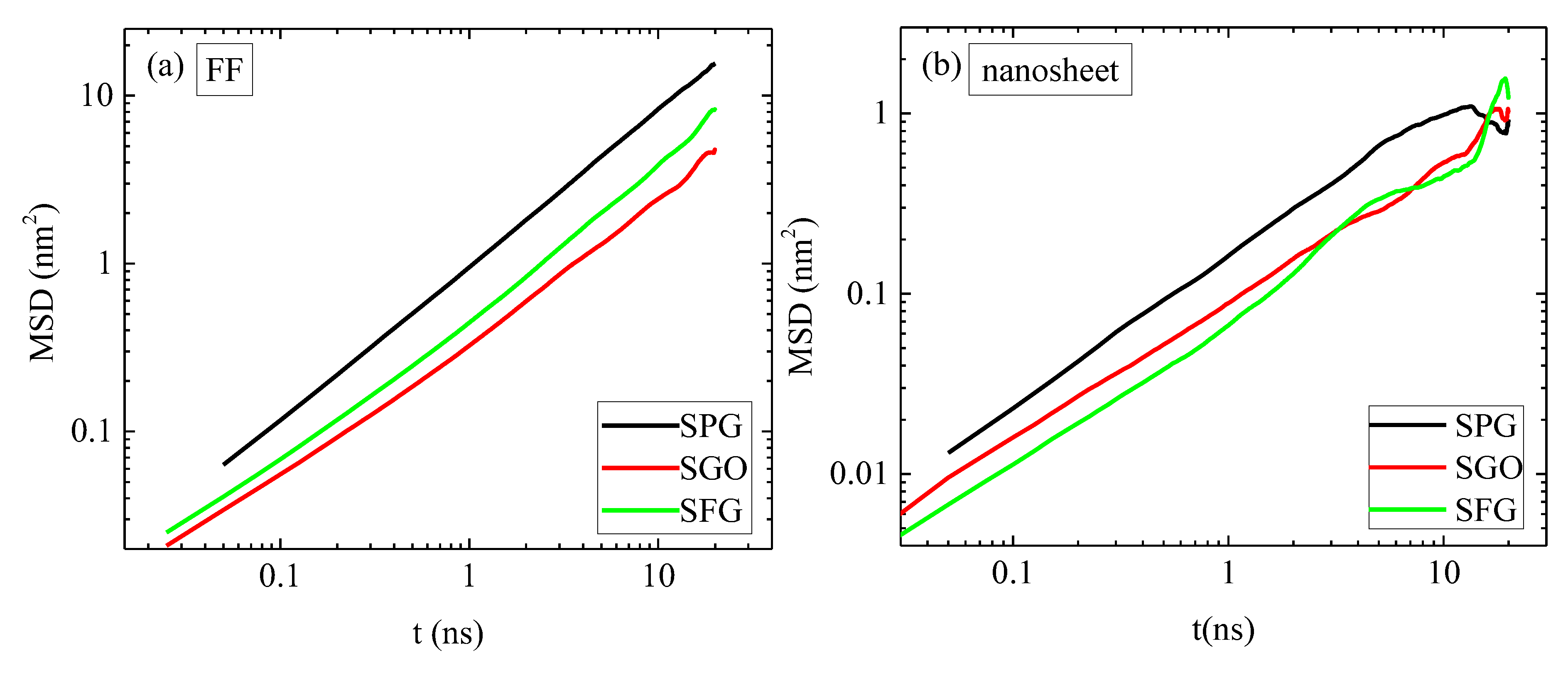
2.3. Dynamics
3. Systems and Simulation Method
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ishraq, S.; Liu, Y. Synthesis, Characterization and Bioapplications of Pristine Graphene: A Review. Univers. J. Carbon Res. 2023, 1, 1–21. [Google Scholar] [CrossRef]
- Zhao, S.; Zhao, Z.; Yang, Z.; Ke, L.L.; Kitipornchai, S.; Yang, J. Functionally Graded Graphene Reinforced Composite Structures: A Review. Eng. Struct. 2020, 210, 110339. [Google Scholar] [CrossRef]
- Dasari, B.L.; Nouri, J.M.; Brabazon, D.; Naher, S. Graphene and Derivatives—Synthesis Techniques, Properties and Their Energy Applications. Energy 2017, 140, 766–778. [Google Scholar] [CrossRef]
- Sang, M.; Shin, J.; Kim, K.; Yu, K.J. Electronic and Thermal Properties of Graphene and Recent Advances in Graphene Based Electronics Applications. Nanomaterials 2019, 9, 374. [Google Scholar] [CrossRef]
- Liang, M.; Luo, B.; Zhi, L. Application of Graphene and Graphene-Based Materials in Clean Energy-Related Devices. Int. J. Energy Res. 2009, 33, 1161–1170. [Google Scholar] [CrossRef]
- Shtein, M.; Nadiv, R.; Buzaglo, M.; Regev, O. Graphene-Based Hybrid Composites for Efficient Thermal Management of Electronic Devices. ACS Appl. Mater. Interfaces 2015, 7, 23725–23730. [Google Scholar] [CrossRef]
- Das, T.; Sharma, B.K.; Katiyar, A.K.; Ahn, J.-H. Graphene-Based Flexible and Wearable Electronics. J. Semicond. 2018, 39, 11007. [Google Scholar] [CrossRef]
- Burdanova, M.G.; Kharlamova, M.V.; Kramberger, C.; Nikitin, M.P. Applications of Pristine and Functionalized Carbon Nanotubes, Graphene, and Graphene Nanoribbons in Biomedicine. Nanomaterials 2021, 11, 3020. [Google Scholar] [CrossRef]
- Han, S.; Sun, J.; He, S.; Tang, M.; Chai, R. The Application of Graphene-Based Biomaterials in Biomedicine. Am. J. Transl. Res. 2019, 11, 3246. [Google Scholar]
- Zhang, H.; Grüner, G.; Zhao, Y. Recent Advancements of Graphene in Biomedicine. J. Mater. Chem. B 2013, 1, 2542–2567. [Google Scholar] [CrossRef]
- Ferreras, A.; Matesanz, A.; Mendizabal, J.; Artola, K.; Nisina, Y.; Acedo, P.; Jorcano, J.; Ruiz Estrada, A.; Reina, G.; Martín Jiménez, C. Light-Responsive and Antibacterial Graphenic Materials as a Holistic Approach to Tissue Engineering. ACS Nanosci. Au 2024, 4, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, W.; Yu, X.; Wang, Z.; Su, Z.; Wei, G. When Biomolecules Meet Graphene: From Molecular Level Interactions to Material Design and Applications. Nanoscale 2016, 8, 19491–19509. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Tian, J.; Xing, F.; Feng, Y. Optical Biosensor Based on Graphene and Its Derivatives for Detecting Biomolecules. Int. J. Mol. Sci. 2022, 23, 838. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Yang, H.; Wu, Y.; Wang, Z.; Yu, C.; Tang, Y.; Wang, G. Recent Advances in Biological Molecule Detection Based on a Three-Dimensional Graphene Structure. Analyst 2024, 149, 1364–1380. [Google Scholar] [CrossRef]
- Ye, J.; Fan, M.; Zhan, J.; Zhang, X.; Lu, S.; Chai, M.; Zhang, Y.; Zhao, X.; Li, S.; Zhang, D. In Silico Bioactivity Prediction of Proteins Interacting with Graphene-Based Nanomaterials Guides Rational Design of Biosensor. Talanta 2024, 277, 126397. [Google Scholar] [CrossRef]
- Kakatkar, A.; Abhilash, T.; De Alba, R.; Parpia, J.; Craighead, H. Detection of DNA and Poly-l-Lysine Using CVD Graphene-Channel FET Biosensors. Nanotechnology 2015, 26, 125502. [Google Scholar] [CrossRef]
- Texter, J. Graphene Dispersions. Curr. Opin. Colloid. Interface Sci. 2014, 19, 163–174. [Google Scholar] [CrossRef]
- Su, R.; Zhang, X. Wettability and Surface Free Energy Analyses of Monolayer Graphene. J. Therm. Sci. 2018, 27, 359–363. [Google Scholar] [CrossRef]
- Bourdo, S.E.; Al Faouri, R.; Sleezer, R.; Nima, Z.A.; Lafont, A.; Chhetri, B.P.; Benamara, M.; Martin, B.; Salamo, G.J.; Biris, A.S. Physicochemical Characteristics of Pristine and Functionalized Graphene. J. Appl. Toxicol. 2017, 37, 1288–1296. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, C.; Zhang, J. Interactions of Graphene and Graphene Oxide with Proteins and Peptides. Nanotechnol. Rev. 2013, 2, 27–45. [Google Scholar] [CrossRef]
- Margani, F.; Magrograssi, M.; Piccini, M.; Brambilla, L.; Galimberti, M.; Barbera, V. Facile Edge Functionalization of Graphene Layers with a Biosourced 2-Pyrone. ACS Sustain. Chem. Eng. 2022, 10, 4082–4093. [Google Scholar] [CrossRef]
- Bačová, P.; Rissanou, A.N.; Harmandaris, V. Edge-Functionalized Graphene as a Nanofiller: Molecular Dynamics Simulation Study. Macromolecules 2015, 48, 9024–9038. [Google Scholar] [CrossRef]
- Mathesh, M.; Liu, J.; Nam, N.D.; Lam, S.K.H.; Zheng, R.; Barrow, C.J.; Yang, W. Facile Synthesis of Graphene Oxide Hybrids Bridged by Copper Ions for Increased Conductivity. J. Mater. Chem. C 2013, 1, 3084–3090. [Google Scholar] [CrossRef]
- Trapani, G.; Caruso, V.C.L.; Cucci, L.M.; Attanasio, F.; Tabbì, G.; Forte, G.; La Mendola, D.; Satriano, C. Graphene Oxide Nanosheets Tailored with Aromatic Dipeptide Nanoassemblies for a Tuneable Interaction with Cell Membranes. Front. Bioeng. Biotechnol. 2020, 8, 427. [Google Scholar] [CrossRef]
- Baweja, L.; Balamurugan, K.; Subramanian, V.; Dhawan, A. Effect of Graphene Oxide on the Conformational Transitions of Amyloid Beta Peptide: A Molecular Dynamics Simulation Study. J. Mol. Graph. Model. 2015, 61, 175–185. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, N.; Fu, B.; Huang, F.; Liu, J. Self-Assembling Peptide-Based Nanodrug Delivery Systems. Biomater. Sci. 2019, 7, 4888–4911. [Google Scholar] [CrossRef]
- Yin, L.; Yuvienco, C.; Montclare, J.K. Protein Based Therapeutic Delivery Agents: Contemporary Developments and Challenges. Biomaterials 2017, 134, 91–116. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, L.; Zhang, X.; Zhou, Z.; Shen, X.; Hu, H.; Sun, R.; Tang, J. Advances in Self-Assembled Peptides as Drug Carriers. Pharmaceutics 2023, 15, 482. [Google Scholar] [CrossRef]
- Naskar, J.; Roy, S.; Joardar, A.; Das, S.; Banerjee, A. Self-Assembling Dipeptide-Based Nontoxic Vesicles as Carriers for Drugs and Other Biologically Important Molecules. Org. Biomol. Chem. 2011, 9, 6610–6615. [Google Scholar] [CrossRef]
- Lee, S.; Trinh, T.H.T.; Yoo, M.; Shin, J.; Lee, H.; Kim, J.; Hwang, E.; Lim, Y.B.; Ryou, C. Self-Assembling Peptides and Their Application in the Treatment of Diseases. Int. J. Mol. Sci. 2019, 20, 5850. [Google Scholar] [CrossRef]
- Saeedimasine, M.; Brandt, E.G.; Lyubartsev, A.P. Atomistic Perspective on Biomolecular Adsorption on Functionalized Carbon Nanomaterials under Ambient Conditions. J. Phys. Chem. B 2021, 125, 416–430. [Google Scholar] [CrossRef] [PubMed]
- De La Rica, R.; Matsui, H. Applications of Peptide and Protein-Based Materials in Bionanotechnology. Chem. Soc. Rev. 2010, 39, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Yan, C.; Wang, C.; Wang, C.; Cao, Y.; Zhou, Y.; Guan, P.; Hu, X.; Zhu, W.; Ding, S. Advanced Nanomaterials for Modulating Alzheimer’s Related Amyloid Aggregation. Nanoscale Adv. 2022, 5, 46–80. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kaplan, J.A.; Sun, Y.; Shieh, A.; Sun, H.-L.; Croce, C.M.; Grinstaff, M.W.; Parquette, J.R. The Self-Assembly of Anticancer Camptothecin–Dipeptide Nanotubes: A Minimalistic and High Drug Loading Approach to Increased Efficacy. Chem.-Eur. J. 2015, 21, 101–105. [Google Scholar] [CrossRef]
- Yang, Z.; Ge, C.; Liu, J.; Chong, Y.; Gu, Z.; Jimenez-Cruz, C.A.; Chai, Z.; Zhou, R. Destruction of Amyloid Fibrils by Graphene through Penetration and Extraction of Peptides. Nanoscale 2015, 7, 18725–18737. [Google Scholar] [CrossRef]
- Li, C.; Adamcik, J.; Mezzenga, R. Biodegradable Nanocomposites of Amyloid Fibrils and Graphene with Shape-Memory and Enzyme-Sensing Properties. Nat. Nanotechnol. 2012, 7, 421–427. [Google Scholar] [CrossRef]
- Xiuhua, Y.; Baoyu, L.; Liu, S.; Gu, Z.; Zhou, B.; Yang, Z. Effect of the Surface Curvature on Amyloid-β Peptide Adsorption for Graphene. RSC Adv. 2019, 9, 10094–10099. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Akhavan, O.; Ghavami, M.; Rezaee, F. Graphene Oxide Strongly Inhibits Amyloid Beta Fibrillation. Nanoscale 2012, 4, 7322–7325. [Google Scholar] [CrossRef]
- Li, Q.; Liu, L.; Zhang, S.; Xu, M.; Wang, X.; Wang, C.; Besenbacher, F.; Dong, M. Modulating Aβ33–42 Peptide Assembly by Graphene Oxide. Chem. Eur. J. 2014, 20, 7236–7240. [Google Scholar] [CrossRef]
- Jie, W.; Cao, Y.; Li, Q.; Liu, L.; Dong, M. Size Effect of Graphene Oxide on Modulating Amyloid Peptide Assembly. Chem. Eur. J. 2015, 21, 9632–9637. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Z.; Sun, Y.; Lei, J.; Wei, G. Mechanistic Insights into the Inhibition and Size Effects of Graphene Oxide Nanosheets on the Aggregation of an Amyloid-β Peptide Fragment. Nanoscale 2018, 10, 8989–8997. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Sun, Y.; Chen, Y.; Lei, J.; Wei, G. Molecular Dynamics Simulations Reveal the Mechanism of Graphene Oxide Nanosheet towards the Inhibition of Aβ1-42 Peptide Aggregation. Phys. Chem. Chem. Phys. 2019, 21, 10981–10991. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Xu, J.; Li, C.; Gao, T.; Jiang, P.; Jiang, F.-L.; Liu, Y. Insights into Mechanism of Aβ42 Fibril Growth on Surface of Graphene Oxides: Oxidative Degree Matters. Adv. Health Mater. 2021, 10, 2100436. [Google Scholar] [CrossRef] [PubMed]
- No, Y.H.; Kim, N.H.; Gnapareddy, B.; Choi, B.; Kim, Y.T.; Dugasani, S.R.; Lee, O.S.; Kim, K.H.; Ko, Y.S.; Lee, S.; et al. Nature-Inspired Construction of Two-Dimensionally Self-Assembled Peptide on Pristine Graphene. J. Phys. Chem. Lett. 2017, 8, 3734–3739. [Google Scholar] [CrossRef]
- Silva-Alves, D.A.; Camara, M.V.S.; Chaves-Neto, A.M.J.; Gester, R.; Andrade-Filho, T. Theoretical Study of the Adsorption of Diphenylalanine on Pristine Graphene. Theor. Chem. Acc. 2020, 139, 83. [Google Scholar] [CrossRef]
- Camden, A.N.; Barr, S.A.; Berry, R.J. Simulations of Peptide-Graphene Interactions in Explicit Water. J. Phys. Chem. B 2013, 117, 10691–10697. [Google Scholar] [CrossRef]
- Akdim, B.; Pachter, R.; Kim, S.S.; Naik, R.R.; Walsh, T.R.; Trohalaki, S.; Hong, G.; Kuang, Z.; Farmer, B.L. Electronic Properties of a Graphene Device with Peptide Adsorption: Insight from Simulation. ACS Appl. Mater. Interfaces 2013, 5, 7470–7477. [Google Scholar] [CrossRef]
- Cheng, Y.; Koh, L.-D.; Li, D.; Ji, B.; Zhang, Y.; Yeo, J.; Guan, G.; Han, M.-Y.; Zhang, Y.-W. Peptide–Graphene Interactions Enhance the Mechanical Properties of Silk Fibroin. ACS Appl. Mater. Interfaces 2015, 7, 21787–21796. [Google Scholar] [CrossRef]
- Huang, R.; Qi, W.; Su, R.; Zhao, J.; He, Z. Solvent and Surface Controlled Self-Assembly of Diphenylalanine Peptide: From Microtubes to Nanofibers. Soft Matter 2011, 7, 6418–6421. [Google Scholar] [CrossRef]
- Yan, X.; Zhu, P.; Li, J. Self-Assembly and Application of Diphenylalanine-Based Nanostructures. Chem. Soc. Rev. 2010, 39, 1877–1890. [Google Scholar] [CrossRef]
- Kim, J.; Han, T.H.; Kim, Y.I.; Park, J.S.; Choi, J.; Churchill, D.G.; Kim, S.O.; Ihee, H. Role of Water in Directing Diphenylalanine Assembly into Nanotubes and Nanowires. Adv. Mater. 2010, 22, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Tamamis, P.; Adler-Abramovich, L.; Reches, M.; Marshall, K.; Sikorski, P.; Serpell, L.; Gazit, E.; Archontis, G. Self-Assembly of Phenylalanine Oligopeptides: Insights from Experiments and Simulations. Biophys. J. 2009, 96, 5020–5029. [Google Scholar] [CrossRef] [PubMed]
- Rissanou, A.N.; Georgilis, E.; Kasotakis, E.; Mitraki, A.; Harmandaris, V. Effect of Solvent on the Self-Assembly of Dialanine and Diphenylalanine Peptides. J. Phys. Chem. B 2013, 117, 3962–3975. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Lake, P.T.; McCullagh, M. Initial Aggregation and Ordering Mechanism of Diphenylalanine from Microsecond All-Atom Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 12331–12341. [Google Scholar] [CrossRef]
- da Silveira, G.D.; Izabelle, C.; Saubamea, B.; Varenne, A.; d’Orlyé, F. Insights into Diphenylalanine Peptide Self-Assembled Nanostructures for Integration as Nanoplatforms in Analytical and Medical Devices. Int. J. Pharm. 2023, 648, 123559. [Google Scholar] [CrossRef]
- Jeon, J.; Mills, C.E.; Shell, M.S. Molecular Insights into Diphenylalanine Nanotube Assembly: All-Atom Simulations of Oligomerization. J. Phys. Chem. B 2013, 117, 3935–3943. [Google Scholar] [CrossRef]
- Nguyen, V.; Zhu, R.; Jenkins, K.; Yang, R. Self-Assembly of Diphenylalanine Peptide with Controlled Polarization for Power Generation. Nat. Commun. 2016, 7, 13566. [Google Scholar] [CrossRef]
- Ravikumar, A.; Natraj, V.; Verma, A.; Sivagnanam, S.; Sivalingam, Y.; Das, P.; Surya, V.J.; Han, W.H.; Liu, N. Wearable Sensors for Real-Time Physiological Monitoring Based on Self-Assembled Diphenylalanine Peptide Nanostructures. Surf. Interfaces 2023, 39, 102986. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Cheng, J.; Bhosale, S.; Aphale, A.; Macwan, I.; Patra, P.K.; Mukerji, I. UV Resonance Raman Characterization of Diphenylalanine-Graphene Nanotubes. Spectroscopy 2012, 27, 40. [Google Scholar]
- Ryan, K.; Neumayer, S.M.; Maraka, H.V.R.; Buchete, N.V.; Kholkin, A.L.; Rice, J.H.; Rodriguez, B.J. Thermal and Aqueous Stability Improvement of Graphene Oxide Enhanced Diphenylalanine Nanocomposites. Sci. Technol. Adv. Mater. 2017, 18, 172–179. [Google Scholar] [CrossRef]
- Rissanou, A.N.; Keliri, A.; Arnittali, M.; Harmandaris, V. Self-Assembly of Diphenylalanine Peptides on Graphene: Via Detailed Atomistic Simulations. Phys. Chem. Chem. Phys. 2020, 22, 27645–27657. [Google Scholar] [CrossRef] [PubMed]
- van der Spoel, D.; van Maaren, P.J.; Larsson, P.; Tîmneanu, N. Thermodynamics of Hydrogen Bonding in Hydrophilic and Hydrophobic Media. J. Phys. Chem. B 2006, 110, 4393–4398. [Google Scholar] [CrossRef] [PubMed]
- Tamamis, P.; Kasotakis, E.; Mitraki, A.; Archontis, G. Amyloid-like Self-Assembly of Peptide Sequences from the Adenovirus Fiber Shaft: Insights from Molecular Dynamics Simulations. J. Phys. Chem. B 2009, 113, 15639–15647. [Google Scholar] [CrossRef]
- Steele, W.A. The Physical Interaction of Gases with Crystalline Solids: I. Gas-Solid Energies and Properties of Isolated Adsorbed Atoms. Surf. Sci. 1973, 36, 317–352. [Google Scholar] [CrossRef]
- Bellido, E.P.; Seminario, J.M. Molecular Dynamics Simulations of Folding of Supported Graphene. J. Phys. Chem. C 2010, 114, 22472–22477. [Google Scholar] [CrossRef]
- Sinclair, R.C. Make-Graphitics-Github. GitHub. Available online: https://github.com/velocirobbie/make-graphitics (accessed on 8 January 2025).
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Walther, J.H.; Jaffe, R.; Halicioglu, T.; Koumoutsakos, P. Carbon Nanotubes in Water: Structural Characteristics and Energetics. J. Phys. Chem. B 2001, 105, 9980–9987. [Google Scholar] [CrossRef]
- Pandey, Y.N.; Brayton, A.; Burkhart, C.; Papakonstantopoulos, G.J.; Doxastakis, M. Multiscale Modeling of Polyisoprene on Graphite. J. Chem. Phys. 2014, 140, 054908. [Google Scholar] [CrossRef]
- Gao, W.; Alemany, L.B.; Ci, L.; Ajayan, P.M. New Insights into the Structure and Reduction of Graphite Oxide. Nat. Chem. 2009, 1, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Lorentz, H.A. Ueber Die Anwendung Des Satzes Vom Virial in Der Kinetischen Theorie Der Gase. Ann. Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef]
- García, A.E.; Sanbonmatsu, K.Y. Helical Stabilization by Side Chain Shielding of Backbone Hydrogen Bonds. Proc. Natl. Acad. Sci. USA 2002, 99, 2782–2787. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SPG | SGO | SFG | |
|---|---|---|---|
| d (nm) | 0.577 ± 0.002 | 0.573 ± 0.002 | 0.578 ± 0.002 |
| Rg (nm) | 0.387 ± 0.001 | 0.386 ± 0.001 | 0.388 ± 0.001 |
| SASA (nm2) | SPG | SGO | SFG |
|---|---|---|---|
| FF | 3.191 ± 0.024 | 2.446 ± 0.029 | 2.547 ± 0.021 |
| nanosheet | 73.121 ± 2.996 | 99.905 ± 0.459 | 85.499 ± 3.151 |
| Energy (kJ/mol) | SPG | SGO | SFG |
|---|---|---|---|
| Coulomb (FF-FF) | −625,472.82 ± 883.42 | −624,726.43 ± 1520.14 | −642,118.48 ± 7.23 |
| Coulomb (FF-nanosheet) | −464.06 ± 52.46 | −24,688.66 ± 1134.26 | −11,218.56 ± 962.41 |
| Coulomb (FF-Water) | −302,032.66 ± 1889.66 | −251,514.51 ± 4254.17 | −241,920.87 ± 2162.55 |
| Coulomb (nanosheet-nanosheet) | −8556.72 ± 14.13 | −34,489.96 ± 114.83 | −5593.73 ± 37.19 |
| Coulomb (nanosheet-Water) | −2364.74 ± 109.90 | −10,262.29 ± 1949.36 | −4947.82 ± 244.69 |
| L-J (FF-FF) | −27,684.42 ± 222.96 | −31,415.91 ± 602.40 | −30,318.18 ± 243.81 |
| L-J (FF-nanosheet) | −15,623.13 ± 133.15 | −19,547.94 ± 276.86 | −19,150.44 ± 196.88 |
| L-J (FF-Water) | −5137.34 ± 492.37 | −4929.80 ± 754.56 | −5367.93 ± 422.77 |
| L-J (nanosheet-nanosheet) | 34,822.62± 118.66 | −8838.69 ± 40.71 | −5223.51 ± 92.93 |
| L-J (nanosheet-Water) | −2601.64 ± 81.13 | −464.31 ± 185.02 | −1261.99 ± 104.92 |
| HBs | SPG | SGO | SFG |
|---|---|---|---|
| FF-FF/FF | 0.733 ± 0.024 | 0.923 ± 0.032 | 1.048 ± 0.027 |
| FF-nanosheet/FF | - | 0.372 ± 0.024 | 0.281 ± 0.009 |
| FF-W/FF | 8.1215 ± 0.054 | 7.257 ± 0.094 | 7.082 ± 0.066 |
| Nanosheet-W/nanosheet | - | 208.865 ± 7.746 | 55.419 ± 2.272 |
| HBs | SGO |
|---|---|
| FF-COO−/FF | 0.349 ± 0.022 |
| FF-OH/FF | 0.017 ± 0.004 |
| FF-epoxy/FF | 0.007 ± 0.003 |
| s | |||||
|---|---|---|---|---|---|
| SPG | 4128 | 110,542 | 361,554 | 0.119 | 15.5 × 15.5 × 15.5 |
| SGO | 5136 | 51,953 | 186,967 | 0.2004 | 12.5 × 12.5 × 12.5 |
| SFG | 5208 | 51,047 | 184,149 | 0.204 | 12.4 × 12.4 × 12.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markopoulou, E.; Nikolakis, P.; Savvakis, G.; Rissanou, A.N. Employing Molecular Dynamics Simulations to Explore the Behavior of Diphenylalanine Dipeptides in Graphene-Based Nanocomposite Systems. Inorganics 2025, 13, 92. https://doi.org/10.3390/inorganics13030092
Markopoulou E, Nikolakis P, Savvakis G, Rissanou AN. Employing Molecular Dynamics Simulations to Explore the Behavior of Diphenylalanine Dipeptides in Graphene-Based Nanocomposite Systems. Inorganics. 2025; 13(3):92. https://doi.org/10.3390/inorganics13030092
Chicago/Turabian StyleMarkopoulou, Elena, Panagiotis Nikolakis, Gregory Savvakis, and Anastassia N. Rissanou. 2025. "Employing Molecular Dynamics Simulations to Explore the Behavior of Diphenylalanine Dipeptides in Graphene-Based Nanocomposite Systems" Inorganics 13, no. 3: 92. https://doi.org/10.3390/inorganics13030092
APA StyleMarkopoulou, E., Nikolakis, P., Savvakis, G., & Rissanou, A. N. (2025). Employing Molecular Dynamics Simulations to Explore the Behavior of Diphenylalanine Dipeptides in Graphene-Based Nanocomposite Systems. Inorganics, 13(3), 92. https://doi.org/10.3390/inorganics13030092