Abstract
Among many metal ions in biological systems, iron plays a fundamental role. Transferrins are iron-binding glycoproteins responsible for transporting Fe3+ in vertebrate blood. Neisseria meningitidis, a Gram-negative pathogen causing meningitis, relies on iron for survival and acquires it from human transferrin (hTf) using two surface proteins, TbpA and TbpB. These proteins interact with hTf to form a ternary TbpA–TbpB–hTf complex, enabling iron capture from the host. The absence of an experimental crystal structure for this complex has hindered computational studies, a detailed understanding of Fe3+ dissociation, and designing efficient therapeutics. This study presents the first computational model of the ternary complex, its validation, and molecular dynamics simulations. Structural analyses revealed key electrostatic interactions regulating Fe3+ coordination and essential contact regions between proteins. The role of Lys359 from TbpA was investigated via QM/MM calculations by evaluating Fe3+ binding energies of isolated hTf, the ternary complex, and Lys359Ala, Lys359Arg, Lys359Asp mutant models. Results revealed that the proton transfer from Lys359 leads to disruption of Tyr517–Fe3+ coordination, facilitating iron transfer to the bacterial system. Natural bond orbital analysis confirmed this mechanism. The findings provide new molecular insight into N. meningitidis iron acquisition and identify Lys359 as a potential target for covalent inhibitor design, guiding the development of novel therapeutics against meningococcal infection.
1. Introduction
Metal ions such as Na+1, K+1, Ca+2, Mg+2, Cu+1/Cu+2, Zn+2, Fe+2/Fe+3 are involved in essential processes in living organisms such as homoeostasis, excitability, signal transduction and enzymatic reaction [1,2]. Iron plays a particularly fundamental role due to its involvement in oxygen transport, electron transfer, and enzyme activity. Given the critical roles of Fe(II)- and Fe(III)-containing proteins in diverse biological processes, disruptions in iron homeostasis can lead to a range of pathological condition [3,4]. Iron ions cannot simply pass through the phospholipid cell membrane independently and need transmembrane ion channels and transporter proteins to be transferred in and out of the cell. However, the mechanism of most ion transporters has not been elucidated at the molecular level except for the few well-studied systems. Interrupting the function of these ion channel proteins in human pathogens is among the primary strategies for treating various diseases such as meningitis, gonococcal infections, influenza, and Bartter syndrome. Meningitis is a serious disease that can be fatal if not treated promptly and effectively. Due to rising drug resistance and limited treatment options, there is an urgent need for novel therapeutics and vaccines. Neisseria meningiditis, a Gram-negative bacterium that leads to meningitis, travels in oropharynx, circulation system, and cerebrospinal fluid of the human host [5]. N. meningitidis acquires the Fe3+ ion needed for its survival and virulence directly from human transferrin protein (hTf) present in the blood plasma [6]. The role of the protein in iron storage and transport makes it an excellent target for this pathogen. For this task, N. meningitidis devises a neisserial transport system, comprising transferrin binding proteins A (TbpA) and B (TbpB). TbpA, a 100 kDa weighing TonB-dependent transporter, is a transmembrane protein located in the outer membrane of the bacterium. It is a 915 amino acid long protein and due to its barrel shape structure, the abstracted Fe3+ ion can be directly transferred into the periplasm of the bacterium [7]. While TbpA is proven to be enough for the Fe3+ transfer from human host to the bacterium, TbpB, an 85 kDa protein, increases the efficiency and rate of the transfer [8]. TbpB protein is anchored to the membrane and only approaches to TbpA during the formation of the neisserial transport system, TbpA-TbpB-hTf ternary complex. Visualization of the ternary complex is very critical to understand the iron ion transfer mechanism at the atomistic level. There are only two X-ray structures of TbpA protein, complexed with C-lobe of holo hTf (PDB ID: 3V89) and complexed with full length apo hTf (PDB ID: 3V8X) [9]. This study presents the first computational model of the ternary protein complex elucidating protein–protein interactions essential for Fe3+ transfer. Subsequent Fe3+ binding energy calculations provide chemical insights into the destabilization of Fe coordination. Moreover, the bacterium only infects humans; thus, it is a particular interest to understand the specificity of these interactions within the neisserial transport system [10].
Among computational modeling tools, molecular dynamics (MD) simulations are based on the molecular mechanics (MM) method utilizing classical Newton’s equations of motion and simulating the motions of individual particles as a function of time. MD simulations are essential to understand the dynamics of biological systems and play a major role in sampling dynamic molecular structures and time-averaged refinement. Nearly five decades after the first protein simulations [11,12], advancements in hardware, software, and algorithms have established molecular dynamics simulations as a vital tool for investigating protein structures and their dynamic behavior. These simulations offer high-resolution atomistic insights into protein conformations and interactions in aqueous environments, enabled by the significant progress in computational technologies. However, the major drawback is that they cannot model electronic structures and chemical bonds.
On the other hand, density functional theory (DFT)-based quantum mechanics (QM) calculations are superior to MM in terms of accuracy. DFT methods can model electronic structures in explicit detail, providing both qualitative and quantitative insights into enzymatic reactions which may include binding energies, non-covalent interactions, prediction of reaction energies, and activation barriers [13]. Nevertheless, the major disadvantage of employing DFT calculations is that they are computationally too expensive for large complex molecules and impossible to apply for the full enzyme/protein systems. Consequently, smaller and simplified active site models are generally constructed to apply DFT calculations, which is denoted as quantum cluster approach [14,15,16,17,18,19,20]. However, this may raise the inevitable question of how realistic this active site model can be without the three-dimensional presence and constraints of all surrounding residues. To overcome this problem, hybrid approaches using quantum mechanics (QM) and molecular mechanics (MM), such as our own N-layered integrated molecular orbital and molecular mechanics (ONIOM), can be used in the calculations [21,22,23,24,25,26]. Atoms directly involved in chemical changes can be modeled by DFT methods, and the surrounding amino acid residues can be treated by molecular mechanics methods to include non-covalent interactions which provides a realistic scheme around the active sites [27]. The same approach was employed in this work on a large QM/MM cluster model to explore the trends in Fe3+ release from the wild type TbpA-TbpB-hTf ternary complex and its mutant models.
Majority of studies in the literature on Fe3+ release mechanism focus on hTf, which stores and carries Fe3+ ions through its N-lobe and C-lobe domains. The Fe3+ ion carried by serum hTf is transported into the intracellular space by binding of transferrin receptors (TfR) (receptor-mediated endocytosis) [28,29]. Since two Fe3+ ions are strongly bound to Tf in human serum at pH~7.4, the mechanism of iron release is of interest. The pH drop in the endosome during iron uptake into the cell (pH~5.6) facilitates iron release [30]. In serum hTf, iron coordination in both lobes (N-lobe and C-lobe) is mediated by similar residues: one aspartic acid, two tyrosines, and one histidine. The octahedral coordination of iron in both lobes is complemented by a synergistic anion identified as carbonate (CO32−) or bicarbonate (HCO3−) (Figure 1) [31].
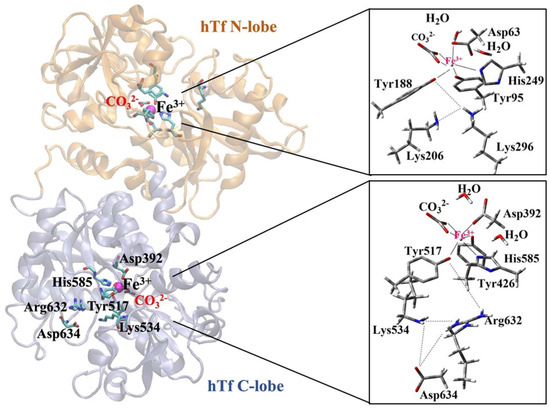
Figure 1.
Coordination shell of Fe3+ ion in serum hTf N-lobe and C-lobe.
Although the iron-binding ligands in each lobe of hTf are fully conserved, the two lobes differ in their response to pH [32], anions [32], TfR [33], and the conformation of the other lobe [34]. This is largely due to differences in “second shell” residues (residues that do not coordinate Fe3+ directly but form a complex hydrogen bond network with primary iron ligands).
Recent DFT calculations confirmed that not only the residues forming the Fe3+ coordination sphere but also those in the second shell are essential for the release mechanism [35]. Among them, the amino acids Lys206 and Lys296, the so-called “dilysine triggers” shown in Figure 1 for the N-lobe, play a key role in Fe3+ ion release [35,36,37,38]. Although a similar situation is expected in the C-lobe, fewer studies have been conducted on C-lobe. Dewan et al. identified a pH-sensitive triad of residues (Lys534-Asp634-Arg632) in the C-lobe equivalent to the dilysin trigger in the N-lobe [39]. These triad residues interact with each other via H-bonds, but iron release from the hTf C-lobe is slower than from the N-lobe and occurs at lower pH (pH = ~4.8) [30]. According to the hypothesis that the negatively charged Asp634 in the Lys534-Asp634-Arg632 triad of the C-lobe stabilizes the positive charge of residues Lys534 and Arg632, point mutation experiments in the hTf protein revealed prolonged Fe3+ ion release in vitro for Lys534Ala, Arg632Ala, and Asp634Ala [30]. These results highlight the crucial role of the triad residues in the mechanism through which the C-lobe releases the Fe3+ ion.
Computational studies in the literature have mainly focused on the N-lobe [40], as it is the primary site of iron release from serum hTf. Using the quantum cluster model of the first coordination shell, Sakajiri et al. computed the binding affinities of several metals to the N-lobe of hTf employing the DFT B3LYP/6-31G(d,p) approach [41]. Using the complete hTf protein bound to Al3+ and Fe3+ at the N-lobe, Mujika et al. conducted QM/MM MD simulations in neutral and acidic environments and suggested that His249 protonation plays a role in metal release [42]. The same team then performed MD simulations with the CHARMM force field and observed that the dilysin trigger alone did not lead to a conformational change and that the unfolding could only occur with the protonation of Tyr188 [36]. According to another study employing BP86/TZVP calculations on the quantum cluster model, metal release is only feasible at acidic pH since Tyr188 is strongly bound to Fe3+ at physiological pH and must be protonated (acidic pH) to weaken this bond [43]. A very new DFT and metadynamics calculations on the N-lobe proposed proton-coupled electron transfer modulating the iron release at physiological pH [44].
Recently the C-lobe of hTf has also started to receive attention. Using MD simulations, Abdizadeh et al. examined the conformational dynamics and crosstalk of the N- and C-lobes in the hTf [45,46,47]. The results showed that the dissociation process is similar in both lobes, but the iron dissociation in the N-lobe is thermodynamically more favorable than in the C-lobe. It was proposed that protonation of Tyr188 in the N-lobe could occur by proton transfer from Lys206 (protonated in acidic medium) of dilysine, whereas protonation of Tyr517 in the C-lobe could take place by proton transfer from the triad Lys534, Arg632, Asp634. Furthermore, computational studies on iron transporter proteins were reviewed and collectively described by Abdizadeh et al. [48].
As outlined above, the published work in the literature mainly concentrated on understanding the function of hTf in iron transportation in human serum and its cellular uptake. On the other hand, in the present study, we aim to explore how N. meningitidis binds to hTf and removes the iron ion from the C-lobe. N. meningitidis is known to bind to hTf from the C-lobe [45]. The published literature suggests that certain amino acid residues may play a critical role in iron release from the C-lobe as the pH decreases in the endosome. However, the chemical mechanism and the specific role of these residues in bacterial iron uptake at physiological pH remain unclear. Despite several experimental studies in the literature, molecular-level investigations using computational methods are very limited [6,45].
Noinaj et al. elucidated the structures of TbpA-hTf and TbpB-hTf binary complexes from the bacteria N. meningitidis by X-ray scattering and provided valuable clues for the mechanism of Fe3+ ion release, which remains unclear at neutral pH [9,49]. Upon binding of TbpA to hTf, the C-lobe of hTf assumes a half-open conformation, and the protonated Lys359 (helix finger of loop 3) in TbpA gets close to Asp634 in hTf (Figure 2). Thus, it was proposed that the charge balancing provided by Asp634 to the two basic amino acids in the Asp634-Arg632-Lys534 triad is disrupted (Figure 2) [6]. The resulting charge repulsion deforms the C-lobe, facilitating the release of Fe3+ ion. Utilizing MD simulations on TbpA, our recent study hypothesized that this disruption would affect the hydrogen bonding between Arg632 and Tyr517 (bound to Fe3+), leading to the relaxation of the Fe3+ coordination [50].
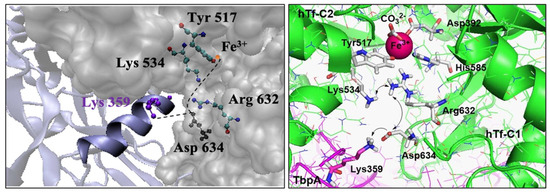
Figure 2.
Helix finger Lys359 at the interface of TbpA-hTF (left) and charge–charge repulsion proposed in the C-lobe of TbpA-hTf (right).
In this study, the ternary TbpA-TbpB-hTf protein complex was constructed and subjected to molecular dynamics simulations for the first time in the literature. The aim of these simulations was to obtain equilibrated ternary protein complex structure, which will be utilized in subsequent QM/MM ONIOM optimizations to calculate Fe3+ binding energies for the C-lobe. A parallel analysis was performed on hTf alone for comparison, aiming to confirm the expected reduction in Fe binding affinity upon formation of the TbpA–TbpB–hTf ternary complex and thus, to estimate the extent to which Fe3+ relaxation is facilitated. Our main focus is to explore the role of Lys359 which is the key residue from TbpA. Therefore, the Fe3+ binding (or dissociation) energies of the TbpA–TbpB–hTf complex and its Lys359Ala, Lys359Arg, and Lys359Asp mutant models were computed to systematically evaluate the structural and energetic consequences of neutral and charged substitutions at position 359 on iron coordination stability. This comprehensive approach aims to provide computational insights into how N. meningitidis initiates Fe3+ dissociation from hTf under physiological pH conditions.
2. Materials and Methods
2.1. MD Simulations
Modeling and simulations of ternary protein complex: The 3-D model of the ternary protein complex was generated by precisely aligning three available crystal structures. First, the crystal structure of the TbpA-hTf C-lobe complex (PDB ID: 3V89) was aligned with crystal structure of the full length holo hTf protein (PDB ID: 3V83). Upon addition of missing residues and deletion of hTf C-lobe, the full length TbpA-hTf complex was obtained. Then, this complex structure was aligned with the crystal structure of TbpB-hTf complex (PDB ID: 3VE1). After the addition of missing TbpB residues and deletion of hTf protein from TbpB-hTf complex, the 3-D structure of the full length TbpA-TbpB-hTf complex was obtained (Figure 3). The accuracy of this complex structure was checked at the ProCheck [51,52] and ProSA [53,54] servers, which validated the structure with decent Z-scores and Ramachandran plot values.
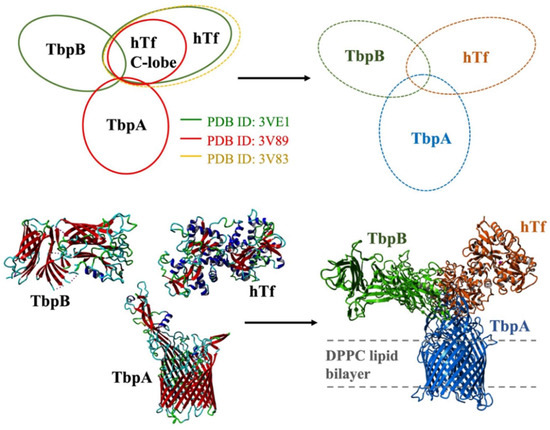
Figure 3.
Modeling of the ternary TbpA-TbpB-hTf complex through alignment of the crystal structures and the structure of model system embedded in lipid bilayer.
Then, the ternary complex model was embedded into lipid bilayer from transmembrane TbpA protein by a Perl script, inflating and packing the lipid molecules around the protein structure. The position of lipid bilayer was obtained from the OPM server [55] utilized on the crystal structure (PDB ID: 3V89) and it was projected onto the ternary complex. The modeling of the lipid bilayer comprising DPPC model lipid molecules was described in detail below.
This ternary protein complex embedded in the lipid bilayer was then subjected to classical MD simulations. All MD simulations were performed with GROMACS 5.1.4 software [56], deploying GROMOS 53A6 force field [57]. Protonation states of titratable residues were predicted at a physiological pH of 7.0 on the PROPKA server [58]. Residues in the Fe coordination shell (Tyr426, Tyr517, His585, Asp392 for C-lobe) were set to their deprotonated (negatively charged) states, in line with the previous QM studies [35,43]. The Lys359 residue was maintained in its protonated form to mimic its state in TbpA [50]. Parameters for CO32− and Fe3+ ions were obtained as described in the next paragraph. Periodic boundary conditions were applied at all dimensions of the cubic simulation box, and the box was filled with water, TIP4P model, and Na+, Cl− ions to neutralize the total charge of the system. Steepest descent method up to 50,000 steps was applied to minimize the structure, followed by 500 ps long NVT and NPT equilibration steps. The pressure was raised to 1.0 bar and the temperature was set to 323 K, which was above the phase transition temperature of DPPC [59] with a coupling constant of 0.1 ps. SETTLE algorithm [60] was employed to fix the water atom bond lengths/angles and LINCS algorithm to restrain the bond distance of proteins [61]. Long-range electrostatic interactions were treated with Particle-Mesh Ewald (PME) method [62]. Leap-Frog algorithm with 2 ps time steps were utilized for system iteration. MD simulations were run for 150 nano seconds (ns). All the analysis were performed with tools integrated into GROMACS 5.1.4 software.
Modeling and simulations of the sole hTf: One of the aims of the current study is to compare the ternary complex with the isolated hTf in terms of structural features and the Fe binding/release ability. Thus, parallel to ternary protein complex MD simulations, sole hTf protein MD simulations were also run. In order to obtain the initial structure of the full hTf protein, an X-ray crystal structure (PDB ID: 3V83, 2.1 Å) containing two Fe3+ ions and lacking amino acids at the N- and C-termini was aligned with an X-ray crystal structure of hTf complex with the TbpB protein, which has complete N- and C-termini (PDB ID: 3VE1, 2.9 Å). In this structure, missing residues (Val1, Glu89, Glu333, Lys414, Ser415, Asp416, Asn417, Ser437, Asp438, Tyr440, Asn472, Asp565, Thr567, Lys569, Asn611, Val612, Tyr613, Asp614, Ser616, Glu625, Glu653, Lys657, and Arg677 amino acids) were completed in the YASARA Structure v.21.12.19 program [63,64]. CO32− ions coordinated with Fe3+ ions were also retained in the system. Parameters for CO32− ion, located at the Fe3+ binding site in hTf protein, was obtained from the PRODRG server [65]. The parameters of the iron (Fe3+) atom were not defined in the force field used. First, the 12-6 Lennard–Jones (LJ) parameters were calculated using the epsilon values of the iron atom in the literature [66] and placed into the force field employed. The hTf protein was then placed in a cubic box measuring 15.0 × 15.0 × 15.0 nm, filled with water, and 36 sodium and 30 chloride ions were added to neutralize the system. The same algorithms and parameters were utilized as in the ternary protein complex simulations, discussed in detail above. Simulations were run for 150 ns.
Modeling of the lipid membrane: Parameters for 128 DPPC molecules were obtained from the research article by Tieleman and colleagues [67]. As the size of the ternary protein structure was larger than the lipid structure, this already equilibrated lipid model structure was extended to a model containing 768 lipid molecules. This model was subjected to MD simulations to obtain equilibrated, larger lipid structure. MD simulations for the lipid molecules were performed utilizing GROMOS 53A6 force field and simulation parameters were kept the same from the study by Tieleman and Berendsen to obtain a 128-molecule-containing system at 323 K and 1 bar [68]. This extended lipid model was placed into a box with dimensions 6.63 × 13.10 × 6.77 nm. This box was filled with SPC model water molecules and 20 ns long classical MD simulation was performed. The most representative lipid bilayer structure from the last 10 ns of the simulations was obtained. The area per lipid value was found out to be 63.9 Å2, within the experimental values (62.9–64.0) [59]. This bilayer structure was employed for the MD simulations of ternary protein complex.
2.2. QM/MM Calculations
All QM and QM/MM calculations were performed with Gaussian 09 [69]. When selecting a DFT method for the QM region, achieving a reasonable balance between computational cost and accuracy is crucial [70]. The M06 functional was proposed in the literature for both main group elements and transition metals as well as Fe. Due to its broad applicability, it is recommended for studying energy changes in reactions where both organic and transition metal bonds are altered simultaneously [71,72]. Furthermore, using the 6–31G(d,p) basis set with the quantum cluster approach, Sakajiri et al. [41] calculated the binding energies of various metals to hTf, including Fe3+. They obtained a very good correlation with the experimental binding constants, revealing the good performance of 6–31G(d,p). Moreover, since the size of the protein structures to be optimized limits the use of larger basis sets, the performance of the 6–31G(d,p) basis set with the M06 functional, which can also take into account dispersion interactions, was tested on a model reaction.
The experimental −logK value for the model hydrolysis reaction of octahedral Fe3+ in Equation (1) was reported as 2.2 at room temperature [73]. The conversion of this data to the experimental Gibbs free energy of the reaction yielded a value of 3 kcal/mol [74].
As a benchmark study, we calculated the Gibbs free energy change for this reaction in aqueous medium (as dielectric continuum) using the M06/6-31G(d,p), M06/6-31+G(d,p), and M06/6-311+G(d,p) methods which gave rise to Gibbs free energy changes of 3.44 kcal/mol, 5.25 kcal/mol, and 4.42 kcal/mol, respectively. Thus, M06/6-31G(d,p) was chosen as the appropriate method, which provided excellent agreement with the experimental data compared to other methods with high computational costs. Therefore, the M06/6-31G(d,p) method was used for the QM calculations in this study.
The average structure obtained from the 75–125 ns timeframe of classical MD simulations of the ternary holo protein complex TbpA-TbpB-hTf at physiological pH conditions was used as the initial structure of the QM/MM system (Figure 4). In this structure, Lys359 of TbpA was protonated (positively charged), mimicking the situation in bacterial TbpA where pKa of Lys359 was calculated as 10.2 [50]. Spin multiplicity of Fe3+ was chosen as sextet since it is known that, in transferrin protein, Fe3+ is high-spin (sextet) and the energy of high-spin Fe3+ is lower than that of low-spin Fe3+ [75].
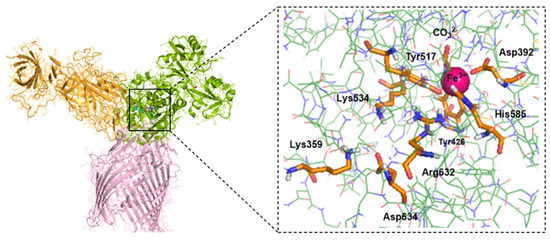
Figure 4.
Average MD structure of the TbpA-TbpB-hTf ternary complex and enlarged C-lobe selected for QM/MM calculations. Atoms in the QM region (Fe3+ octahedral coordination shell and the release channel) are shown by the tube model, while the remaining parts in the MM region are shown by the wire model.
QM/MM calculations were employed using ONIOM technique [24]. Amino acid residues of primary importance for Fe3+ ion release were included in the QM region and calculated at the M06/6-31G(d,p) level. The rest of the protein was treated as the MM region with AMBER [76] force field. Nevertheless, the large size of the ternary protein complex precluded geometry optimization of the entire structure. To maintain a reasonable computational cost, the system was truncated to include only atoms within 12 Å of the Fe3+ center (Figure 4) as well as the release channel. This represents a suitable size to mimic the steric and electrostatic surrounding of the QM region [77,78]. The obtained cluster structure consisted of the 65 amino acids (1057 atoms) as listed in Supplementary Information (SI) Table S1, carbonate, Fe, and 6 water molecules. The incomplete valances of the amino acid residues remaining in the final truncated part were complemented with hydrogen atoms. Thus, the QM region consisted of Asp392, Tyr426, Tyr517, His585, Fe3+, CO32−, and two water molecules in the Fe3+ coordination shell, and Lys534, Arg632, Asp634, Lys359 in the Fe3+ release channel (158 atoms in total) (Table S1 of SI). Among them, Arg632 and Lys359 were protonated (positively charged) while the residues in coordination with Fe (Asp392, Tyr426, Tyr517, His585) and CO32− were deprotonated (negatively charged) as in the previous computational studies [35,43] (Table S2 of SI). Second and third shell Fe coordination residues were included in MM region. This initial protein structure was used to construct Lys359Ala, Lys359Arg, and Lys359Asp mutant protein models by replacing the Lys side chain of the wild type with Ala, Arg, and Asp manually.
The QM/MM model of the isolated hTf protein was constructed using the average structure derived from MD simulations, following the same procedure applied to the wild-type TbpA–TbpB–hTf complex. However, this model included only the amino acid residues from the hTf unit present in the ternary complex. As a result, the Lys359 residue from the TbpA protein was absent in this model. The corresponding apo protein structures were prepared by removing the Fe3+ ion from the holo proteins.
Protein setup was performed following the previous protocols [16,17,27,35,77,78]. To avoid structural deviations from the real structure during QM/MM geometry optimizations, the coordinates of the α-carbon atom at the backbone of the outer-shell amino acids in the MM region were fixed (Table S1 in SI). Geometrical parameters of the remaining atoms in the MM region and all atoms in the QM region were fully optimized. Details of the protein setup were given in SI. Hydrogens were used as the link atoms at the QM/MM intersection points. Geometry optimizations were applied to all structures and their energies were calculated with ONIOM (M06/6-31G(d,p):Amber) QM/MM method. Each structure was verified as a true minimum on the potential energy surface, confirmed by the presence of all positive vibrational frequencies in the normal mode analysis. The binding energy of Fe3+ to hTf and the ternary protein complexes were calculated by the energy change in Equations (2)–(4). Thus, aqueous de-solvation of Fe3+(H2O)6 was taken into account with six explicit water molecules.
2.3. Natural Bond Orbital Analysis
Natural bond orbital (NBO) second-order perturbation theory analysis [79] was employed on the QM region of the optimized structures with single-point energy calculations at the M06/6-31G(d,p) level. Stabilization energies E(2) due to donor–acceptor interactions were calculated from NBO Fock matrix.
3. Results and Discussions
3.1. Results from MD Simulations
The MD simulations were run for 150 ns for the ternary complex, embedded in the lipid bilayer and sole hTf protein. The RMSD, RMSF, and radius of gyration graphs were provided in SI Figures S1–S4. Here, it should be noted that there was a slight increase in RMSD after 130 ns for the ternary complex. Thus, two clusters, one from the 75–125 ns timeframe and one from the 125–150 ns timeframe, were obtained. The structures were superimposed and aligned with an RMSD of 1.29 Å, showing no significant structural changes at the protein–protein interfaces or the Fe3+ coordination site. Therefore, all the analyses were performed on the cluster average structure obtained from the 75–125 ns timeframe of the simulations. On the other hand, The RMSD plot of the isolated hTf protein indicated that structural equilibration was achieved after approximately 100 ns of simulation. Thus, the cluster structure was obtained from the last 50 ns timeframe.
To validate both the accuracy of constructed ternary protein complex structure and results from MD simulations, a series of analyses were performed. The superposition of TbpA protein in ternary complex with its corresponding counterpart from the double-protein complex crystal structure (PDB ID: 3V89) provided a root mean square deviation (RMSD) value of 0.43 Å. Likewise, the superposition of TbpB and hTf proteins with their counterparts from the crystal structure (PDB ID: 3VE1) resulted in RMSD values of 0.03 Å and 0.22 Å, respectively. Later, the initial ternary complex structure was subjected to Z-score, energy per residue, and Ramachandran analyses in ProSA [53,54] and ProCheck [51,52] servers. These analyses were performed for pre- and post-MD simulation structures validating both the rationale for the initial construct and MD simulations parameters. Z-scores calculated in the ProSA server indicate the quality of protein models. In the plot, Z-scores from crystal structures in the literature with similar length were provided with a shaded region and the Z-score of the query protein (black point) is expected to fall within this range. Z-score for TbpA protein in the crystal structure (PDB ID: 3V89) fell outside of the shaded area, which stayed almost at the same point for the TbpA protein in the initial ternary protein construct (pre-MD) and in the equilibrated average ternary protein complex structure after MD simulations (post-MD) (Figure S5 in SI). The Z-score did not change significantly, and was only displaced by −0.45 and +0.34. The same comparison for the TbpB protein was made and the Z-score for the TbpB protein in the pre-MD structure was found to decrease and fell into a better shaded range. After MD simulations, the Z-score became −4.9, very similar to that of TbpB in the crystal structure (PDB ID:3VE1) (Figure S6 in SI). The Z-score of hTf protein remained similar throughout the modeling and MD simulations. The pre-MD hTf protein in the ternary complex provided a Z-score of −14.1, which is only higher than the Z-score of hTf in the double-protein complex (PDB ID:3VE1) by 0.01. After MD simulations, the Z-score increased by 1.05 and became −13.05. The relative position of the Z-score in the plot did not change; it was still close to the shaded area (Figure S7 in SI). The ProSA server also provides energies per residue, in which positive values mostly indicate problematic or poor-quality fragments in the model. The values for TbpA protein in the ternary complex remained mostly in the positive region for the initial pre-MD model, like the crystal structure (Figure S5 in SI). Slight decrease was observed for the C-terminal region peak observed in the crystal structure. However, in the post-MD structure, most of the values came closer to the zero line, with a huge decrease in the positive region of the C-terminal as well. These results clearly indicated an improvement and relaxation of the protein structure. In the TbpB protein case, values for the pre-MD ternary complex structure clearly diminished in comparison to these from the crystal structure (Figure S6 in SI). Two high peaks in the C-terminal region disappeared and most of the values became negative. In the post-MD structure, there was an increase in the values in general; however, when compared to the initial crystal structure, they remained lower. Moreover, two positive high peaks in the C-terminal turned into one small peak. For the hTf protein, there was almost no difference between the values for hTf protein from the crystal double-protein complex and the initial pre-MD ternary complex (Figure S7 in SI). Although there was a slight increase in general, except for a few residues in the C-terminal, all the residues possessed negative energies. ProSA analyses indicated that the modeling of the ternary protein complex and the equilibrated average structure obtained from MD simulations did not reveal any poor quality or faulty modeling. Nevertheless, the quality and the accuracy of the models and the post-MD structure was also assessed through Ramachandran analyses by the ProCheck server. The percentage of amino acids in the disallowed regions were compared for TbpA-hTf, TbpB-hTf, and TbpA-TbpB protein complexes in the crystal and in pre-MD and post-MD structures. There was only a 0.2% increase for amino acids in the disallowed region for the TbpA-hTf complex between the crystal (PDBID:3V89) and post-MD structures (Figure 5).
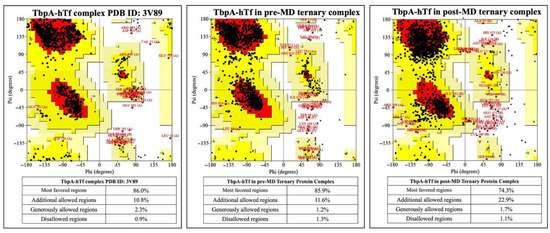
Figure 5.
Ramachandran plots for TbpA and hTf proteins in the crystal structure and in the ternary complexes.
1.0% increment was observed for the TbpB-hTf protein complex upon modeling the ternary protein complex, followed by MD simulations (Figure 6).
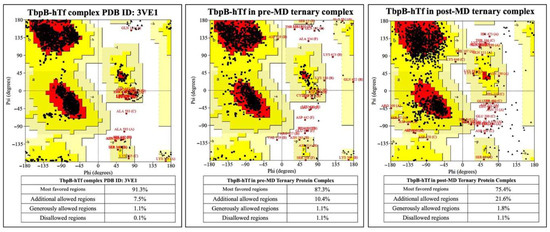
Figure 6.
Ramachandran plots for TbpB and hTf proteins in the crystal structure and in the ternary complexes.
The comparison for the TbpA-TbpB protein complex was made only between pre- and post-MD structures as there is no crystal structure of this complex available. There was 0.6% decrement for the amino acids in the disallowed region after MD simulations (Figure 7).
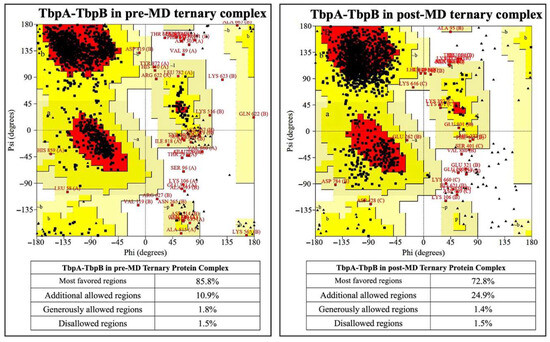
Figure 7.
Ramachandran plots for TbpA and TbpB proteins in the crystal structure and in the ternary complexes.
The percentages of amino acids in the disallowed regions vary between 0.9 and 1.1% in the post-MD simulation structures. This value is below the accepted threshold of 2.0% for good quality structure [80]. These analyses also validated the accuracy of the models and the MD simulation parameters.
It is noteworthy that, here, the TbpA-TbpB-hTf ternary protein complex was modeled and subjected to MD simulations for the first time in the literature. Thus, one of the very important findings of the study was to elucidate protein–protein interactions formed at the TbpA-hTf, TbpB-hTf and TbpA-TbpB interfaces. The interacting residues were obtained from the cluster structure at Protein Protein Interaction Server [81]. These residues were provided in Tables S3–S5 in SI. Targeting the interface residues of hTf with rationally designed ligands to block bacterial receptor binding represents a promising strategy for the development of new therapeutics.
Root mean square fluctuation (RMSF) plots of TbpA, TbpB and hTf moieties of the ternary complex were given in SI Figure S3, together with the view of the fluctuating loop regions. RMSF graph of isolated hTf was also shown (Figure S2 in SI) for comparison. The fluctuation of water-exposed amino acids were higher than those buried, as expected. The fluctuations observed in the Ser362–Lys365, Asp416–Ala424, and Cys543–asp558 loops of isolated hTf were largely reduced in the ternary complex. As shown in SI Tables S3 and S4, these loops correspond to interface regions between hTf and TbpA/TbpB, where intermolecular interactions appear to dampen their mobility.
The radius of gyration (Rg) of the ternary system (SI Figure S4) was measured as 4.81 nm at the beginning of the simulation and was observed to stabilize around 4.67 nm after 90 ns. Therefore, the ternary system became more compact during the MD simulation compared to its initial structure. Rg analysis of hTf revealed slight decrease at the beginning of the simulation, but a subsequent slight increase to retain the overall compactness of the protein.
Another important finding of the ternary complex is that six Fe3+ coordination bonds remained intact throughout the simulations, making the averaged MD structure a suitable starting point for the subsequent QM/MM optimizations. Fluctuations in Fe3+ coordination distances of first-shell residues in C-lobe were given in Figure S8. In the averaged structure, six coordination distances varied from 1.92 Å (CO32−) to 2.12 Å (His585), indicating stable coordination around Fe (Table S6).
Moreover, our modeled and equilibrated ternary protein complex structure clearly revealed the electrostatic network, which is believed to play a significant role in the disruption of Fe3+ from its coordination shell [6]. Noinaj et al. also highlighted the significance of protonated Lys359 in TbpA protein, depicted as the helix finger (Figure 2), and its probable involvement in Fe3+ transfer in the TbpA-hTf double-protein complex crystal structure that they identified [6]. In the current study, we shed light on the whole electrostatic network comprising TbpA residue Lys359 and hTf residues Asp634, Lys534, and Arg632. Initial formation of TbpA Lys359 and hTf Asp634 H-bond brought Lys534 and Arg632 in a H-bonding conformation elongating up to Tyr517, which coordinated to Fe3+ directly (Figure 8). As seen in Figure 8, insertion of TbpA Lys359 into hTf domain of the ternary protein complex brought Arg632 and Tyr517 in a strong H-bond distance. This might have been the near-attack conformation for the proton transfer to Tyr517, which, in turn, could disrupt the Tyr517–Fe3+ coordination. The dramatic turn of events would finally free the Fe3+ ion from its coordination shell. This hypothesis was supported by calculating the Fe3+ binding energies as discussed in the QM/MM section.
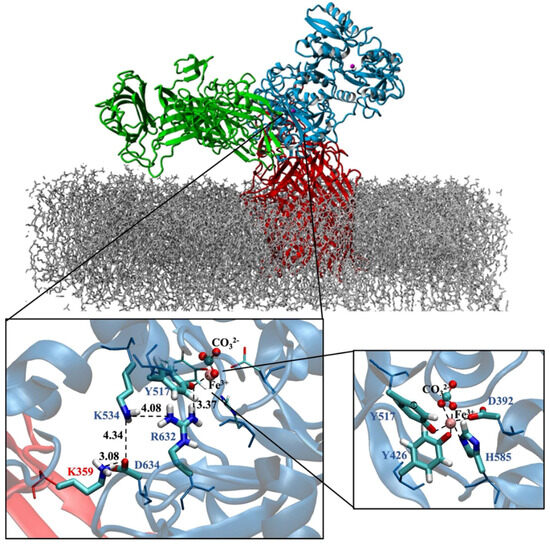
Figure 8.
TbpA-TbpB-hTf ternary protein complex (the most representative structure of the equilibrated part of the trajectory) and zoom in view of the electrostatic network (Distances are in Å). Fe3+ ion coordination shell is provided on the bottom right corner.
Meanwhile, 150 ns long MD simulations for sole hTf protein provided the equilibrated structure of the protein. There was no substantial difference between the pre- and post-MD structures. Both Fe3+ ions remained in their coordination sites (SI, Figure S9) and the electrostatic network, mentioned above, was intact. Here it is noteworthy to mention that in the averaged hTf structure, Asp634, was located in between Lys534 and Arg632, closer to the residues (SI, Figure S10) compared to that in the ternary TbpA-TbpB-hTf complex (Figure 8). Moreover, it directly creates a H-bond with Arg632 pulling it away from Fe3+-coordinating Tyr517. Thus, the proton transfer from Arg632 to Tyr517 hypothesized in this study (next section) for the ternary complex seemed improbable in the structural arrangement of sole hTf. This, yet again, highlighted the electrostatic impact of TbpA Lys359 residue for the disruption of the network.
3.2. Results from QM/MM Calculations
A two-layer ONIOM (QM/MM) approach was employed to explore the tendency of Fe3+ dissociation by calculating Fe3+ binding energies in the TbpA-TbpB-hTf ternary complex and its mutant variants. The results were interpreted based on relative trends rather than absolute binding energies. Accordingly, lower binding energy values indicate stronger binding affinity and reduced propensity for dissociation. First, we calculated the binding energies of Fe3+ in the C-lobe of sole hTf and the TbpA-TbpB-hTf protein complex to verify how bacterial binding would influence Fe3+ binding/dissociation. Then, based on the existing literature and our findings from MD simulations, we focused on the role of Lys359 which is in the key position from TbpA. Mutation constructs, Lys359Ala, Lys359Arg, and Lys359Asp, were generated to investigate the effects of neutral, positively charged, and negatively charged residues, respectively.
The first step of the QM/MM study was to formulate a model system. In this context, the TbpA–TbpB–hTf complex structure derived from MD simulations was utilized, offering key insights into amino acid interactions within critical regions (Figure 8). Figure 9 demonstrates the QM/MM model of the protein structure, which was truncated from the ternary complex as described in Materials and Methods Section. The same procedure was applied to model hTf. Thus, C-lobe Fe3+, its coordinating ligands (CO32− Asp392, Tyr426, Tyr517, His585), two water molecules near Fe3+ and the other critical residues Arg632, Lys534, Asp634 were placed in the QM region (Figure 1) while the remaining residues were kept in the MM region.
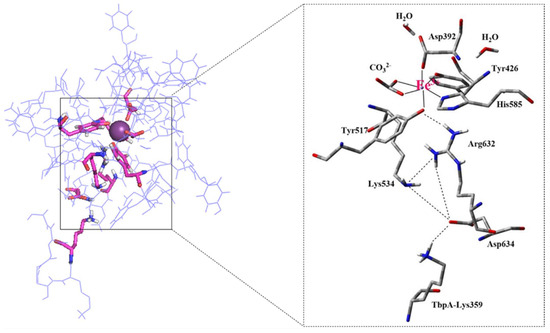
Figure 9.
Pre-optimized structure of the ternary complex QM/MM model and enlarged view of the QM region containing Asp392, Tyr426, Tyr517, Lys534, His585, Arg632, Asp634, Lys359 residues with their backbones, Fe3+, CO32− and two water molecules.
3.2.1. QM/MM Calculations for hTf and TbpA-TbpB-hTf Protein Complex
Binding energies calculated for hTf and bacterial ternary complex TbpA-TbpB-hTf are provided in Table 1 revealing highly exothermic reactions for Fe3+ binding. The experimentally measured binding constants for the hTf protein (≈1020–1023 M−1) indicated that the Fe3+ ion binds very tightly to hTf [47,82,83]. In parallel, Sakajiri et al. calculated the binding energies of various metal ions to hTf, including Fe3+, using DFT (B3LYP/6-31G) [41]. Although a direct comparison between experimental binding constant and the computed hTf binding energy is challenging due to the complexity of experimental conditions, the exceptionally low binding energy calculated in this study confirms the tight binding of Fe3+ to hTf and is consistent in magnitude with values reported in the literature [41].

Table 1.
Absolute electronic energies a,b, binding energies (ΔE), and relative binding energies c (ΔΔE) in explicit water for hTf, TbpA-TbpB-hTf, and ternary mutant protein complexes.
It was logically expected that dissociation of Fe3+ would be more favorable in the ternary complex than in isolated hTf, as bacterial binding facilitates iron removal. Indeed, Fe3+ binding energy for hTf was 171 kcal/mol smaller (stronger binding) than that of the TbpA-TbpB-hTf ternary complex, justifying that Fe3+ release from ternary complex was more favorable (by 171 kcal/mol) as expected. To rationalize this behavior, the conformational and geometrical differences between the two structures were systematically analyzed. Three-dimensional views of the QM regions and the geometry of the first-shell Fe coordination are illustrated in Figure 10. Important distances and angles in the optimized structures are listed in SI Table S7.
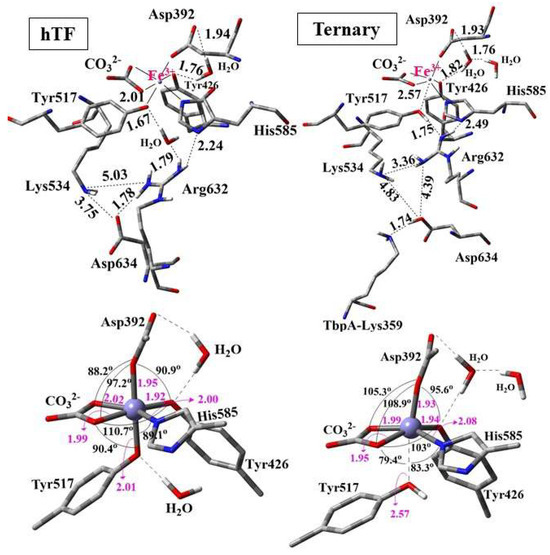
Figure 10.
Optimized structures of the QM regions (above) and geometry of the Fe coordination (below) for hTf and ternary TbpA–TbpB–hTf complex obtained from ONIOM(M06/6-31G(d,p):Amber) optimizations. Distances are in Å.
In the optimized structure of hTf, the first-shell coordination distances of Fe3+ ranged from 1.92 Å to 2.02 Å, indicating strong coordination with residues His585, Asp392, Tyr517, Tyr426 and the synergistic ion CO32− (Figure 10). These distances were consistent with the computational data in the literature (Table S6 of SI). All the coordinating residues were negatively charged as suggested in previous reports [35,43] where Fe3+ adopted an expected octahedral geometry. During the geometry optimization of hTf, Asp634 shifted upward, approaching Arg632 and Lys534, and formed a strong hydrogen bond with Arg632 at a distance of 1.78 Å. Additionally, one of the water molecules in the QM region was positioned between Tyr517 and Arg632, establishing strong hydrogen bonds with them at distances of 1.67 Å and 1.79 Å, respectively. Thus, a continuous H-bond network was established between Asp634-Arg632-H2O-Try517. The second water molecule in the QM region was located between Asp392 and Tyr426, forming H-bonds to these first-shell residues (Figure 10).
The QM region of the geometry-optimized TbpA–TbpB–hTf ternary complex incorporating the Lys359 residue is also depicted in Figure 10. Interestingly, the proton of positively charged Lys359 was transferred to Asp634, which triggered the transfer of Arg632 proton to Tyr517. As a result, Tyr517 was protonated and its coordination bond to Fe3+ was elongated remarkably from the initial O-Fe distance of 2.09 Å to 2.57 Å. As expected, no such proton transfer occurred in hTf due to the absence of Lys359, leading to a shorter O–Fe coordination distance of 2.01 Å. The elongation of the Tyr517–Fe coordination bond distorted the octahedral geometry of the Fe3+ center, leading to enlargements in the Asp392–Fe–Tyr426 and Asp392–Fe–CO32− angles, while the Tyr517–Fe–CO32−, Tyr517–Fe–His585, and Tyr517–Fe–Tyr426 angles were markedly reduced relative to hTf (Figure 10 and Table S7).
As a result, TbpA–TbpB–hTf exhibited distinct structural features compared to native hTf. For instance, the Asp634–Arg632 and Asp634–Lys534 distances were significantly elongated, likely due to the neutralization of Asp634, which weakened its interactions with Arg632 and Lys534, positioning it closer to Lys359. Thus, neutral Asp634 formed a strong H-bond with Lys359 nitrogen while neutral Arg632 acted as a H-bond acceptor of OH proton from Tyr517 (Figure 10). Based on these observations, we hypothesized the following mechanism (Scheme 1) for the Fe3+ release at physiological pH, triggered by N. Meningitidis. Upon binding of TbpA to hTf, protonated Lys359 from TbpA acts as an acid catalyst, initiating a cascade of events that ultimately may lead to Fe3+ dissociation from the C-lobe: (i) Lys359 transfers its proton to negatively charged Asp634 in hTf. (ii) The neutralization of Asp634 disturbs the electrostatic balance within the Lys534–Asp634–Arg632 triad, causing charge repulsion and reorganization. (iii) This neutralization weakens the interaction between Asp634 and positively charged Arg632, leaving Arg632 with an unbalanced positive charge. (iv) To restore charge balance, Arg632 transfers its proton to Tyr517 in the Fe3+ coordination shell. (v) As a result, the Tyr517–Fe3+ coordination bond is substantially disrupted, which may initiate dissociation of Fe3+. The schematic representation of these changes in the ternary protein complex is shown in Scheme 1.
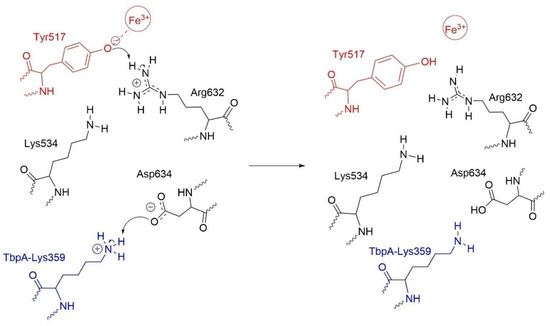
Scheme 1.
Disruption of the Tyr517-Fe3+ coordination bond triggered by proton transfer from Lys359 of TbpA.
3.2.2. QM/MM Calculations for the Lys359 Mutants of TbpA-TbpB-hTf Protein Complex
To investigate the role of the key TbpA residue Lys359 in Fe3+ dissociation from the ternary complex, point mutations substituting Lys359 with alanine, arginine, and aspartic acid were manually introduced. Apo and holo structures of each protein complex were optimized and the binding energies were calculated. The 3-D views and important distances in QM regions of the optimized structures are shown in Figure 11. Calculated energies and the geometrical parameters are given in Table 1 and Table S7, respectively.
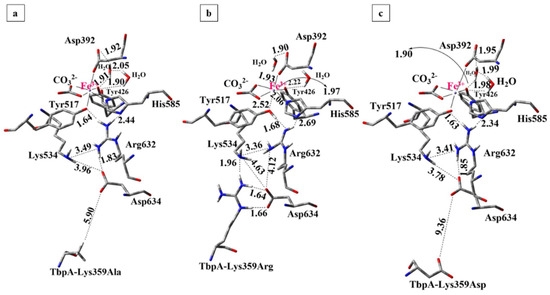
Figure 11.
Optimized structures of mutant protein complexes obtained from ONIOM(M06/6-31G(d,p):Amber calculations (Distances are in Å). (a) Lys359Ala, (b) Lys359Arg, (c) Lys359Asp.
The effect of Lys359 alanine mutation: The alanine mutation was introduced to prevent the Lys359–Asp634 interaction observed in the wild-type complex and to assess the impact of a neutral, significantly shorter side chain on the Fe3+ binding energy. As demonstrated in Table 1, the calculated Fe3+ binding energy for the Lys359Ala mutant was 96 kcal/mol lower than that of the wild-type ternary complex, indicating a significantly higher Fe3+ affinity and reduced propensity for Fe3+ release. This result supports our hypothesis that the positively charged Lys359 in the wild-type complex disrupts the electrostatic balance of the Lys534–Arg632–Asp634 triad, thereby facilitating Fe3+ dissociation. As anticipated, substituting Lys359 with a neutral alanine residue stabilized Fe3+ binding by preserving the charge balance. The structures of the optimized mutant complex (Figure 11a) and wild-type TbpA-TbpB-hTf complex (Figure 10) were compared along with the important distances in Table S7 in SI.
The most notable observation was the absence of proton transfer from Arg632 to Tyr517 in the mutant complex, with the Fe3+ coordination remaining intact, as evidenced by a shorter Tyr517–Fe3+ distance of 2.09 Å, in contrast to the 2.57 Å observed in the wild type. In the mutant structure, the deprotonated Asp634 residue retained its negative charge, effectively stabilizing the cationic Arg632 via electrostatic interaction. Consequently, Arg632 did not donate its proton to Tyr517, preserving the integrity of the Fe3+ coordination sphere, contrary to the case in the wild-type complex where Lys359 triggered the transfer of Arg632 proton to Tyr517. During geometry optimization of the Lys359Ala complex, Asp634 shifted upward toward Lys534 and Arg632, likely due to the absence of stabilizing interactions by Lys359Ala residue. Consequently, as Lys534, Asp634 and Arg632 moved closer together, Asp634 formed a strong hydrogen bond with Arg632. Notably, a similar structural feature was also observed in hTf having no stabilization effect on Asp634 due to the absence of Lys359, which is likely to be the common feature for the structures with tight Fe3+ binding. These structural rearrangements stabilize the Lys534–Asp634–Arg632 triad, maintaining charge balance around Arg632 and preserving electron density at the Fe3+ coordination site, thereby favoring Fe3+ binding. In contrast, disruption of this triad in the wild-type complex likely leads to electron withdrawal by Arg632 from the Fe site, weakening Fe3+ coordination.
The effect of Lys359 arginine mutation: the Lys359Arg mutation introduces a longer, positively charged Arg side chain to enhance interaction with Asp634, promote proton transfer, and facilitate Fe3+ dissociation. Consistently, its Fe3+ binding energy was 30 kcal/mol higher than the wild type and 126 kcal/mol higher than the neutral Lys359Ala mutant (Table 1), indicating significantly weaker Fe3+ binding and supporting the electrostatic repulsion mechanism proposed by Noinaj et al. [6,49].
In the optimized Lys359Arg structure, Arg359 formed strong H-bonds with Asp634 (1.64 Å, 1.66 Å) and an additional bond with Lys534 (1.96 Å) (Figure 11b). On the other hand, the shorter Lys359 side chain in the wild type was ~5 Å from Lys534, preventing H-bonding (Figure 10). As a result, Lys359’s positive charge remained unbalanced, leading to proton transfer to Asp634 during optimization. In contrast, no proton transfer from Lys359Arg to Asp634 occurred in the mutant during optimization (Table S7, Figure 11b). This is likely due to the lower acidity of Arg (pKa 12.48) compared to Lys (pKa 10.53) [84] making it less prone to proton donation. Instead, Arg359 stabilizes its positive charge via three strong H-bonds. Meanwhile, deprotonated Asp634 retains its negative charge, contributing to the electrostatic balance of Arg632. However, when the structure was re-optimized with the proton positioned equidistant between Arg632 and Tyr517, it preferentially bonded to Tyr517. This suggests that proton transfer from Arg632 to Tyr517 is energetically favorable but may not occur spontaneously, unlike in the wild-type complex. In the re-optimized structure, the distance between Tyr517 and the Fe3+ ion increased to 2.52 Å, resulting in remarkably weaker Fe3+ coordination and slightly distorted octahedral geometry (Table S7 in SI). No other major structural differences were observed compared to the initial optimization. The second structure, featuring neutralized Tyr517 and Arg632, was 3.2 kcal/mol more stable and was therefore selected as the representative model for the Lys359Arg mutant. The Fe3+ binding energy was 30 kcal/mol higher than that of the wild type, indicating weaker binding. This reduction is attributed to charge separation between Arg359 and Asp634, which lowers structural stability. In contrast, the wild-type structure gains stability through complete charge neutralization via proton transfer from Lys359 to Asp634.
The overall structure of the Lys359Arg mutant resembled the wild type, notably with a similarly elongated Tyr517–Fe coordination (2.52 Å). Asp634 remained positioned lower and distant from Arg632 (4.17 Å) and Lys534 (4.61 Å), unlike in hTf and Lys359Ala, where it was closer to these key residues (Figure 10 and Figure 11). Both the wild type and Lys359Arg showed distorted Fe3+ octahedral geometry, indicated by reduced CO3–Fe–Tyr426 and CO3–Fe–Tyr517 angles and enlarged CO3–Fe–Asp392 angles (Table S7). Overall, since both Lys and Arg are protonated at physiological pH, the mutant and wild type similarly promoted Fe3+ dissociation, though the weaker acidity of Arg led to subtle differences.
The effect of Lys359 aspartate mutation: the Lys359Asp mutation replaces the positively charged lysine with a negatively charged aspartate to induce an opposite electrostatic effect relative to the wild type. Due to its shorter side chain and charge repulsion with Asp634, Asp359 is expected to remain spatially distant from Asp634. As shown in Table 1, this mutant exhibited significantly stronger Fe3+ binding, with binding energy 134 kcal/mol lower than the wild type and 38 kcal/mol lower than the neutral Lys359Ala mutant, indicating reduced tendency for Fe3+ dissociation.
In the QM/MM geometry optimizations, the distance between Asp634 and Lys359Asp residues increased to 9.36 Å, which can be attributed to the electrostatic repulsion between two negatively charged Asp residues (Figure 11c). During optimization, the Asp634 residue shifted upward toward the positively charged Arg632, forming a strong hydrogen bond at a distance of 1.85 Å. Meanwhile, it also moved closer to Lys534, forming a weak interaction at a distance of 2.85 Å, thereby contributing to the formation of the Lys534–Asp634–Arg632 triad. As observed in Figure 11, in the Lys359Asp mutant complex, the Lys534–Asp634 (3.78 Å) and Asp634–Arg632 (1.85 Å) distances were shorter and closely resembled those observed in hTf and the Lys359Ala mutant, both of which exhibited less favorable Fe3+ dissociation like Lys359Asp, compared to the wild-type complex.
In contrast to the wild-type ternary complex (Figure 10) and the Lys359Arg mutant, the Lys359Asp variant exhibited a more compact configuration of the critical Lys534–Asp634–Arg632 triad (Figure 11), leading to enhanced stabilizing interactions that did not perturb iron coordination. This structural arrangement implies a higher affinity for Fe3+ and a reduced propensity for its dissociation.
3.3. Natural Bond Orbital Stabilization Energies
To gain further insight into Fe3+ coordination disruption, second-order perturbation theory analysis of the Fock matrix in the NBO basis was performed to evaluate donor–acceptor charge transfer interactions [79]. The stabilization energies E(2), reflecting electron donation from the lone pair (LP) orbitals of donor atoms to the unoccupied LP* orbitals of Fe3+, quantify the strength of each coordination bond. Higher E(2) values indicate stronger interactions and greater charge transfer [17]. As shown in Table 2, hTf exhibited the highest total stabilization energy (275.99 kcal/mol), confirming the strongest Fe3+ coordination in the absence of bacterial proteins while the wild-type bacterial complex and Lys359Arg mutant displayed the lowest E(2) values (234.09 and 227.75 kcal/mol), as expected. The stabilization energy trend—hTf > Lys359Asp > Lys359Ala > wild type ternary complex > Lys359Arg—correlated well with calculated relative binding energies (ΔΔE), yielding a strong linear relationship (R2 = 0.88; SI Figure S11). Lower stabilization energies were associated with higher ΔΔE values, indicating increased Fe3+ dissociation propensity and confirming that ΔΔE reflects the weakening in Fe3+ coordination.
Table 2.
Donor–acceptor stabilization energies, E(2), in kcal/mol, for the first coordination shell of Fe3+, obtained from NBO second-order perturbation theory analysis of hTf, wild-type TbpA-TbpB-hTf and its mutants.
Table 2 further quantifies the contribution of individual Fe3+-coordinating residues to the total stabilization energy in each structure. The significant decrease in the total stabilization energies for the wild-type and Lys359Arg complexes relative to hTf (by 41.9 and 48.2 kcal/mol, respectively), primarily originated from reduced LP(Tyr517–O)→LP*(Fe3+) interactions indicating weakened Tyr517-Fe3+ coordination due to Tyr517 protonation. Across all models, LP(His585–N)→LP*(Fe3+) and LP(Tyr517–O)→LP*(Fe3+) exhibited the lowest E(2) values, suggesting that His585 and Tyr517 are the most susceptible sites for coordination disruption, critical for initiating Fe3+ dissociation from the hTf C-lobe.
4. Conclusions
To date, experimental structures for TbpA-hTf, TbpB-hTf double-protein complexes and single TbpA, hTf protein structures have been resolved while partial ternary and binary complex models have been proposed, which is pivotal to understand the Fe3+ transfer mechanism of N. meningitidis infection leading to fatal meningitis disease [6,9,50]. This study revealed the first computationally derived and validated 3-D structure of the full TbpA-TbpB-hTf ternary complex, responsible for Fe3+ acquisition from the human host. The accuracy of the MD-derived structure was validated through various computational tools, and key protein–protein interfaces were characterized. The TbpA–hTf and TbpB–hTf interfaces contained 67% and 53% polar or charged residues, respectively, indicating that hydrophilic interactions dominate complex formation. TbpA-TbpB interface was very minimal compared to those with hTf, and composed of 41% polar residues.
MD simulations clearly revealed the dynamic reorganization of the electrostatic network, proposed to be vital in loosening and abstraction of Fe3+ ion from its coordination shell. In isolated hTf, Asp634 was positioned between Lys534 and Arg632 forming a H-bond with Arg632 and pulling it away from the Fe3+ coordination site at Tyr517. Upon formation of ternary TbpA-TbpB-hTf complex, TbpA Lys359 formed a new H-bond with Asp634, pulling it slightly away from its original position between Lys534 and Arg632. Thus, the electrostatic network was reoriented and Arg632 moved closer to Tyr517, creating a H-bond which might be the initiation of the proton transfer proposed in this study.
Fe3+ binding energies calculated via ONIOM (M06/6-31G(d,p):Amber) revealed stronger binding in isolated hTf than in all ternary bacterial complexes, consistent with facilitated Fe3+ dissociation upon bacterial binding. Mutational analysis provided important insights into the disruption of Fe3+ coordination. Lys359Ala and Lys359Asp mutants exhibited binding energies 96 and 134 kcal/mol lower than the wild type, indicating more favorable Fe3+ binding. Conversely, Lys359Arg showed a 30 kcal/mol increase, suggesting weaker binding. These QM/MM results highlight the critical role of Lys359 in modulating Fe3+ coordination and initiating its release. The key conclusions are summarized below:
- (1)
- Point mutations Lys359Ala and Lys359Asp, which increase the local electron density by introducing neutral or negative charge in the QM region, significantly enhance Fe3+ binding affinity. In contrast, the positively charged Lys359Arg mutation reduces electron density, weakening Fe3+ coordination. These findings suggest that increased electron density near the Fe3+ center stabilizes its binding, whereas the introduction of positive charge (as in wild-type Lys359 or Lys359Arg mutant) perturbs the electrostatic environment of highly cationic Fe3+, lowering the binding affinity and facilitating Fe3+ release.
- (2)
- Consistent with energetic results, structural analyses emphasize the importance of maintaining charge balance within the Asp634–Arg632–Lys534 triad for stable Fe3+ coordination. In protein models exhibiting stronger Fe3+ binding (e.g., hTf, Lys359Ala, Lys359Asp), Asp634 forms a stabilizing H-bond with Arg632 and is positioned proximally to both Arg632 and Lys534 (Figure 10 and Figure 11). In contrast, in weaker-binding systems (wild-type and Lys359Arg), the positively charged Lys359 or Arg359 interacts electrostatically with Asp634, displacing it from Arg632. This displacement disrupts local charge neutrality, leaving Arg632 insufficiently balanced near the Fe3+ center, thereby inducing electrostatic repulsion and destabilizing Fe3+ binding.
- (3)
- Although both the wild-type and Lys359Arg mutant reduce Fe3+ binding relative to neutral or negatively charged mutants, their mechanisms differ slightly. In the wild type, proton transfer from Lys359 to Asp634 enables charge neutralization, which slightly lowers Fe3+ binding energy but triggers a second, spontaneous proton transfer from Arg632 to Tyr517, weakening the Tyr517–Fe3+ bond. In contrast, no proton transfer from Arg359 occurs in the Lys359Arg mutant due to the lower acidity of Arg, resulting in incomplete charge neutralization and a higher binding energy. These findings suggest that Fe3+ release requires both a destabilizing positive charge and an acidic proton to disrupt coordination.
- (4)
- NBO donor–acceptor stabilization energies revealed that reduced Fe3+ binding in the wild-type and Lys359Arg complexes primarily stems from a significant decrease in LP(Tyr517–O)→LP(Fe3+) interactions. Across all models, LP(His585–N)→LP*(Fe3+) and LP(Tyr517–O)→LP*(Fe3+) consistently exhibited the lowest stabilization energies, identifying Tyr517 and His585 as the most labile coordination sites.
In conclusion, point mutations at the Lys359 position in the wild-type TbpA–TbpB–hTf complex support the hypothesis that Lys359 from TbpA donates an acidic proton essential for disrupting Fe3+ coordination—mimicking endosomal acidification and initiating iron dissociation at physiological pH. Additionally, incorporation of a positively charged residue destabilizes the holo form and reduces Fe3+ affinity. The wild-type complex reflects both effects: proton donation and destabilization. In contrast, the Lys359Arg mutant, despite its positive charge, fails to donate a proton efficiently due to the lower acidity of Arg compared to Lys, preventing spontaneous proton transfer to Asp634. Conversely, substitution with an anionic residue (Asp) enhances holo complex stability and Fe3+ binding, while a neutral residue (Ala) yields an intermediate effect.
Overall, this study highlights the following important aspects about Fe binding/dissociation mechanism from the C-lobe of hTf upon binding of bacterial proteins from N. meningitidis. (i) The first validated ternary model of TbpA–TbpB–hTf complex was obtained. (ii) A Lys359-triggered proton transfer mechanism was proposed for the disruption of iron coordination, awaiting experimental confirmation in future studies. (iii) The data presented here serves as a basis for developing therapeutic strategies. Notably, we propose Lys359 as a potential target for designing covalent drugs against meningitis, as covalent modification of lysine residues has recently gained attention as a promising strategy in the development of targeted covalent inhibitors [18,85]. These insights provide a molecular basis for the rational design of therapeutic agents that disrupt iron uptake in bacterial pathogens by targeting host–pathogen protein interactions.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics13120384/s1, Table S1. Amino acids included within 12 Å from Fe3+ in the QM/MM cluster model. Table S2. Charges of the ionizable residues in QM and MM regions of QM/MM cluster structures Table S3. Interacting residuesa at the TbpA-hTf interface for post-MD ternary protein complex structure. Table S4. Interacting residues at the TbpB-hTf interface for post-MD ternary protein complex structure. Table S5. Interacting residues at the TbpA-TbpB interface for post-MD ternary protein complex structure. Table S6. Fe-X coordination distances (Å) of C-lobe in hTf and ternary complex compared to the available data. Table S7. Important geometrical parameters in the optimized structures of hTf, wild-type holo TbpA-TbpB-hTf protein complex and its mutant complexes Lys359Ala, Lys359Arg, Lys359Asp. Figure S1. Root mean square deviation (RMSD) graphs for ternary TbpA-TbpB-hTf protein complex (left) and sole hTf protein (right). Figure S2. Root mean square fluctuation (RMSF) graph for sole hTf protein. Fluctuating loop regions are shown in the average hTf structure. Figure S3. Root mean square fluctuation (RMSF) graphs for ternary TbpA-TbpB-hTf complex (TbpA:red, TbpB:green, hTF:blue). Fluctuating loop regions are shown in the average structure. Figure S4. Radius of gyration (Rg) graph for sole hTf (blue) and ternary TbpA-TbpB-hTF protein complex (black). Figure S5. Z-scores and energy per residue values for TbpA protein in the crystal structure and in the ternary protein complex. Figure S6. Z-scores and energy per residue values for TbpB protein in the crystal structure and in the ternary protein complex. Figure S7. Z-scores and energy per residue values for hTf protein in the crystal structure and in the ternary protein complex. Figure S8. Fe-X coordination distances in ternary complex throughout the simulations. Figure S9. Fe-X coordination distances in hTF throughout the simulations. Figure S10. (A) Equilibrated hTf protein structure (B) zoom in view of the electrostatic network. (C) Fe3+ ion coordination shell. Figure S11. Linear correlation between relative binding energies and donor-acceptor stabilization energies E(2). (Correlation coefficient R2 = 0.88).
Author Contributions
C.D.Ö.: Methodology, investigation, data curation, formal analysis, visualization, project administration, funding acquisition, writing—original draft. G.N.D.: Methodology, investigation, data curation, formal analysis, visualization. V.F.: Methodology, formal analysis, review and editing. M.Ö.: Conceptualization, methodology, formal analysis, supervision, project administration, funding acquisition, writing—original draft, review and editing. S.S.E.: Conceptualization, methodology, formal analysis, supervision, writing—original draft, review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Scientific and Technological Research Council of Türkiye (TÜBİTAK), Project no: 118Z353 and Project no: 223Z201.
Data Availability Statement
The most representative structures for TbpA-TbpB-hTf ternary protein complex and sole hTf protein obtained from equilibrated time windows of MD simulations are available upon request.
Acknowledgments
This study was funded by the Scientific and Technological Research Council of Türkiye (TÜBİTAK), Project no: 118Z353 and Project no: 223Z201. CDÖ acknowledges to BİDEB 2211-A National PhD Scholarship Program. Calculations were partially performed at TÜBİTAK ULAKBİM High Performance and Grid Computing Center, referred to as the Turkish National e-Science e-Infrastructure (TRUBA).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Peters, K.; Staehlke, S.; Rebl, H.; Jonitz-Heincke, A.; Hahn, O. Impact of Metal Ions on Cellular Functions: A Focus on Mesenchymal Stem/Stromal Cell Differentiation. Int. J. Mol. Sci. 2024, 25, 10127. [Google Scholar] [CrossRef]
- Majorek, K.A.; Gucwa, M.; Murzyn, K.; Minor, W. Metal Ions in Biomedically Relevant Macromolecular Structures. Front. Chem. 2024, 12, 1426211. [Google Scholar] [CrossRef]
- Zhong, M.; Wang, Y.; Min, J.; Wang, F. Iron Metabolism and Ferroptosis in Human Health and Disease. BMC Biol. 2025, 23, 263. [Google Scholar] [CrossRef] [PubMed]
- SantaMaria, A.M.; Rouault, T.A. Regulatory and Sensing Iron–Sulfur Clusters: New Insights and Unanswered Questions. Inorganics 2024, 12, 101. [Google Scholar] [CrossRef]
- Perkins-Balding, D.; Ratliff-Griffin, M.; Stojiljkovic, I. Iron Transport Systems in Neisseria Meningitidis. Microbiol. Mol. Biol. Rev. 2004, 68, 154–171. [Google Scholar] [CrossRef]
- Noinaj, N.; Buchanan, S.K.; Cornelissen, C.N. The Transferrin-Iron Import System from Pathogenic Neisseria Species. Mol. Microbiol. 2012, 86, 246–257. [Google Scholar] [CrossRef]
- Irwin, S.W.; Averil, N.; Cheng, C.Y.; Schryvers, A.B. Preparation and Analysis of Isogenic Mutants in the Transferrin Receptor Protein Genes, TbpA and TbpB, from Neisseria Meningitidis. Mol. Microbiol. 1993, 8, 1125–1133. [Google Scholar] [CrossRef]
- Anderson, J.E.; Sparling, P.F.; Cornelissen, C.N. Gonococcal Transferrin-Binding Protein 2 Facilitates but Is Not Essential for Transferrin Utilization. J. Bacteriol. 1994, 176, 3162–3170. [Google Scholar] [CrossRef] [PubMed]
- Noinaj, N.; Easley, N.C.; Oke, M.; Mizuno, N.; Gumbart, J.; Boura, E.; Steere, A.N.; Zak, O.; Aisen, P.; Tajkhorshid, E. Structural Basis for Iron Piracy by Pathogenic Neisseria. Nature 2012, 483, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Schryvers, A.B.; Gonzalez, G.C. Receptors for Transferrin in Pathogenic Bacteria Are Specific for the Host’s Protein. Can. J. Microbiol. 1990, 36, 145–147. [Google Scholar] [CrossRef]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of Folded Proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Berendsen, H.J.C. Algorithms for Macromolecular Dynamics and Constraint Dynamics. Mol. Phys. 1977, 34, 1311–1327. [Google Scholar] [CrossRef]
- Voth, G.A. Computational Approaches for Studying Enzyme Mechanism; Methods in Enzymology; Academic Press/Elsevier: Cambridge, MA, USA, 2016; Volume 577–578, ISBN 9780128053478. [Google Scholar]
- Akyüz, M.A.; Erdem, S.S.; Edmondson, D.E. The Aromatic Cage in the Active Site of Monoamine Oxidase B: Effect on the Structural and Electronic Properties of Bound Benzylamine and p-Nitrobenzylamine. J. Neural Transm. 2007, 114, 693–698. [Google Scholar] [CrossRef]
- Lovell, T.; Himo, F.; Han, W.G.; Noodleman, L. Density Functional Methods Applied to Metalloenzymes. Coord. Chem. Rev. 2003, 238–239, 211–232. [Google Scholar] [CrossRef]
- Akyüz, M.A.; Erdem, S.S. Computational Modeling of the Direct Hydride Transfer Mechanism for the MAO Catalyzed Oxidation of Phenethylamine and Benzylamine: ONIOM (QM/QM) Calculations. J. Neural Transm. 2013, 120, 937–945. [Google Scholar] [CrossRef]
- Cakir, K.; Erdem, S.S.; Atalay, V.E. ONIOM Calculations on Serotonin Degradation by Monoamine Oxidase B: Insight into the Oxidation Mechanism and Covalent Reversible Inhibition. Org. Biomol. Chem. 2016, 14, 9239–9252. [Google Scholar] [CrossRef] [PubMed]
- Fındık, V.; Varınca Gerçik, B.T.; Sinek, Ö.; Erdem, S.S.; Ruiz-López, M.F. Mechanistic Investigation of Lysine-Targeted Covalent Inhibition of PI3Kδvia ONIOM QM:QM Computations. J. Chem. Inf. Model. 2022, 62, 6775–6787. [Google Scholar] [CrossRef]
- Sheng, X.; Himo, F. The Quantum Chemical Cluster Approach in Biocatalysis. Acc. Chem. Res. 2023, 56, 938–947. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Chen, S.; Liao, R. Theoretical Studies of Nickel-Dependent Enzymes. Inorganics 2019, 7, 95. [Google Scholar] [CrossRef]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. A Practical Guide to Modelling Enzyme-Catalysed Reactions. Chem. Soc. Rev. 2012, 41, 3025–3038. [Google Scholar] [CrossRef]
- Vreven, T.; Byun, K.S.; Komáromi, I.; Dapprich, S.; Montgomery, J.A.; Morokuma, K.; Frisch, M.J. Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM. J. Chem. Theory Comput. 2006, 2, 815–826. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Zhang, Q.; Chen, K.; Zhu, W. Probing Origin of Binding Difference of Inhibitors to MDM2 and MDMX by Polarizable Molecular Dynamics Simulation and QM/MM-GBSA Calculation. Sci. Rep. 2015, 5, 17421. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed]
- Morokuma, K.L.M. Multi-Scale Quantum Models for Biocatalysis; York, D.M., Lee, T.-S., Eds.; Challenges and Advances in Computational Chemistry and Physics; Springer: Dordrecht, The Netherlands, 2009; Volume 7, ISBN 978-1-4020-9955-7. [Google Scholar]
- Mihaljević-Jurič, P.; Sousa, S.F. A QM/MM Evaluation of the Missing Step in the Reduction Mechanism of HMG-CoA by Human HMG-CoA Reductase. Processes 2021, 9, 1085. [Google Scholar] [CrossRef]
- Yildiz, I.; Yildiz, B.S.; Kirmizialtin, S. Comparative Computational Approach to Study Enzyme Reactions Using QM and QM-MM Methods. ACS Omega 2018, 3, 14689–14703. [Google Scholar] [CrossRef] [PubMed]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. Cell Biology PH and the Recycling of Transferrin during Receptor-Mediated Endocytosis (Endocytic Vesicle/Apotransferrin/Transferrin Receptor). Proc. Natl Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef] [PubMed]
- Benjamín-Rivera, J.A.; Cardona-Rivera, A.E.; Vazquez-Maldonado, A.L.; Dones-Lassalle, C.Y.; Pabon-Colon, H.L.; Rodriguez-Rivera, H.M.; Rodriguez, I.; Gonzalez-Espiet, J.C.; Pazol, J.; Perez-Rios, J.D. Exploring Serum Transferrin Regulation of Nonferric Metal Therapeutic Function and Toxicity. Inorganics 2020, 8, 48. [Google Scholar] [CrossRef]
- Halbrooks, P.J.; He, Q.Y.; Briggs, S.K.; Everse, S.J.; Smith, V.C.; MacGillivray, R.T.A.; Mason, A.B. Investigation of the Mechanism of Iron Release from the C-Lobe of Human Serum Transferrin: Mutational Analysis of the Role of a PH Sensitive Triad. Biochemistry 2003, 42, 3701–3707. [Google Scholar] [CrossRef]
- Mason, A.B.; Halbrooks, P.J.; James, N.G.; Connolly, S.A.; Larouche, J.R.; Smith, V.C.; MacGillivray, R.T.A.; Chasteen, N.D. Mutational Analysis of C-Lobe Ligands of Human Serum Transferrin: Insights into the Mechanism of Iron Release. Biochemistry 2005, 44, 8013–8021. [Google Scholar] [CrossRef]
- Baldwin, D.A. The Kinetics of Iron Release from Human Transferrin by EDTA. Effect of Salts and Detergents. BBA Protein Struct. 1980, 623, 183–198. [Google Scholar] [CrossRef]
- Zak, O.; Aisen, P. Iron Release from Transferrin, Its C-Lobe, and Their Complexes with Transferrin Receptor: Presence of N-Lobe Accelerates Release from C-Lobe at Endosomal PH. Biochemistry 2003, 42, 12330–12334. [Google Scholar] [CrossRef]
- Steere, A.N.; Byrne, S.L.; Chasteen, N.D.; Mason, A.B. Kinetics of Iron Release from Transferrin Bound to the Transferrin Receptor at Endosomal PH. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Koca Fındık, B.; Cilesiz, U.; Bali, S.K.; Atilgan, C.; Aviyente, V.; Dedeoglu, B. Investigation of Iron Release from the N- and C-Lobes of Human Serum Transferrin by Quantum Chemical Calculations. Org. Biomol. Chem. 2022, 20, 8766–8774. [Google Scholar] [CrossRef] [PubMed]
- Mujika, J.I.; Escribano, B.; Akhmatskaya, E.; Ugalde, J.M.; Lopez, X. Molecular Dynamics Simulations of Iron- and Aluminum-Loaded Serum Transferrin: Protonation of Tyr188 Is Necessary to Prompt Metal Release. Biochemistry 2012, 51, 7017–7027. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, D.; Field, M.J. A Computational Study of the Open and Closed Forms of the N-Lobe Human Serum Transferrin Apoprotein. Biophys. J. 2003, 85, 3485–3501. [Google Scholar] [CrossRef]
- Baker, H.M.; Nurizzo, D.; Mason, A.B.; Baker, E.N. Structures of Two Mutants That Probe the Role in Iron Release of the Dilysine Pair in the N-Lobe of Human Transferrin. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 408–414. [Google Scholar] [CrossRef]
- Dewan, J.C.; Mikami, B.; Sacchettini, J.C.; Hirose, M. Structural Evidence for a PH-Sensitive Dilysine Trigger in the Hen Ovotransferrin N-Lobe: Implications for Transferrin Iron Release. Biochemistry 1993, 32, 11963–11968. [Google Scholar] [CrossRef]
- Reilley, D.J.; Fuller, J.T.; Nechay, M.R.; Victor, M.; Li, W.; Ruberry, J.D.; Mujika, J.I.; Lopez, X.; Alexandrova, A.N. Toxic and Physiological Metal Uptake and Release by Human Serum Transferrin. Biophys. J. 2020, 118, 2979–2988. [Google Scholar] [CrossRef]
- Sakajiri, T.; Yajima, H.; Yamamura, T. Density Functional Theory Study on Metal-Binding Energies for Human Serum Transferrin-Metal Complexes. ISRN Biophys. 2012, 2012, 124803. [Google Scholar] [CrossRef]
- Mujika, J.I.; Lopez, X.; Rezabal, E.; Castillo, R.; Marti, S.; Moliner, V.; Ugalde, J.M. A QM/MM Study of the Complexes Formed by Aluminum and Iron with Serum Transferrin at Neutral and Acidic PH. J. Inorg. Biochem. 2011, 105, 1446–1456. [Google Scholar] [CrossRef]
- Mishra, L.; Sundararajan, M. Quantum Chemical Studies of Structures and Spin Hamiltonian Parameters of Iron Transferrin Using Isolated and Embedded Clusters Models. J. Chem. Sci. 2019, 131, 15. [Google Scholar] [CrossRef]
- Sundararajan, M.; Mishra, L.; Bharti, N.K.; Mantry, S.P. Proton-Coupled Electron Transfer Modulates the Metal Release of Blood Serum Iron Transferrin. Dalt. Trans. 2025, 54, 14677–14686. [Google Scholar] [CrossRef]
- Abdizadeh, H.; Atilgan, C. Predicting Long Term Cooperativity and Specific Modulators of Receptor Interactions in Human Transferrin from Dynamics within a Single Microstate. Phys. Chem. Chem. Phys. 2016, 18, 7916–7926. [Google Scholar] [CrossRef]
- Abdizadeh, H.; Atilgan, A.R.; Atilgan, C. Detailed Molecular Dynamics Simulations of Human Transferrin Provide Insights into Iron Release Dynamics at Serum and Endosomal PH. JBIC J. Biol. Inorg. Chem. 2015, 20, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Abdizadeh, H.; Atilgan, A.R.; Atilgan, C.; Dedeoglu, B. Computational Approaches for Deciphering the Equilibrium and Kinetic Properties of Iron Transport Proteins. Metallomics 2017, 9, 1513–1533. [Google Scholar] [CrossRef] [PubMed]
- Abdizadeh, H.; Atilgan, A.R.; Atilgan, C. Mechanisms by Which Salt Concentration Moderates the Dynamics of Human Serum Transferrin. J. Phys. Chem. B 2017, 121, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- Noinaj, N.; Guillier, M.; Barnard, T.J.; Buchanan, S.K. TonB-Dependent Transporters: Regulation, Structure, and Function. Annu. Rev. Microbiol. 2010, 64, 43–60. [Google Scholar] [CrossRef]
- Duran, G.N.; Ozbil, M. Structural Rearrangement of Neisseria Meningitidis Transferrin Binding Protein A (TbpA) Prior to Human Transferrin Protein (HTf) Binding. Turkish J. Chem. 2021, 45, 1146–1154. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for Checking the Quality of Protein Structures Solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Sippl, M.J. Recognition of Errors in Three-dimensional Structures of Proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 355–362. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson–Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Nagle, J.F. Area/Lipid of Bilayers from NMR. Biophys. J. 1993, 64, 1476–1481. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. Models@Home: Distributed Computing in Bioinformatics Using a Screensaver Based Approach. Bioinformatics 2002, 18, 315–318. [Google Scholar] [CrossRef]
- Krieger, E.; Elmar, G.V. YASARA—Yet Another Scientific Artificial Reality Application. YASARA.Org. 2013. Available online: http://www.yasara.org (accessed on 19 November 2025).
- Schüttelkopf, A.W.; Van Aalten, D.M.F. PRODRG: A Tool for High-Throughput Crystallography of Protein-Ligand Complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Li, P.; Song, L.F.; Merz, K.M. Parameterization of Highly Charged Metal Ions Using the 12-6-4 LJ-Type Nonbonded Model in Explicit Water. J. Phys. Chem. B 2015, 119, 883–895. [Google Scholar] [CrossRef]
- Benett, W.F.D.; MacCallum, J.L.; Tieleman, D.P. Thermodynamic Analysis of the Effect of Cholesterol on Dipalmitoylphosphatidylcholine Lipid Membranes. J. Am. Chem. Soc. 2009, 131, 1972–1978. [Google Scholar]
- Tieleman, D.P.; Berendsen, H.J.C. Molecular Dynamics Simulations of a Fully Hydrated Dipalmitoylphosphatidylcholine Bilayer with Different Macroscopic Boundary Conditions and Parameters. J. Chem. Phys. 1996, 105, 4871–4880. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Hirao, H.; Thellamurege, N.; Zhang, X. Applications of Density Functional Theory to Iron-Containing Molecules of Bioinorganic Interest. Front. Chem. 2014, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Flynn, C.M. Hydrolysis of Inorganic Iron (III) Salts. Chem. Rev. 1984, 84, 31–41. [Google Scholar] [CrossRef]
- De Abreu, H.A.; Guimaraes, L.; Duarte, H.A. Density-Functional Theory Study of Iron (III) Hydrolysis in Aqueous Solution. J. Phys. Chem. A 2006, 110, 7713–7718. [Google Scholar] [CrossRef]
- Rinaldo, D.; Field, M.J. A Density Functional Theory Study of the Iron-Binding Site of Human Serum Transferrin. Aust. J. Chem. 2004, 57, 1219–1222. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, I. Computational Analysis of Hydride and Proton Transfer Steps in L-Lactate Oxidase Based on QM and QM–MM Methods. J. Mol. Struct. 2024, 1295, 136706. [Google Scholar] [CrossRef]
- Yildiz, I. Computational Insights on the Hydride and Proton Transfer Mechanisms of L-Proline Dehydrogenase. PLoS ONE 2023, 18, e0290901. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, B.H.; Song, S.H.; Kim, M.K. Revisiting the Ramachandran plot based on statistical analysis of static and dynamic characteristics of protein structures. J. Struct. Biol. 2023, 215, 107939. [Google Scholar]
- Tina, K.G.; Bhadra, R.; Srinivasan, N. PIC: Protein Interactions Calculator. Nucleic Acids Res. 2007, 35, W473–W476. [Google Scholar] [CrossRef]
- Aisen, P.; Leibman, A.; Zweier, J. Stoichiometric and Site Characteristics of the Binding of Iron to Human Transferrin. J. Biol. Chem. 1978, 253, 1930–1937. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Sadler, P.J. Transferrin as a Metal Ion Mediator. Chem. Rev. 1999, 99, 2817–2842. [Google Scholar] [CrossRef]
- Schmidt, C.L.A.; Kirk, P.L.; Appleman, W.K. The Apparent Dissociation Constants of Arginine and of Lysine and the Apparent Heats of Ionization of Certain Amino Acids. J. Biol. Chem. 1930, 88, 285–293. [Google Scholar] [CrossRef]
- Findik, V.; Ruiz-López, M.F.; Erdem, S.S. Mechanistic Insights into Lysine-Targeting Covalent Inhibition through a Theoretical Study of Ester Aminolysis. Org. Biomol. Chem. 2021, 19, 9996–10004. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).