2. Results and Discussion
Synthesis and characterization of ligands. The proligands [H]
PhPyalk (
L1) and [H]
PyrPyalk (
L2) were synthesized using the previously reported lithiation reactions of 2-bromopyridine and their respective ketone precursors [
3,
4]. The proligands [H]
ThioPyalk (
L3), [H]
FePyalk (
L4), and [H]
FeOMePyalk (
L5) were synthesized in a similar method using 2-acetyl thiophene and acetylferrocene as their ketone precursors (
Scheme 1). For the synthesis of
L5, 2-methyoxy-5-bromopyridine was used as the pyridine precursor (
Table 1). The proligand
L3 was isolated cleanly as a white solid, and the
1H NMR spectrum of
L3 in CDCl
3 shows seven resonances in the aromatic region, corresponding to four pyridine and three thiophene protons. The synthesis and purification of
L4 and
L5 gave reddish-brown solids. The
1H NMR spectra in CDCl
3 of
L4 and
L5 indicated one cyclopentadienyl (Cp) ring on the ferrocenyl moiety that has five equivalent proton resonances. The pyalk-substituted Cp rings of
L4 and
L5 show four distinct
1H NMR peaks due to the chiral center at the tertiary carbon of the pyalk compound. The
13C{
1H} NMR spectra of
L4 and
L5 further support the structures of
L4 and
L5 with five distinct carbon peaks, as opposed to three if there were mirror symmetry. Compounds
L3-
L5 were each isolated in moderate yields and characterized by
1H and
13C{
1H} NMR, single crystal X-ray diffraction (
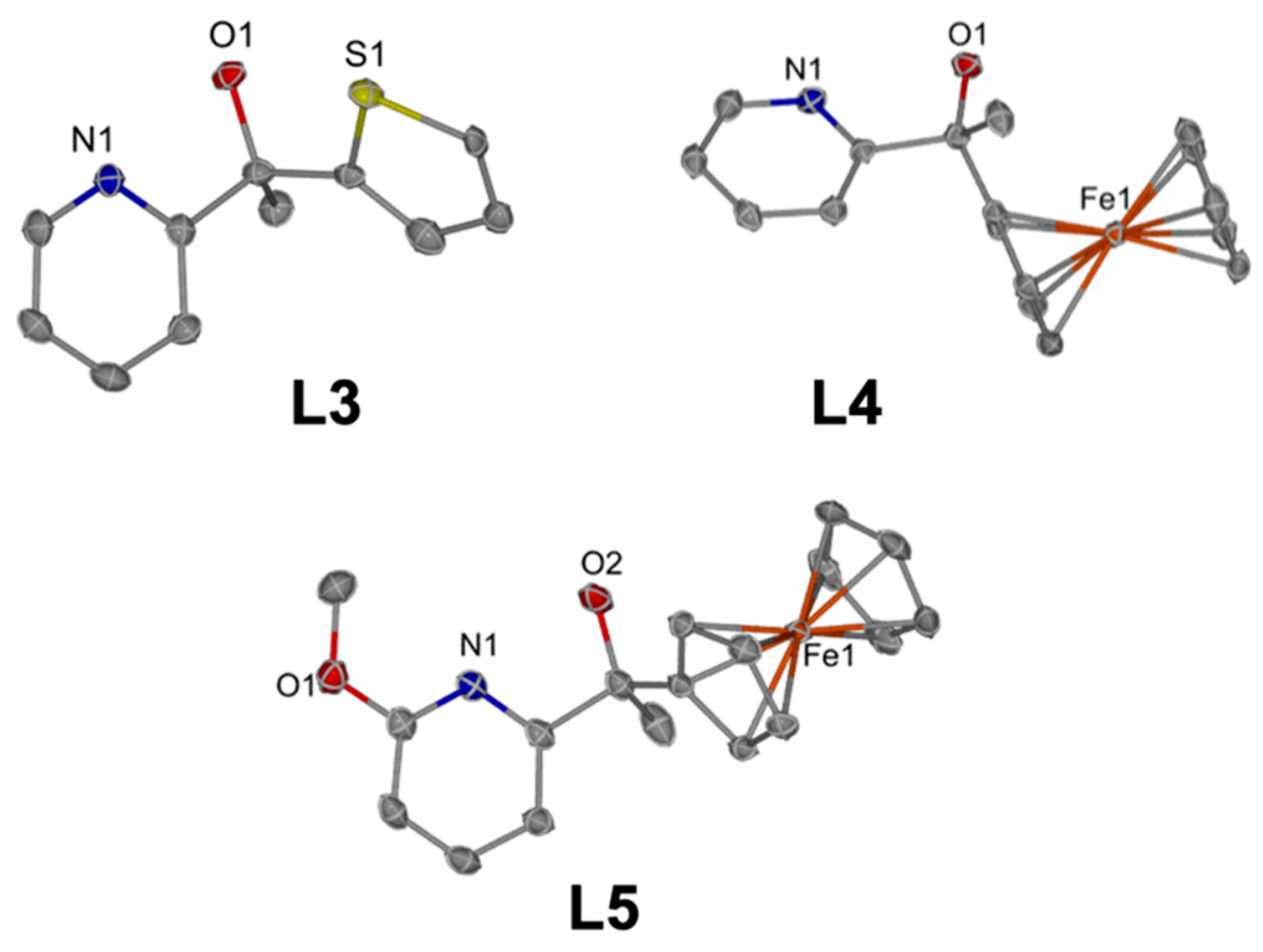
Figure 2), and elemental analysis.
Synthesis and Characterization of Complexes. Monocopper complexes Cu(
PhPyalk)
2 (
1a), Cu(
PyrPyalk)
2 (
2a), Cu(
ThioPyalk)
2 (
3a), and Cu(
FePyalk)
2 (
4a) (the label “a” indicates a mononuclear copper complex) were synthesized by treating CuCl
2·2H
2O with two equivalents of ligand in the presence of base, resulting in mononuclear Cu
II complexes with two ligands coordinated (
Scheme 2a). The ligand
L5 did not form a complex with Cu
II under any conditions attempted. Upon the addition of 1 M KOH/MeOH into methanolic solutions of two equivalents of
L1,
L2, and
L3 with CuCl
2·2H
2O, the solutions immediately turn purple, and
1a and
2a slowly began forming a pinkish/purple precipitate (
3a was more soluble in methanol than
1a or
2a). Solutions of
L4 and CuCl
2·2H
2O began as brown/red solutions that turned yellow upon the addition of base and likewise began precipitating a yellow solid. The solid-state structures of
1a-
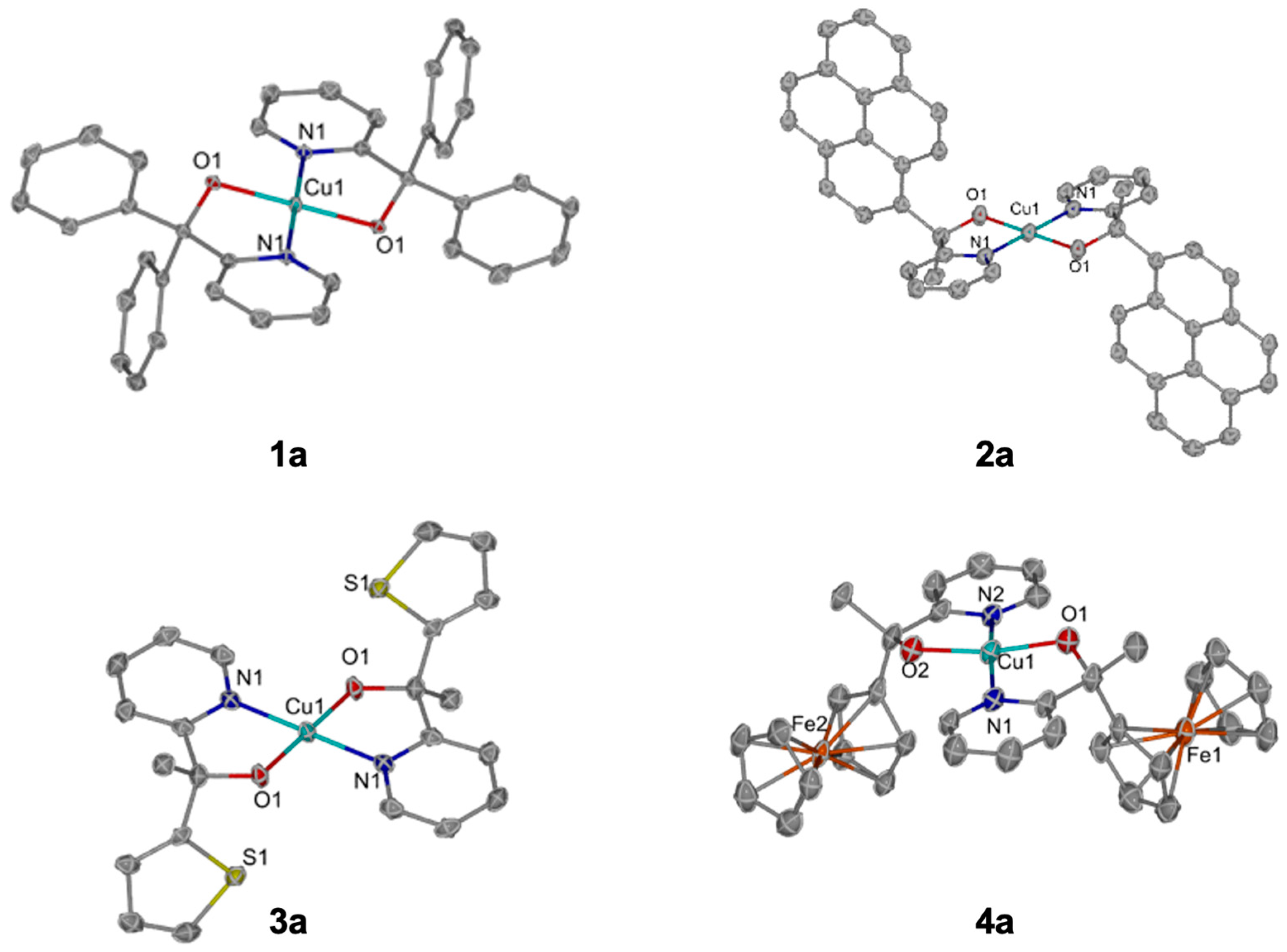
4a are shown in
Figure 3. The solid-state structure of
2a exhibited close aryl–aryl interatomic distances, potentially indicating non-covalent interactions in the solid state between pyrene groups (
Figure S31). In the unit cell, the ferrocene group in
4a is likewise interlocked between the ferrocene moieties of another
4a molecule, indicating a potential non-covalent interaction as well (
Figure S32). Also, there are interstitial molecules of methanol and water in hydrogen bonding interactions with the basic alkoxides of
4a (
Figure S33).
Each of the monocopper complexes
1a–
4a exhibit square planar geometries that are typical of Cu
II [
21]. The Cu–N bond distances range from 1.9598(12) Å to 1.980(3) Å, and the Cu–O bond distances range from 1.8832(16) Å to 1.9158(8) Å, which are in close agreement with the bond lengths of similar Cu
II complexes previously reported by Crabtree and Brudvig (selected bond lengths and angles shown in
Table 2) [
5,
8]. These complexes were paramagnetic and insoluble in most solvents except chlorocarbons and 2,2,2-trifluoroethanol (TFE). Due to their paramagnetic nature, NMR spectra were not useful.
The formation of bis–copper complexes can be accomplished with control of the ligand/Cu stoichiometry. Thus, the bis–copper complexes {CuCl}
2(µ-O-
PhPyalk)
2 (
1b), {CuCl}
2(µ-O-
PyrPyalk)
2 (
2b), {CuCl}
2(µ-O-
ThioPyalk)
2 (
3b), and {CuCl}
2(µ-O-
FePyalk)
2 (
4b) (the label “
b” indicates dinuclear complexes) were synthesized in the same manner as
1a–
4a but with 0.9 equivalent of ligand resulting in a dinuclear alkoxide-bridged complex (
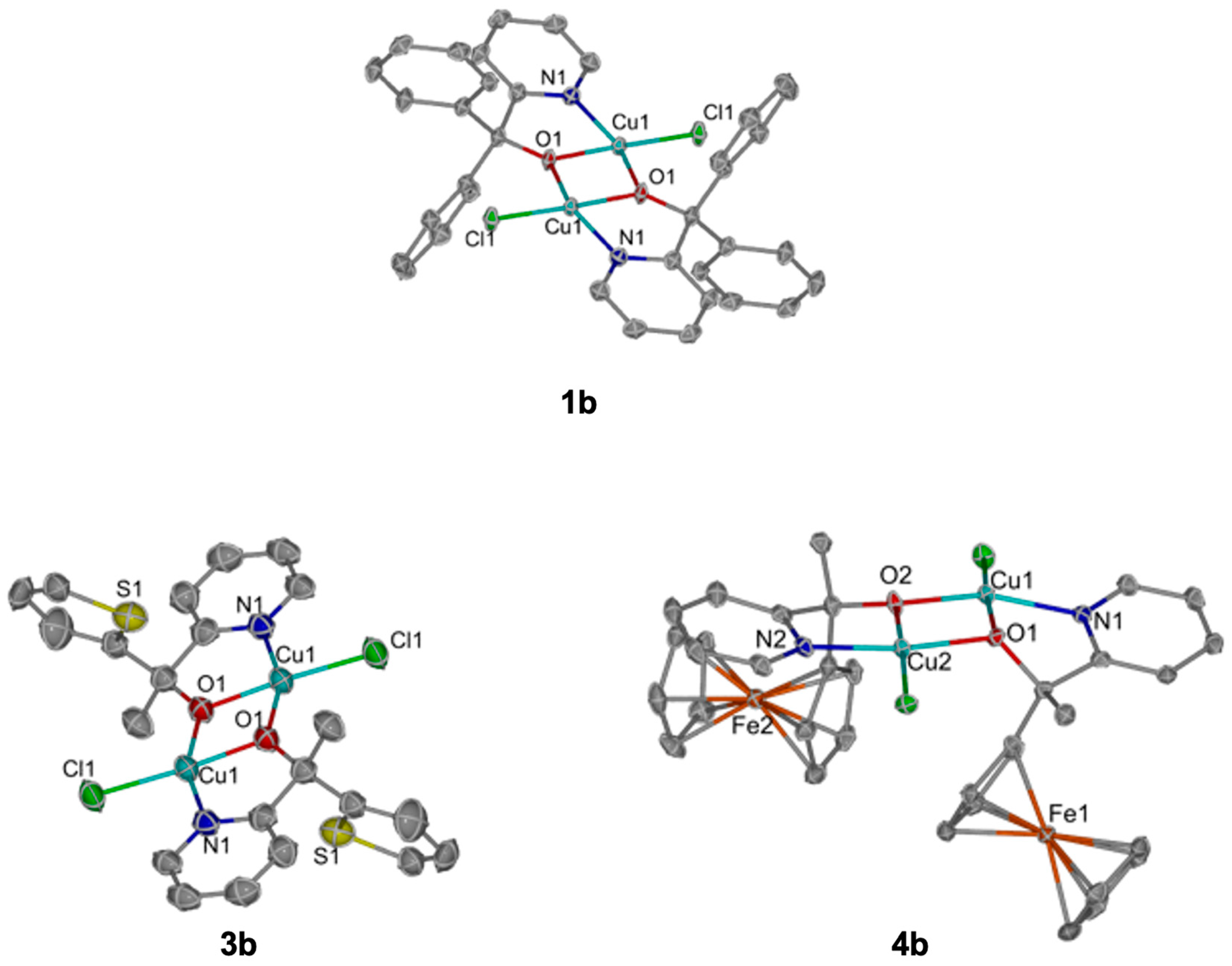
Scheme 2b). The reactions occurred similarly to the monocopper complexes; however, in this case, the color changed from blue to dark green upon the slow addition of base into the methanolic solution, and the product precipitated as green solids. The solid-state structures of
1b–
4b demonstrate the flexibility of alkoxide moieties to bridge two metal centers (
Figure 4-
1b,
3b,
4b;
Figure S34-2b). These structures closely resemble recently reported structures of the potential decomposition of Cu
II pyridine-alkoxide water oxidation catalyst products [
14]. Similar to
1a–
4a, due to the paramagnetic nature of
1b–
4b, NMR spectra were not useful. The local geometry at Cu does not change significantly between monocopper and bis–copper pyalk complexes, as demonstrated by similar Cu–ligand bond lengths (
Table 2).
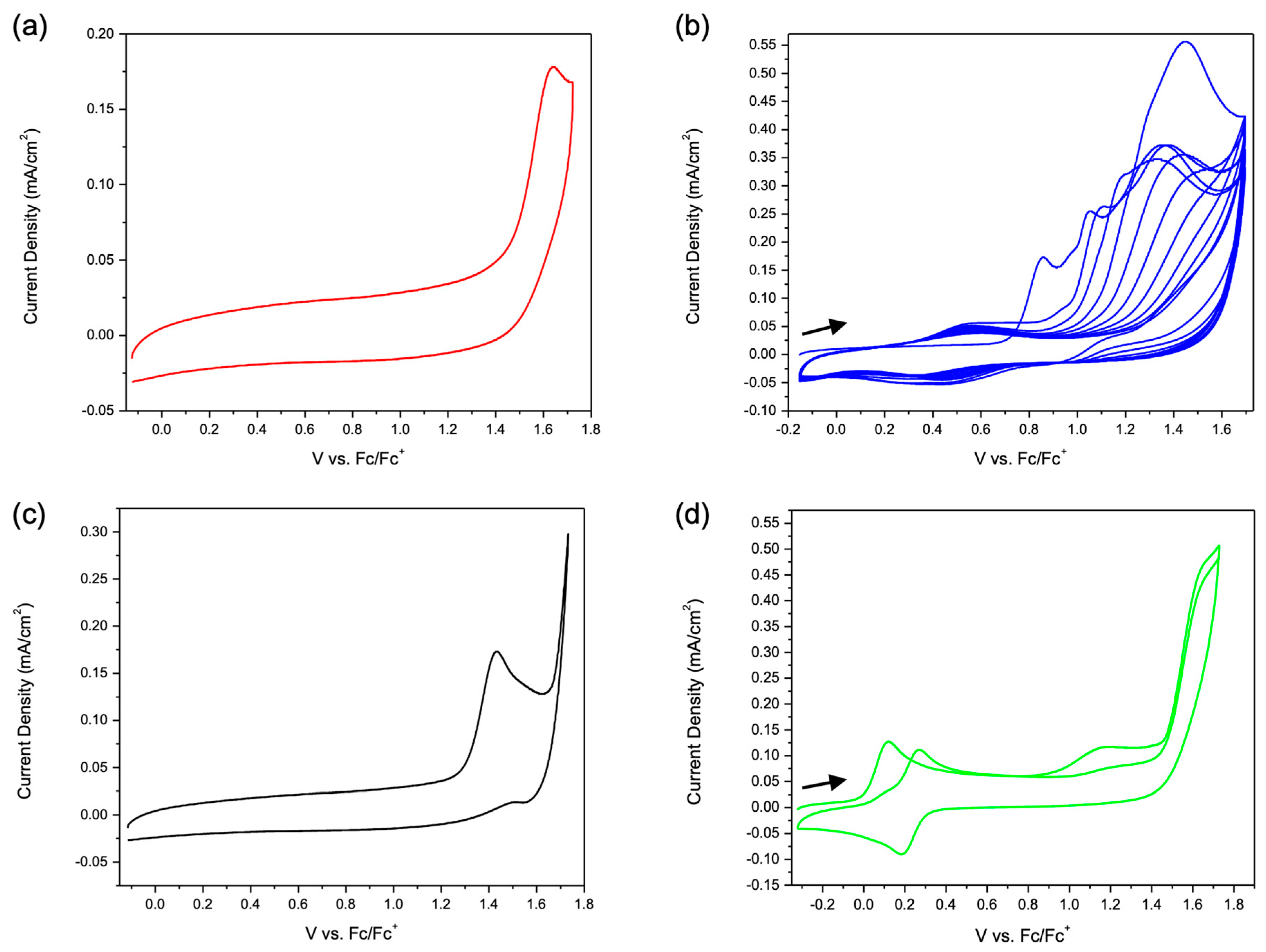
Cyclic voltammetry. The relative oxidative stability of the ligands and complexes were probed via cyclic voltammetry in 2,2,2-trifluoroethanol (TFE). The compound
L1 exhibited an irreversible oxidation at
Ep,a = 1.64 V vs. Fc/Fc
+ (Fc/Fc
+ =
E1/2 of the Fe
II/Fe
III of ferrocene) (
Figure 5a). The pyrene containing ligand
L2 displayed a broad irreversible oxidation peak when scanned anodically (
Figure 5b). After cycling through the irreversible oxidation multiple times, a reversible peak formed with an
E1/2 of 0.43 V vs. Fc/Fc
+. This reversible peak was retained after rinsing the working electrode and scanning anodically in a fresh electrolyte solution containing no
L2. Pyrene has been demonstrated to form polymers when placed under oxidative potentials due to a coupling reaction between radical pyrenes [
15]. The cyclic voltammogram of
L3 shows a redox event at
Ep,a = 1.44 V vs. Fc/Fc
+ (
Figure 5c). This oxidation does not result in the formation of a new reversible peak as was observed with
L2. Although it is common for thiophene-containing molecules to electropolymerize [
16], the steric bulk of the pyalk potentially prevents the polymerization reaction. The compound
L4 exhibits an Fe
II/Fe
III E
1/2 at 0.08 V vs. Fc/Fc
+ that scan rate studies indicate is a freely diffusing process (
Figure S7). Upon scanning more anodic of the Fe
II/Fe
III oxidation, an irreversible peak appeared at
Ep,a = 1.20 V vs. Fc/Fc
+, and the subsequent scan of the ferrocene peak showed the formation of a new reversible peak with an E
1/2 = 0.23 V vs. Fc/Fc
+, which lies between the Fe
II/Fe
III redox feature of ferrocene and acetylferrocene (
Figure 5d). The peak-to-peak separation for this feature is 0.09 V, and the current ratio is 1.23. The compound
L5 had oxidative responses similar to
L4, with an Fe
II/Fe
III E
1/2 = 0.05 V vs. Fc/Fc
+ as well as an irreversible peak at
Ep,a = 1.42 V vs. Fc/Fc
+ and a degradation species
E1/2 of 0.20 V vs. Fc/Fc
+.
Previously reported mechanisms of tertiary alcohol oxidation include radical mechanisms in which a C–H bond is cleaved. For example, when tert-butanol is oxidized by hydroxyl radicals, a C–H bond is cleaved to form acetone and methyl radical, which can then be oxidized to formaldehyde [
22]. Tertiary alcohols also can be oxidized via dehydration mechanisms and the subsequent oxidation of the product. Triphenyl methanol, which bears similarity to the ligands described here, does not contain easily abstracted C–H bonds, which results in the formation of stable peroxide compounds when treated with hydrogen peroxide [
23]. The presence of an Fe
II/Fe
III oxidation after the oxidative decomposition of
L4 with an E
1/2 in between that of ferrocene and acetylferrocene indicates that the methyl substituent is likely lost, and the alcohol is oxidized to a ketone.
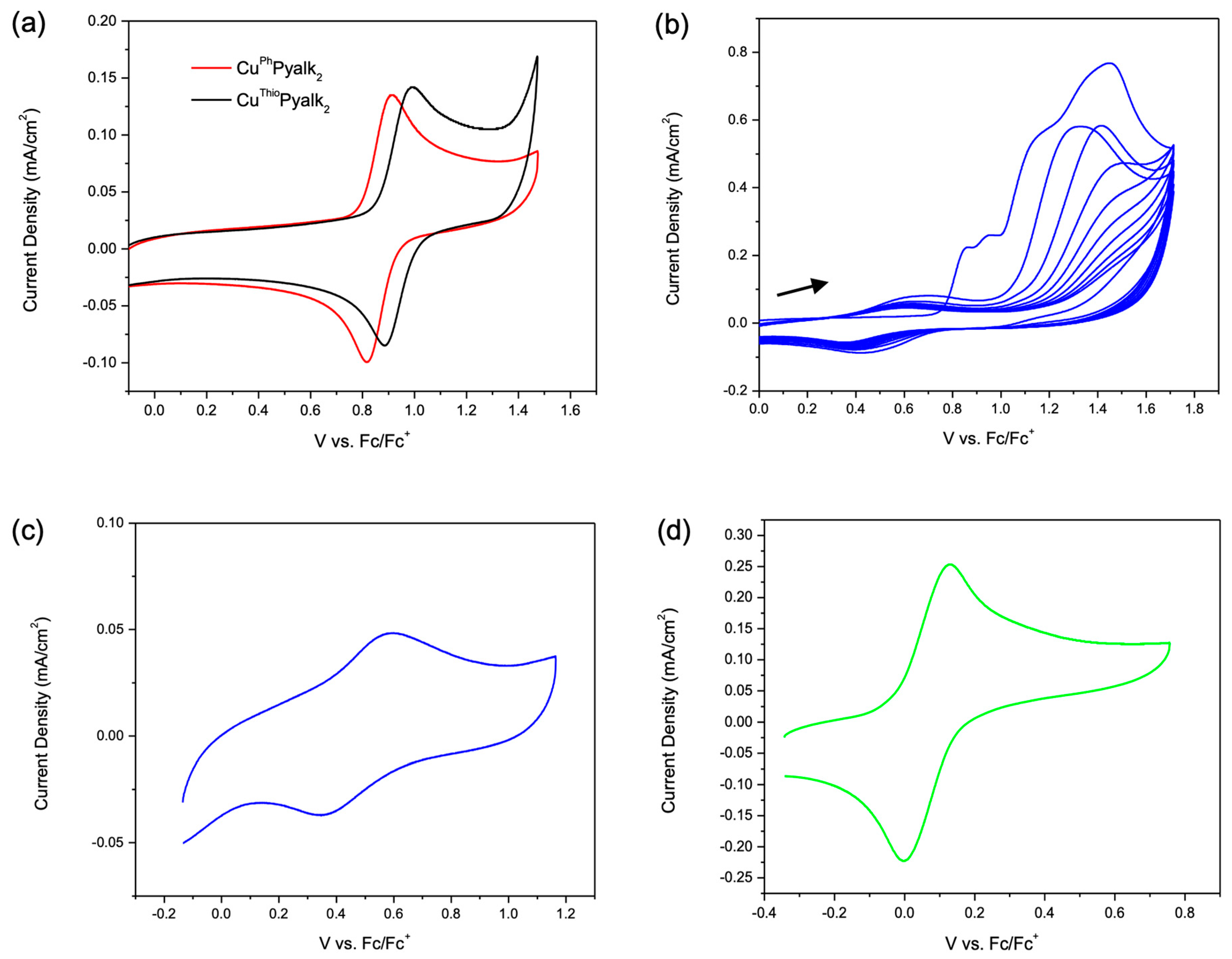
The mononuclear complexes
1a and
3a each have a reversible Cu
II/Cu
III event in the oxidative window (E
1/2 = 0.87 V and 0.94 V vs. Fc/Fc
+, respectively) (
Figure 6a). The Cu
II/Cu
III oxidation potentials are similar to the oxidation potentials reported by the Brudvig and Crabtree groups of a similar complex, but direct comparisons cannot be made due to our use of TFE (as opposed to water) as the cyclic voltammetry solvent [
5]. When oxidized past the reversible peak at 0.94 V,
3a gives an irreversible peak at
Ep,a = 1.61 V vs. Fc/Fc
+, and the reversibility of the Cu
II/Cu
III cycle is lost, indicating oxidative decomposition (
Figure S24). When scanned to the same potential, complex
1a did not display an irreversible peak, indicating stability under oxidizing conditions. The increase in the stability of
1a compared to
3a may be due to the lack of easily oxidizable methyl C–H bonds in
1a compared to
3a. When scanned to oxidative potentials,
2a shows an irreversible peak that is similar to
L2, indicating that
2a likely electropolymerizes (
Figure 6b,c). When swept multiple times, the solution of
2a turned from pink to yellow, potentially indicating the complex is protonated by freed protons released during the polymerization reaction (
Figure S18). In previous work, it has been demonstrated that electropolymerization can impact pH by releasing free protons into the electrolyte [
24]. A reversible peak similar to the peak formed by the electropolymerization of
L4 was observed after rinsing of the electrode and scanning in fresh electrolyte solution containing no
2a (
Figure 6c). Cyclic voltammetry of
4a shows a typical Fe
II/Fe
III oxidation event at 0.06 V vs. Fc/Fc
+ (
Figure 6d); however, when scanned to more positive potentials, two irreversible oxidations appeared at
Ep,a = 1.12 and
Ep,a = 1.41 V vs. Fc/Fc
+ (
Figure S25). After scanning through the irreversible peaks, a new species forms with an
E1/2 = 0.22 V vs. Fc/Fc
+, which is similar to the degradation of
L4 and
L5. The current density of the Fe
II/Fe
III oxidation of
4a is approximately double the current density of the Fe
II/Fe
III oxidation of the ligand
L4 at the same scan rates and concentrations, which may indicate that both ferrocenes are oxidized. A summary of the oxidative stability of
1a-
4a is included in
Table 3.
The mononuclear complexes were studied under reducing conditions to −1.3 V (vs. Fc/Fc
+). The cathodic scans of each mononuclear complex were quasi-reversible and led to complicated broad reoxidation peaks that ranged from 0.1–0.29 V vs. Fc/Fc
+ depending on the complex used (
Figures S17, S19, S23 and S29). Upon scanning anodically after reducing, a broad oxidation wave appears with
Ep,a’s ranging from 0.1 to 0.29 V vs. Fc/Fc
+. The low reducing potentials required likely result from the donating nature of the pyalk ligand. The complicated nature of the reduction and subsequent reoxidation process may be due to disproportionation or the direct two-electron reduction of Cu
II to Cu
0 [
25].
The dinuclear 1b, 3b, and 4b complexes did not show CuII/III oxidations within the stable oxidative window of TFE, indicating that the presence of two pyridine-alkoxide ligands per Cu are necessary to lower CuII/CuIII potentials into observable electrochemical windows. Complex 2b is insoluble in TFE.
3. Experimental Section
Materials, methods, and instrumentation. Reagent-grade chemicals were purchased from Ambeed (Arlington Heights, IL, USA), TCI Chemical (Japan) or Sigma-Aldrich (St. Louis, MO, USA) and used for synthetic procedures without further purification. Standard Schlenk and glovebox techniques were utilized for the synthesis of ligands
L1–
L5. Glovebox N
2 purity was maintain by periodic purges, and O
2 and H
2O levels were kept below 20 ppm. For NMR experiments, either a Varian (Palo Alto, CA, USA) 600 MHz or a Bruker (Billerica, MA, USA) 800 MHz spectrometer was used. All reported chemical shifts were referenced to residual
1H resonances (
1H NMR) or
13C{
1H} resonances (
13C{
1H} NMR) of chloroform-
d.
1H NMR: 7.26 ppm,
13C{
1H} NMR: 77.2 ppm [
26]. Tetrahydrofuran was purified by passage through an alumina oxide column and stored under N
2 over 4 Å molecular sieves prior to use. The solvent 2,2,2-trifluoroethanol was purchased from Oakwood Chemical (Estill, SC, USA), refluxed over CaSO
4 (purchased from Sigma-Aldrich), and stored in an N
2-filled glovebox prior to use. Tetrabutylammonium hexafluorophosphate (TBAPF
6) was purchased from a commercial source and used as received (purchased from Sigma-Aldrich). All cyclic voltammetry data were recorded on a CH Instruments (Austin, TX, USA) CHI630E potentiostat. All scans are referenced to the
E1/2 of the Fe
II/Fe
III redox event of ferrocene (purchased from Millipore Corporation, Burlington, MA, USA), added at the end of experiments. In the case of overlap between a ligand or complex with the Fe
II/Fe
III of ferrocene, acetylferrocene was used (using our conditions, the
E1/2 of the Fe
II/Fe
III oxidation was 0.35 V vs. Fc/Fc
+). An Ag/AgCl pseudo-reference electrode stored in a TFE solution containing 0.1 M TBAPF
6 was used as an internal reference. All cyclic voltammetry scans were taken of 1 mM solutions of the analyte. The scans shown in the main text were all taken at a scan rate of 100 mV/s. A 3.00 mm (0.0707 cm
2) geometric-diameter glassy carbon (GC) working electrode purchased from BASi (West Lafayette, IN, USA) was used. Elemental analyses were performed in-house using a Perkin-Elmer (Waltham, MA, USA) CHNS/O series 2 elemental analyzer. Details of the single-crystal diffraction measurements are given in the SI. Ligands
L1 and
L2 were synthesized as described previously [
3,
4].
L4 and
1a were previously reported using a different synthetic pathway [
27,
28].
[H]ThioPyalk (L3). To a 250 mL Schlenk flask inside an N2-filled glovebox, 1.20 mL (1.99 g, 12.5 mmol) of 2-bromopyridine and ~100 mL THF were added. The flask was then sealed and placed under N2 on a Schlenk line and cooled to −78 °C. Next, 5.5 mL of 2.5 M n-BuLi (solution in hexanes) were added dropwise to the reaction, which resulted in a color change from colorless to yellow and then red. The reaction was stirred at −78 °C for two hours. After two hours, a separate 50 mL round-bottom flask was charged with 2.20 mL (2.56 g, 12.5 mmol) of acetyl thiophene and diluted to ~25 mL with THF inside an N2-filled glovebox and sealed. The acetyl thiophene solution was then added via cannula into the reaction vessel under N2, and the reaction mixture was allowed to return to room temperature overnight while stirring. The next day, the reaction was removed from N2, and 100 mL of H2O were added. The organic layer was then extracted three times with DCM, dried over Na2SO4, and then filtered through a Celite-packed medium-porosity frit. The filtrate was then evaporated, and the resulting brown oil was placed in a freezer at −15 °C. Crystals were crushed and filtered from the oil and washed with 10 mL hexanes (3x) and then dried to form a white powder (0.598 g, 23%). Single crystals suitable for X-ray diffraction were found after the slow evaporation of a solution of benzene and hexanes containing L3. 1H NMR (600 MHz, CDCl3) δ 8.53 (d, 3JHH = 5 Hz, 1H, pyridine proton), 7.69 (dd, 3JHH = 8 Hz, 1H, pyridine proton), 7.38 (d, 3JHH = 8 Hz, 1H, pyridine proton), 7.24–7.18 (m, 2H, thiophene/pyridine proton), 6.98 (m, 1H, thiophene proton), 6.94 (m, 1H, thiophene proton), 6.20 (s, 1H, alkoxyl proton), 1.97 (s, 3H, methyl protons). 13C{1H} NMR (201 MHz, CDCl3) δ 163.8, 153.0, 147.3, 137.4, 126.8, 125.0, 123.6, 122.5, 120.1, 73.9, 30.8. Anal. Calcd. for C11H11NOS: C, 64.36; H, 5.40; N, 6.82. Found: C, 64.20; H, 5.26; N, 6.83.
[H]FePyalk (L4). To a 250 mL Schlenk flask inside an N2-filled glovebox, 2.99 g (18.9 mmol) of 2-bromopyridine and ~100 mL THF were added. The flask was then sealed and placed under N2 on a Schlenk line and cooled to −78 °C. Next, 7.6 mL of 2.5 M n-BuLi (solution in hexanes) were added dropwise to the reaction, and the mixture changed from colorless to yellow and then red. The reaction was then stirred at −78 °C for two hours. After two hours, a separate 100 mL round-bottom flask was charged with 3.98 g (17.5 mmol) of acetylferrocene and ~25 mL THF inside an N2-filled glovebox and sealed. The acetylferrocene solution was then added via cannula, and the mixture was allowed to return to room temperature overnight while stirring. The next day, the reaction was removed from N2, and 150 mL of H2O were added. The organic layer was extracted three times with DCM, dried over Na2SO4, and then filtered through a Celite-packed medium-porosity frit. The solvent was then evaporated by vacuum, and crystals formed from the reddish-brown oil. The crystals were crushed and washed with minimal pentanes and then dried, leaving a red/brown powder as the product in a (3.319 g, 62%). Single crystals suitable for X-ray diffraction were found after the slow evaporation of DCM solutions containing L4. 1H NMR (800 MHz, CDCl3) δ 8.52 (d, J = 5 Hz, 1H, pyridine proton), 7.64 (dd, J = 8, 8 Hz, 1H, pyridine proton), 7.36 (d, J = 8 Hz, 1H, pyridine proton), 7.17 (dd, J = 7, 5 Hz, 1H, pyridine proton), 5.29 (s, 1H, alkoxyl proton), 4.30–4.28 (m, 1H ferrocene proton), 4.18 (s, 5H, ferrocene protons), 4.16–4.14 (m, 1H, ferrocene proton), 4.11–4.09 (m, 1H, ferrocene proton), 4.04–4.02 (m, 1H, ferrocene proton), 1.83 (s, 3H, methyl proton). 13C{1H} NMR (201 MHz, CDCl3) δ 164.9, 147.3, 136.7, 122.0, 119.7, 98.4, 72.6, 68.7, 68.0, 67.6, 66.8, 65.8, 30.3. Anal. Calcd. for C17H17FeNO: C, 66.47; H, 5.58; N, 4.56. Found: C, 66.20; H, 5.47; N, 4.41.
[H]FeOMePyalk (L5). Inside an N2-filled glovebox, to a 250 mL Schlenk flask, 1.16 g (6.15 mmol) of 2-bromo-6-methoxy pyridine and ~100 mL THF were added. The flask was then sealed and placed under N2 on a Schlenk line and cooled to −78 °C. Next, 2.7 mL of 2.5 M n-BuLi (solution in hexanes) were added dropwise to the reaction, and the mixture changed from colorless to yellow and then red. The mixture was then stirred at −78 °C for two hours. After two hours, a separate 100 mL round-bottom flask was charged with 1.41 g (6.16 mmol) acetylferrocene and ~50 mL THF inside an N2-filled glovebox and sealed. The acetylferrocene solution was added via cannula, and the mixture was allowed to return to room temperature overnight while stirring. The next day, the reaction was removed from N2, and 200 mL of DI H2O were added. The organic layer was extracted three times with DCM, dried over Na2SO4, and then filtered through a Celite-packed medium-porosity frit. The filtrate was evaporated, leaving a reddish-brown oil which crystallized upon standing at room temperature. The crystals were then pulverized with a mortar and pestle and washed with minimal pentanes, leaving an orange solid as the product (1.541 g, 74%). Single crystals suitable for X-ray diffraction were found by allowing a solution of DCM containing L5 to slowly evaporate. 1H NMR (800 MHz, CDCl3) δ 7.53 (dd, 3JHH = 8 Hz, 4JHH = 8 Hz 1H, pyridine proton), 6.88 (d, 3JHH = 7 Hz, 1H, pyridine proton), 6.61 (d, 3JHH = 8 Hz, 1H, pyridine proton), 4.92 (s, 1H, alkoxy proton), 4.27–4.24 (m, 1H, ferrocene proton), 4.18 (s, 5H ferrocene proton), 4.16–4.13 (m, 1H ferrocene proton), 4.12–4.08 (m, 2H ferrocene proton), 3.97 (s, 3H methoxy protons), 1.82 (s, 3H, methyl protons). 13C{1H} NMR (201 MHz, CDCl3) δ 163.1, 162.6, 139.5, 112.1, 108.8, 98.1, 72.7, 68.8, 68.0, 67.6, 66.9, 66.0, 53.5, 30.3. Anal. Calcd. for C18H19FeNO2: C, 64.12; H, 5.68; N, 4.15. Found: C, 64.13; H, 5.64; N, 4.11.
Cu(PhPyalk)2 (1a). In a 100 mL round-bottom flask, CuCl2·2H2O (78 mg, 0.456 mmol), L1 (251 mg, 0.961 mmol), and a 2 mL aliquot of 1M KOH/MeOH were stirred in 50 mL MeOH to give a pink precipitate after stirring overnight. The next day, the solvent was decanted off, and the solid was transferred to a vial with fresh MeOH and washed with MeOH, pentanes, and diethyl ether and dried in vacuo to give a pink powder (214 mg, 80%). Crystals suitable for single crystal X-ray diffraction were found from evaporating a solution of chloroform and MeOH containing 1a. Anal. Calcd. for C36H28CuN2O2: C, 74.02; H, 4.83; N, 4.80. Found: C, 73.67; H, 4.74; N, 4.76.
{CuCl}2(µ-O-PhPyalk)2 (1b). In a 100 mL round-bottom flask, CuCl2·2H2O (110 mg, 0.647 mmol), and L1 (152 mg, 0.582 mmol) were stirred in 30 mL of MeOH to give a light blue solution. An aliquot of 0.58 mL of 1 M KOH/MeOH was added, turning the solution dark green and immediately forming a green precipitate. The reaction was stirred overnight at room temperature. The next day, the dark green solid was collected by filtration and dissolved in DCM and filtered through a Celite-packed medium-porosity frit. The filtrate was reduced to ~25 mL and left to crystallize via slow evaporation. The resulting solid was washed with pentanes and then dried in vacuo to give a green powder (178 mg, 85%). Single crystals suitable for X-ray diffraction were formed by vapor diffusing of diethyl ether into solutions of DCM containing 1b. Anal Calcd. for C36H28Cl2Cu2N2O2: C, 60.17; H, 3.93; N, 3.90. Found: C, 60.33; H, 3.98; N, 3.84.
Cu(PyrPyalk)2 (2a). In a 100 mL round-bottom flask, CuCl2·2H2O (121 mg, 0.708 mmol), L2 (481 mg, 1.49 mmol), and a 3 mL aliquot of 1 M KOH/MeOH were stirred in 150 mL MeOH to give a pink precipitate after stirring overnight. The next day, the solvent was evaporated, and the residue was dissolved in DCM and filtered through a Celite-filled medium-porosity frit. The DCM solution was left to evaporate to produce crystals. The crystals were then washed with pentanes and dried in vacuo to give a pink powder (265 mg, 53%). Single crystals suitable for X-ray diffraction were formed by evaporating solutions of DCM containing 2a. Anal. Calcd. for C46H32CuN2O2: C, 78.00; H, 4.55; N, 3.95. Found: C, 77.38; H, 4.55; N, 3.70.
{CuCl}2(µ-O-PyrPyalk)2 (2b). In a 100 mL round-bottom flask, CuCl2·2H2O (118 mg, 0.690 mmol), L2 (200 mg, 0.620 mmol), and a 0.62 mL aliquot of 1M KOH/MeOH were stirred in 100 mL MeOH to give a brown/green precipitate after stirring overnight. The next day, the solvent was decanted, and the residue was collected on a fine-porosity frit. The green solid was then dissolved in DCM and filtered through a Celite-filled medium-porosity frit. The DCM solution was evaporated, and the solid was washed with pentanes and with diethyl ether and then dried in vacuo to give a green powder (123 mg, 47%). Single crystals suitable for X-ray diffraction were formed by evaporating DCM solutions containing 2b. Anal. Calcd. for C46H32Cl2Cu2N2O2: C, 65.56; H, 3.83; N, 3.32. Found: C, 65.16; H, 3.74; N, 3.20.
Cu(ThioPyalk)2 (3a). In a 100 mL round-bottom flask, CuCl2·2H2O (59 mg, 0.344 mmol), L3 (149 mg, 0.728 mmol), and a 0.73 mL aliquot of 1M KOH/MeOH were stirred in 30 mL MeOH to give a purple solution at room temperature overnight. The next day, the solvent was evaporated, and the purple residue was redissolved in DCM and filtered through a Celite-filled medium-porosity frit. The purple solution was then evaporated, and the purple solid was washed with diethyl ether then dried under vacuum (91 mg, 56%). Single crystals suitable for X-ray diffraction were formed by vapor diffusing pentanes into solutions of DCM containing 3a. Anal. Calcd. for C22H20CuN2O2S2: C, 55.97; H, 4.27; N, 5.93. Found: C, 55.35; H, 4.23; N, 5.74.
{CuCl}2(µ-O-ThioPyalk)2 (3b). In a 100 mL round-bottom flask, CuCl2·2H2O (171 mg, 1.00 mmol), L3 (185 mg, 0.902 mmol), and a 0.91 mL aliquot of 1 M KOH/MeOH were stirred in 30 mL MeOH to give a green precipitate after stirring overnight. Next day the, reaction was dried by vacuum, leaving a green residue which was then dissolved in DCM and passed through a Celite-packed medium-porosity frit. The filtrate was then evaporated, leaving a green powder which was washed with diethyl ether and MeOH (181 mg, 66%). Single crystals suitable for X-ray diffraction were formed by vapor diffusing diethyl ether into solutions of DCM containing 3b. Anal. Calcd. for C22H20Cl2Cu2N2O2S2: C, 43.57; H, 3.32; N, 4.62. Found: C, 43.11; H, 3.11; N, 4.45.
Cu(FePyalk)2 (4a). In a 100 mL round-bottom flask, CuCl2·2H2O (27 mg, 0.160 mmol), L4 (100 mg, 0.327 mmol), and a 0.36 mL aliquot of 1M KOH/MeOH were stirred in 100 mL MeOH to give a brown/red precipitate after stirring overnight. The next day, the solvent was evaporated, leaving a yellow powder. The yellow solid was then dissolved in DCM and filtered through a Celite-filled medium-porosity frit. The DCM solution was then reduced to ~2 mL, and a solid was precipitated out with diethyl ether and washed with diethyl ether and dried in vacuo as a yellow powder (60 mg, 56%). Single crystals suitable for X-ray diffraction were formed by slow evaporation of a MeOH/CHCl3 solution containing 4a. Anal. Calcd. for C34H32CuFe2N2O2: C, 60.42; H, 4.77; N, 4.14. Found: C, 60.46; H, 4.75; N, 4.08.
{CuCl}2(µ-O-FePyalk)2 (4b). In a 100 mL round-bottom flask, CuCl2·2H2O (65 mg, 0.384 mmol), L4 (107 mg, 0.350 mmol), and a 0.35 mL aliquot of 1M KOH/MeOH were stirred in 100 mL MeOH to give a green precipitate after stirring overnight. The next day, the solvent was evaporated, leaving a green powder. The green solid was then dissolved in DCM and filtered through a Celite-filled medium-porosity frit. The DCM solution was then reduced to ~2 mL, and a solid was precipitated out with diethyl ether and washed with diethyl ether and dried in vacuo as a green powder (55 mg, 39%). Single crystals suitable for X-ray diffraction were formed by vapor diffusing pentanes into solutions of 1,2-dichloroethane containing 4b. Anal. Calcd. for C34H32Cl2Cu2Fe2N2O2: C, 50.40; H, 3.98; N, 3.46. Found: C, 49.99; H, 4.01; N, 3.44.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}