
Lu-Lu Bond in Lu2@C60 Metallofullerenes


Abstract
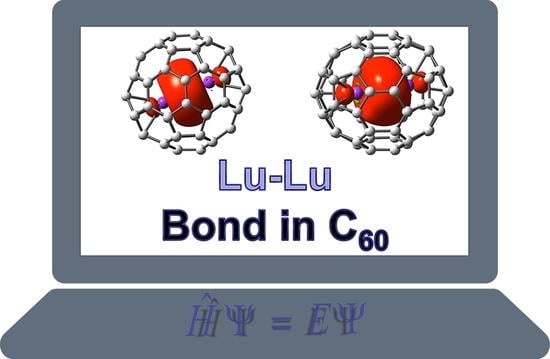
1. Introduction
2. Results and Discussion
2.1. Stability and Geometries of Lu2@C60
2.2. Electronic Structures of Lu2@C60
2.3. Bonding Features of Lu2@C60
2.4. Simulated Spectra of Lu2@C60
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodriguez, J.A.; Goodman, D.W. The nature of the metal-metal bond in bimetallic surfaces. Science 1992, 257, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.A. Metal–metal bonds in biology. J. Inorg. Biochem. 2012, 106, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Powers, I.G.; Uyeda, C. Metal–metal bonds in catalysis. ACS Catal. 2017, 7, 936–958. [Google Scholar] [CrossRef]
- Cotton, F.A. Strong homonuclear metal-metal bonds. Acc. Chem. Res. 1969, 2, 240–247. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C.; Vicha, J.; Marek, R.; Patzschke, M.; Straka, M. Unwilling U-U bonding in U2@C80: Cage-driven metal-metal bonds in di-uranium fullerenes. Phys. Chem. Chem. Phys. 2015, 17, 24182–24192. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Morales-Martínez, R.; Zhang, J.; Wang, Y.; Yao, Y.R.; Pei, C.; Rodríguez-Fortea, A.; Wang, S.; Echegoyen, L.; de Graaf, C.; et al. Characterization of a strong covalent Th3+-Th3+ bond inside an Ih(7)-C80 fullerene cage. Nat. Commun. 2021, 12, 2372. [Google Scholar] [CrossRef]
- Li, M.; Zhao, R.; Dang, J.; Zhao, X. Theoretical study on the stabilities, electronic structures, and reaction and formation mechanisms of fullerenes and endohedral metallofullerenes. Coordin. Chem. Rev. 2022, 471, 214762. [Google Scholar] [CrossRef]
- Moreno-Vicente, A.; Roselló, Y.; Chen, N.; Echegoyen, L.; Dunk, P.W.; Rodríguez-Fortea, A.; de Graaf, C.; Poblet, J.M. Are U–U Bonds Inside Fullerenes Really Unwilling Bonds? J. Am. Chem. Soc. 2023, 145, 6710–6718. [Google Scholar] [CrossRef]
- Yamada, M.; Kurihara, H.; Suzuki, M.; Guo, J.D.; Waelchli, M.; Olmstead, M.M.; Balch, A.L.; Nagase, S.; Maeda, Y.; Hasegawa, T. Sc2@C66 revisited: An endohedral fullerene with scandium ions nestled within two unsaturated linear triquinanes. J. Am. Chem. Soc. 2014, 136, 7611–7614. [Google Scholar] [CrossRef]
- Zhao, S.; Zhao, P.; Cai, W.; Bao, L.; Chen, M.; Xie, Y.; Zhao, X.; Lu, X. Stabilization of Giant Fullerenes C2(41)-C90, D3(85)-C92, C1(132)-C94, C2(157)-C96, and C1(175)-C98 by Encapsulation of a Large La2C2 Cluster: The Importance of Cluster–Cage Matching. J. Am. Chem. Soc. 2017, 139, 4724–4728. [Google Scholar] [CrossRef]
- Li, Q.-Z.; Zheng, J.-J.; He, L.; Nagase, S.; Zhao, X. La–La bonded dimetallofullerenes [La2@C2n]−: Species for stabilizing C2n (2n = 92–96) besides La2C2@C2n. Phys. Chem. Chem. Phys. 2018, 20, 14671–14678. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Roselló, Y.; Yan, Y.; Yao, Y.R.; Fan, X.; Poblet, J.M.; Rodríguez-Fortea, A.; Chen, N. ScY@C3v(8)-C82: Metal-Metal σ2 Bond in Mixed Rare-Earth Di-metallofullerenes. Chin. J. Chem. 2023; early view. [Google Scholar] [CrossRef]
- Popov, A.A.; Yang, S.; Dunsch, L. Endohedral fullerenes. Chem. Rev. 2013, 113, 5989–6113. [Google Scholar] [CrossRef]
- Lu, X.; Akasaka, T.; Nagase, S. Chemistry of endohedral metallofullerenes: The role of metals. Chem. Commun. 2011, 47, 5942–5957. [Google Scholar] [CrossRef]
- Shen, W.; Bao, L.; Lu, X. Endohedral Metallofullerenes: An Ideal Platform of Sub-Nano Chemistry. Chin. J. Chem. 2022, 40, 275–284. [Google Scholar] [CrossRef]
- Shen, W.; Bao, L.; Wu, Y.; Pan, C.; Zhao, S.; Fang, H.; Xie, Y.; Jin, P.; Peng, P.; Li, F.-F. Lu2@C2n (2n = 82, 84, 86): Crystallographic evidence of direct Lu–Lu bonding between two divalent lutetium ions inside fullerene cages. J. Am. Chem. Soc. 2017, 139, 9979–9984. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Bao, L.; Hu, S.; Yang, L.; Jin, P.; Xie, Y.; Akasaka, T.; Lu, X. Crystallographic characterization of Lu2C2n (2n = 76–90): Cluster selection by cage size. Chem. Sci. 2019, 10, 829–836. [Google Scholar] [CrossRef]
- Zheng, H.; Dang, H.; Zhao, Y.; Gu, Y.-X.; Li, M.; Li, Q.-Z.; Zhao, X. Theoretical Investigations of Lu2C84: Unexpected Impact of Metal Electronic Configuration toward the Metal–Metal σ-Bond in Fullerene. Inorg. Chem. 2020, 59, 10113–10122. [Google Scholar] [CrossRef]
- Zhang, K.; Zheng, H.; Li, M.; Li, Q.-Z.; Zhao, Y.; Zhao, X. Significant roles of a particularly stable two-center two-electron Lu–Lu σ bond in Lu2@C86: Electronic structure of Lu and radius of Lu2+. Inorg. Chem. 2021, 60, 2425–2436. [Google Scholar] [CrossRef]
- Stone, A.J.; Wales, D.J. Theoretical studies of icosahedral C60 and some related species. Chem. Phys. Lett. 1986, 128, 501–503. [Google Scholar] [CrossRef]
- Wang, Y.; Tomanek, D.; Bertsch, G.F.; Ruoff, R.S. Stability of C60 fullerite intercalation compounds. Phys. Rev. B 1993, 47, 6711–6720. [Google Scholar] [CrossRef]
- Popov, A.A.; Avdoshenko, S.M.; Pendás, A.M.; Dunsch, L. Bonding between strongly repulsive metal atoms: An oxymoron made real in a confined space of endohedral metallofullerenes. Chem. Commun. 2012, 48, 8031–8050. [Google Scholar] [CrossRef]
- Fu, W.; Zhang, J.; Fuhrer, T.; Champion, H.; Furukawa, K.; Kato, T.; Mahaney, J.E.; Burke, B.G.; Williams, K.A.; Walker, K.; et al. Gd2@C79N: Isolation, characterization, and monoadduct formation of a very stable heterofullerene with a magnetic spin state of S = 15/2. J. Am. Chem. Soc. 2011, 133, 9741–9750. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Morales-Martinez, R.; Zhuang, J.; Yao, Y.R.; Li, X.; Poblet, J.M.; Rodriguez-Fortea, A.; Chen, N. Th@D5h(6)-C80: A highly symmetric fullerene cage stabilized by a single metal ion. Chem. Commun. 2021, 57, 6624–6627. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Abella, L.; Yang, W.; Yao, Y.R.; Liu, X.; Zhuang, J.; Li, X.; Echegoyen, L.; Autschbach, J.; Chen, N. UCN@Cs(6)-C82: An Encapsulated Triangular UCN Cluster with Ambiguous U Oxidation State [U(III) versus U(I)]. J. Am. Chem. Soc. 2021, 143, 16226–16234. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Yu, Y.; Slanina, Z.; Wang, F.; Lian, Y.; Uhlik, F.; Feng, L. Ho2C2 Cluster with Flexible Configurations inside a Large C2(61)-C92 Cage. Inorg. Chem. 2022, 61, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, L.; Liu, B.; Wang, D.; Zhao, Y.; Gao, X. Theoretical study on the ground state structure of uranofullerene U@C82. J. Phys. Chem. A 2012, 116, 11651–11655. [Google Scholar] [CrossRef]
- Mulet-Gas, M.; Abella, L.; Dunk, P.W.; Rodríguez-Fortea, A.; Kroto, H.W.; Poblet, J.M. Small endohedral metallofullerenes: Exploration of the structure and growth mechanism in the Ti@C2n (2n = 26–50) family. Chem. Sci. 2015, 6, 675–686. [Google Scholar] [CrossRef]
- Li, B.; Gu, X.; Jin, P. Overlooked Effects of La-4f Orbitals in Endohedral Metallofullerenes. Inorg. Chem. 2022, 61, 5891–5902. [Google Scholar] [CrossRef]
- Cai, W.; Alvarado, J.; Metta-Magana, A.; Chen, N.; Echegoyen, L. Interconversions between Uranium Mono-metallofullerenes: Mechanistic Implications and Role of Asymmetric Cages. J. Am. Chem. Soc. 2020, 142, 13112–13119. [Google Scholar] [CrossRef]
- Shen, W.; Hu, Z.; Yu, P.; Wei, Z.; Jin, P.; Shi, Z.; Lu, X. An experimental and theoretical study of LuNC@C76,82 revealing a cage-cluster selection rule. Inorg. Chem. Front. 2020, 7, 4563–4571. [Google Scholar] [CrossRef]
- Bao, L.; Li, Y.; Yu, P.; Shen, W.; Jin, P.; Lu, X. Preferential Formation of Mono-Metallofullerenes Governed by the Encapsulation Energy of the Metal Elements: A Case Study on Eu@C2n (2n = 74–84) Revealing a General Rule. Angew. Chem. Int. Ed. 2020, 59, 5259–5262. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Peng, P.; Lu, X. Bonding inside and outside Fullerene Cages. Acc. Chem. Res. 2018, 51, 810–815. [Google Scholar] [CrossRef]
- Xu, D.; Jiang, Y.; Wang, Y.; Zhou, T.; Shi, Z.; Omachi, H.; Shinohara, H.; Sun, B.; Wang, Z. Turning on the Near-Infrared Photoluminescence of Erbium Metallofullerenes by Covalent Modification. Inorg. Chem. 2019, 58, 14325–14330. [Google Scholar] [CrossRef]
- Guan, R.; Chen, Z.-C.; Huang, J.; Tian, H.-R.; Xin, J.; Ying, S.-W.; Chen, M.; Zhang, Q.; Li, Q.; Xie, S.-Y. Self-driven carbon atom implantation into fullerene embedding metal–carbon cluster. Proc. Natl. Acad. Sci. USA 2022, 119, e22025631192022. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Jiang, X.; Yao, Y.-R.; Xin, J.; Jin, H.; Guan, R.; Zhang, Q.; Chen, M.; Xie, S.-Y.; Popov, A.A. Monometallic Endohedral Azafullerene. J. Am. Chem. Soc. 2022, 144, 21587–21595. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, Y.; Yuan, K.; Zhao, R.; Zhao, P.; Zhao, X. Insight into the thermodynamically preferred V3N@Ih(31924)-C80 and acknowledged VxSc3-xN@Ih(31924)-C80 (x = 0, 1 and 2). Carbon 2018, 132, 312–322. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Yuan, K.; Li, M.-Y.; Ehara, M.; Zhao, X. In-Depth Theoretical Probe into Novel Mixed-Metal Uranium-Based Endohedral Clusterfullerenes Sc2UX@Ih(31924)-C80 (X = C, N). Inorg. Chem. 2019, 58, 10769–10777. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, A.; Esper, R.; Dunk, P.W.; Morales-Martínez, R.; Rodríguez-Fortea, A.; Echegoyen, L.; Poblet, J.M. Small Cage Uranofullerenes: 27 Years after Their First Observation. Helv. Chim. Acta 2019, 102, e1900046. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Samoylova, N.A.; Avdoshenko, S.M.; Krylov, D.S.; Thompson, H.R.; Kirkhorn, A.C.; Rosenkranz, M.; Schiemenz, S.; Ziegs, F.; Wolter, A.U.; Yang, S. Confining the spin between two metal atoms within the carbon cage: Redox-active metal–metal bonds in dimetallofullerenes and their stable cation radicals. Nanoscale 2017, 9, 7977–7990. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.A. Redox-active metal–metal bonds between lanthanides in dimetallofullerenes. Curr. Opin. Electrochem. 2018, 8, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bridgeman, A.J.; Cavigliasso, G.; Ireland, L.R.; Rothery, J. The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc. Dalton Trans. 2001, 1, 2095–2108. [Google Scholar] [CrossRef]
- Popov, A.A.; Dunsch, L. Bonding in endohedral metallofullerenes as studied by quantum theory of atoms in molecules. Chem.—Eur. J. 2009, 15, 9707–9729. [Google Scholar] [CrossRef]
- Foster, J.; Boys, S. Canonical configurational interaction procedure. Rev. Modern Phys. 1960, 32, 300. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Atoms | Populations | NPA Charges |
|---|---|---|---|
| Lu2@Ih_C60 | Lu1 | 5d0.376s0.296p0.486d0.887p0.29 | 0.78 |
| Lu2 | 5d0.366s0.376p0.466d0.857p0.28 | 0.78 | |
| Lu2@C2v_C60 | Lu1 | 5d0.366s0.306p0.416d0.977p0.39 | 0.65 |
| Lu2 | 5d0.346s0.306p0.326d0.897p0.40 | 0.83 |
| Molecules | MBO | WBO | Atoms | Hybrid Composition |
|---|---|---|---|---|
| Lu2@Ih_C60 | 0.99 | 0.73 | Lu1 | s(26%)p(46%)d(28%) |
| Lu2 | s(36%)p(43%)d(21%) | |||
| Lu2@C2v_C60 | 0.84 | 0.67 | Lu1 | s(26%)p(44%)d(30%) |
| Lu2 | s(28%)p(45%)d(27%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Shen, W.; Chen, W.; Lu, X. Lu-Lu Bond in Lu2@C60 Metallofullerenes. Inorganics 2023, 11, 277. https://doi.org/10.3390/inorganics11070277
Zhao Y, Shen W, Chen W, Lu X. Lu-Lu Bond in Lu2@C60 Metallofullerenes. Inorganics. 2023; 11(7):277. https://doi.org/10.3390/inorganics11070277
Chicago/Turabian StyleZhao, Yaoxiao, Wangqiang Shen, Weixing Chen, and Xing Lu. 2023. "Lu-Lu Bond in Lu2@C60 Metallofullerenes" Inorganics 11, no. 7: 277. https://doi.org/10.3390/inorganics11070277
APA StyleZhao, Y., Shen, W., Chen, W., & Lu, X. (2023). Lu-Lu Bond in Lu2@C60 Metallofullerenes. Inorganics, 11(7), 277. https://doi.org/10.3390/inorganics11070277