



DFT Analysis of the Electronic and Structural Properties of Lanthanide Nitride Cluster Fullerenes Ln3N@C80


Abstract
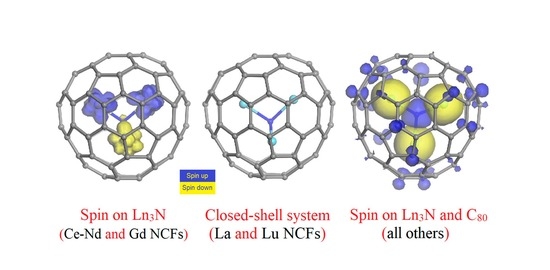
1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
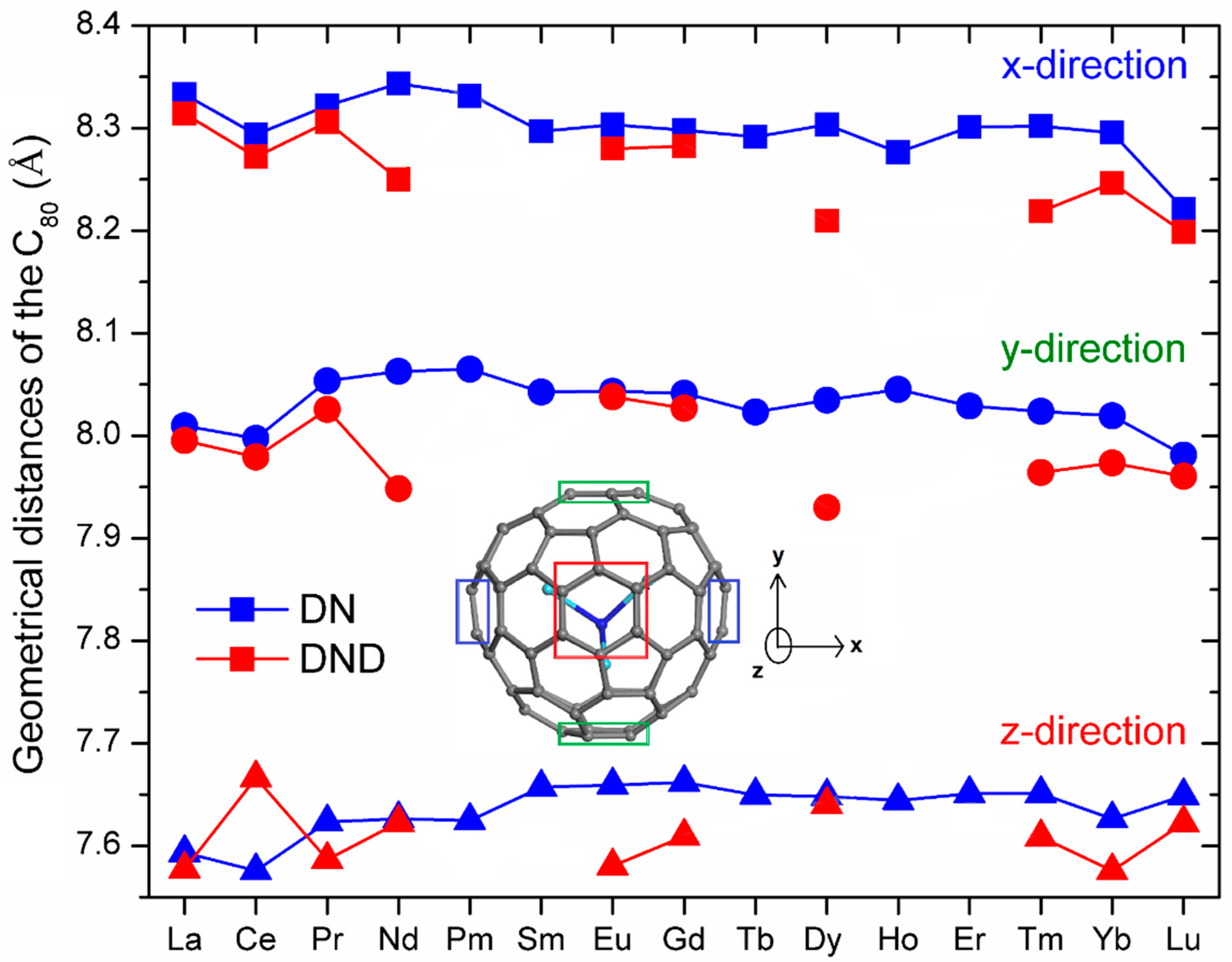{kind=link}
{kind=link}
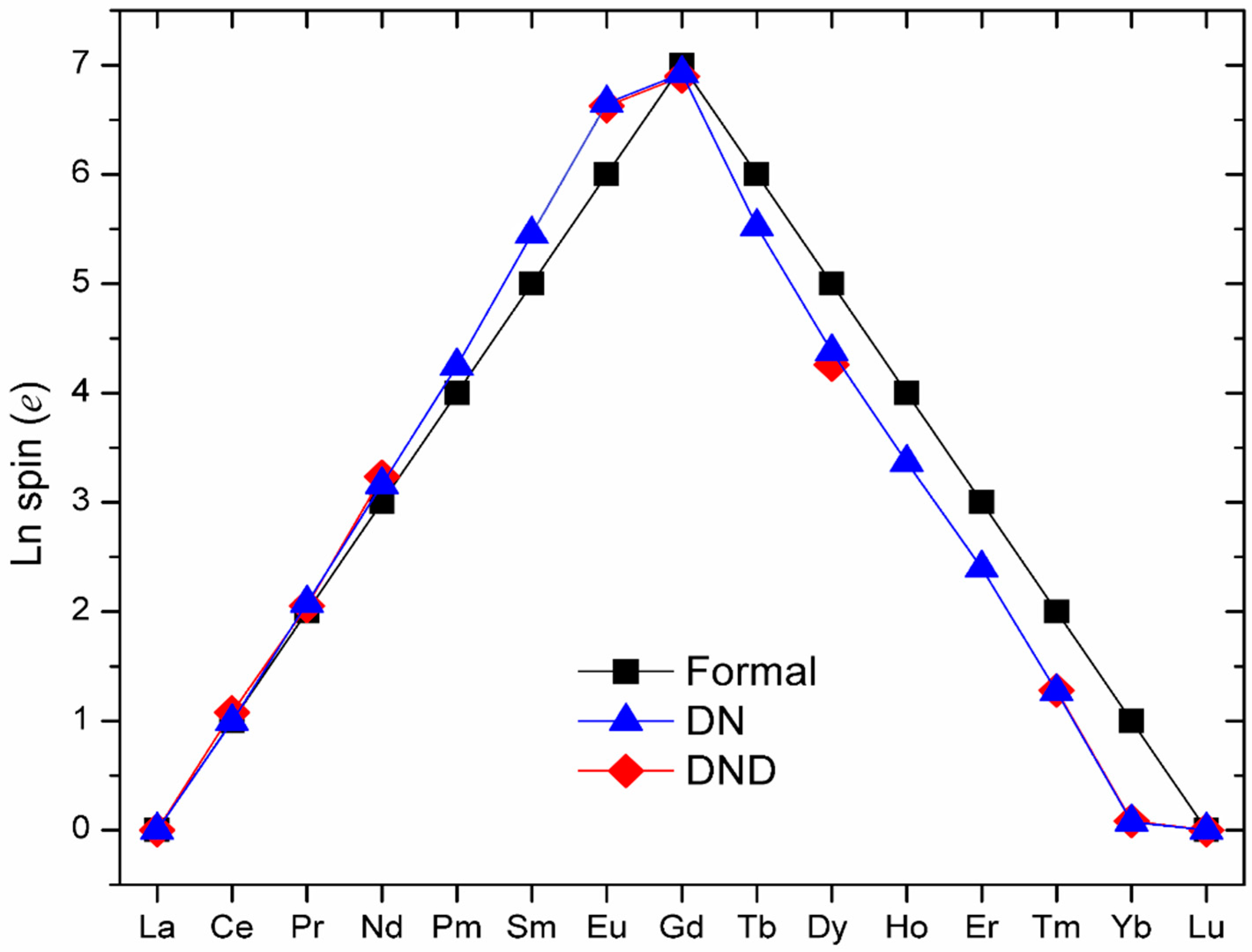{kind=link}
{kind=link}
| NFC | Ln–N (Å) | Ln–N–Ln (°) | θ (°) | Ln⋯Ln (Å) | N⋯CC80 (Å) |
|---|---|---|---|---|---|
| La3N@C80 | 2.247, 2.260, 2.265 | 94.6, 95.8, 96.0 | 31.3 | 3.325, 3.345, 3.352 | 2.949 |
| Ce3N@C80 | 2.179, 2.223, 2.242 | 90.6, 93.5, 95.2 | 33.0 | 3.174, 3.206, 3.266 | 2.962 |
| Pr3N@C80 | 2.068, 2.082, 2.165 | 111.1, 113.5, 117.1 | 14.5 | 3.503, 3.540, 3.540 | 3.597 |
| Nd3N@C80 | 2.079, 2.086, 2.090 | 115.5, 116.7, 117.9 | 10.6 | 3.533, 3.550, 3.568 | 3.758 |
| Pm3N@C80 | 2.076, 2.085, 2.086 | 115.7, 117.0, 118.0 | 10.2 | 3.532, 3.550, 3.568 | 3.770 |
| Sm3N@C80 | 2.058, 2.059, 2.084 | 117.7, 118.8, 119.9 | 6.4 | 3.543, 3.564, 3.566 | 3.925 |
| Eu3N@C80 | 2.064, 2.065, 2.068 | 118.3, 119.2, 119.7 | 5.6 | 3.548, 3.564, 3.569 | 3.948 |
| Gd3N@C80 | 2.064, 2.065, 2.066 | 118.2, 119.1, 119.5 | 5.9 | 3.546, 3.56, 3.567 | 3.957 |
| Tb3N@C80 | 2.090, 2.108, 2.126 | 107.8, 109.4, 112.0 | 19.2 | 3.422, 3.441, 3.481 | 3.443 |
| Dy3N@C80 | 2.097, 2.112, 2.116 | 109.2, 109.2, 111.2 | 18.4 | 3.431, 3.446, 3.477 | 3.461 |
| Ho3N@C80 | 2.081, 2.128, 2.132 | 105.4, 109.3, 112.2 | 20.0 | 3.433, 3.496, 3.889 | 3.423 |
| Er3N@C80 | 2.069, 2.084, 2.086 | 112.0, 112.7, 115.3 | 15.3 | 3.444, 3.472, 3.508 | 3.617 |
| Tm3N@C80 | 2.056, 2.065, 2.092 | 110.8, 110.9, 118.6 | 15.1 | 3.415, 3.423, 3.543 | 3.593 |
| Yb3N@C80 | 2.022, 2.086, 2.088 | 109.0, 114.9, 116.7 | 15.0 | 3.398, 3.464, 3.497 | 3.543 |
| Lu3N@C80 | 2.047, 2.049, 2.050 | 118.8, 119.8, 120.0 | 3.9 | 3.528, 3.545, 3.548 | 3.985 |
| NFC | Ln–N (Å) | Ln–N–Ln (°) | θ (°) | Ln⋯Ln (Å) | N⋯CC80 (Å) |
|---|---|---|---|---|---|
| La3N@C80 | 2.236, 2.251, 2.252 | 95.6, 97.0, 97.2 | 30.4 | 3.360, 3.366, 3.337 | 2.983 |
| Ce3N@C80 | 2.177, 2.223, 2.246 | 88.9, 92.7, 94.0 | 33.9 | 3.126, 3.184, 3.235 | 2.933 |
| Pr3N@C80 | 2.048, 2.120, 2.129 | 111.1, 114.2, 115.7 | 14.8 | 3.503, 3.508, 3.530 | 3.563 |
| Nd3N@C80 | 2.054, 2.310, 2.310 | 78.3, 99.8, 99.8 | 32.5 | 2.917, 3.343, 3.343 | 2.998 |
| Eu3N@C80 | 2.039, 2.090, 2.223 | 105.6, 109.3, 118.4 | 17.6 | 3.446, 3.487, 3.548 | 3.462 |
| Gd3N@C80 | 2.078, 2.093, 2.096 | 115.1, 115.3, 115.3 | 12.8 | 3.520, 3.526, 3.539 | 3.692 |
| Dy3N@C80 | 1.984, 2.304, 2.477 | 63.3, 93.8, 99.4 | 35.9 | 3.275, 3.277, 2.513 | 2.935 |
| Tm3N@C80 | 2.060, 2.123, 2.132 | 104.8, 107.8, 110.2 | 21.3 | 3.370, 3.388, 3.429 | 3.366 |
| Yb3N@C80 | 2.014, 2.084, 2.086 | 117.0, 117.1, 108.0 | 14.4 | 3.373, 3.494, 3.497 | 3.551 |
| Lu3N@C80 | 2.054, 2.054, 2.054 | 119.1, 119.6, 119.8 | 4.2 | 3.541, 3.550, 3.554 | 3.994 |




| NFC | EHOMO (eV) | ELUMO (eV) | Egap (eV) | Charge (e) | Spin (e) a | ||
|---|---|---|---|---|---|---|---|
| Ln | N | Ln | N | ||||
| La3N@C80 | −6.012 | −4.640 | 1.371 | 0.447, 0.467, 0.486 | −0.925 | 0, 0, 0 (0) | 0 |
| Ce3N@C80 | −5.818 | −4.956 | 0.862 | 0.397, 0.415, 0.439 | −0.809 | 0.948, 0.992, 1.044 (1) | 0.042 |
| Pr3N@C80 | −5.500 | −4.798 | 0.702 | 0.303, 0.352, 0.353 | −0.453 | 2.063, 2.072, 2.103 (2) | 0.051 |
| Nd3N@C80 | −5.923 | −4.983 | 0.941 | 0.160, 0.178, 0.180 | −0.634 | 3.147, 3.162, 3.179 (3) | 0.179 |
| Pm3N@C80 | −5.976 | −5.248 | 0.728 | 0.054, 0.070, 0.080 | −0.696 | 4.223, 4.256, 4.268 (4) | 0.066 |
| Sm3N@C80 | −5.956 | −5.660 | 0.296 | 0.424, 0.434, 0.463 | −0.837 | 5.441, 5.446, 5.474 (5) | 0.446 |
| Eu3N@C80 | −5.940 | −5.868 | 0.072 | 0.426, 0.441, 0.469 | −0.857 | 6.646, 6.649, 6.661 (6) | 0.627 |
| Gd3N@C80 | −6.046 | −4.589 | 1.457 | 0.496, 0.512, 0.527 | −0.992 | 6.899, 6.934, 6.935 (7) | 0.028 |
| Tb3N@C80 | −5.979 | −5.791 | 0.189 | 0.409, 0.430, 0.454 | −0.827 | 5.519, 5.520, 5.530 (6) | 0.198 |
| Dy3N@C80 | −5.974 | −5.900 | 0.074 | 0.412, 0.412, 0.435 | −0.824 | 4.338, 4.387, 4.409 (5) | 0.109 |
| Ho3N@C80 | −5.952 | −5.882 | 0.070 | 0.457, 0.499, 0.505 | −0.862 | 3.350, 3.360, 3.384 (4) | 0.058 |
| Er3N@C80 | −5.961 | −5.872 | 0.089 | 0.467, 0.487, 0.542 | −0.878 | 2.377, 2.393, 2.421 (3) | 0.083 |
| Tm3N@C80 | −5.960 | −5.835 | 0.125 | 0.368, 0.375, 0.420 | −0.822 | 1.263, 1.268, 1.273 (2) | 0.005 |
| Yb3N@C80 | −5.942 | −5.859 | 0.083 | 0.417, 0.420, 0.445 | −0.855 | 0.012, 0.099, 0.110, (1) | 0.053 |
| Lu3N@C80 | −6.034 | −4.506 | 1.528 | 0.557, 0.584, 0.595 | −1.074 | 0, 0, 0 (0) | 0 |
| NFC | EHOMO (eV) | ELUMO (eV) | Egap (eV) | Charge (e) | Spin (e) a | ||
|---|---|---|---|---|---|---|---|
| Ln | N | Ln | N | ||||
| La3N@C80 | −5.769 | −4.284 | 1.485 | 0.962, 0.962, 0.962 | −1.021 | 0, 0, 0 (0) | 0 |
| Ce3N@C80 | −5.459 | −4.695 | 0.764 | 0.829, 0.837, 0.838 | −0.889 | 1.052, 1.079, 1.101 (1) | 0.116 |
| Pr3N@C80 | −5.214 | −4.537 | 0.677 | 0.050, 0.082, 0.086 | −0.758 | 2.047, 2.047, 2.065 (2) | 0.051 |
| Nd3N@C80 | −5.579 | −5.140 | 0.438 | 0.060, 0.164, 0.174 | −0.545 | 3.185, 3.220, 3.297 (3) | 0.127 |
| Eu3N@C80 | −5.649 | −5.558 | 0.090 | 0.888, 0.907, 0.907 | −0.933 | 6.567, 6.608, 6.703 (6) | 0.243 |
| Gd3N@C80 | −5.739 | −4.365 | 1.373 | 0.854, 0.857, 0.857 | −1.136 | 6.866, 6.910, 6.913 (7) | 0.031 |
| Dy3N@C80 | −5.634 | −5.550 | 0.084 | 0.696, 0.867, 0.901 | −0.742 | 4.175, 4.159, 4.446 (5) | 0.261 |
| Tm3N@C80 | −5.673 | −5.573 | 0.100 | 0.819, 0.823, 0.832 | −0.964 | 1.266, 1.282, 1.288 (2) | 0.114 |
| Yb3N@C80 | −5.650 | −5.563 | 0.088 | 0.845, 0.862, 0.862 | −1.012 | 0.014, 0.114, 0.122 (1) | 0.085 |
| Lu3N@C80 | −5.758 | −4.272 | 1.485 | 0.882, 0.884, 0.884 | −1.215 | 0, 0, 0 (0) | 0 |



3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaur, M.N.; Melin, F.; Ortiz, A.L.; Echegoyen, L. Chemical, Electrochemical, and Structural Properties of Endohedral Metallofullerenes. Angew. Chem. Int. Ed. 2009, 48, 7514–7538. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.A.; Yang, S.; Dunsch, L. Endohedral fullerenes. Chem. Rev. 2013, 113, 5989–6113. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, T.; Jin, F. When metal clusters meet carbon cages: Endohedral clusterfullerenes. Chem. Soc. Rev. 2017, 46, 5005–5058. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.R.; O’Brien, S.C.; Zhang, Q.; Liu, Y.; Curl, R.F.; Tittel, F.K.; Smalley, R.E. Lanthanum complexes of spheroidal carbon shells. J. Am. Chem. Soc. 1985, 107, 7779–7780. [Google Scholar] [CrossRef]
- Kroto, H.; Heath, J.; O’Brien, S.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Shen, W.; Hu, S.; Lu, X. Endohedral Metallofullerenes: New Structures and Unseen Phenomena. Chem. Eur. J. 2020, 26, 5748–5757. [Google Scholar] [CrossRef]
- Miyazaki, T.; Hino, S. Electronic and Geometric Structures of Cluster Encapsulated Fullerenes. In Physics and Chemistry of Carbon-Based Materials; Kubozono, Y., Ed.; Springer: Singapore, 2019. [Google Scholar] [CrossRef]
- Stevenson, S.; Rice, G.; Glass, T.; Harich, K.; Cromer, F.; Jordan, M.R.; Craft, J.; Hadju, E.; Bible, R.; Olmstead, M.M.; et al. Small-bandgap endohedral metallofullerenes in high yield and purity. Nature 1999, 401, 55–57. [Google Scholar] [CrossRef]
- Dunsch, L.; Yang, S. Metal Nitride Cluster Fullerenes: Their Current State and Future Prospects. Small 2007, 3, 1298–1320. [Google Scholar] [CrossRef]
- Popov, A.A. (Ed.) Endohedral Fullerenes: Electron Transfer and Spin; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Campanera, J.M.; Bo, C.; Poblet, J.M. General Rule for the Stabilization of Fullerene Cages Encapsulating Trimetallic Nitride Templates. Angew. Chem. Int. Ed. 2005, 44, 7230–7233. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Z.; Wu, Y.; Wang, Z. Ln3@C80+ (Ln = lanthanide): A new class of stable metallofullerene cations with multicenter metal-metal bonding in the sub-nanometer confined space. Inorg. Chem. Front. 2022, 9, 2173–2181. [Google Scholar] [CrossRef]
- Bunzli, J.-C.G. Rising Stars in Science and Technology: Luminescent Lanthanide Materials. Eur. J. Inorg. Chem. 2017, 2017, 5058–5063. [Google Scholar] [CrossRef]
- Gaita-Ariño, A.; Luis, F.; Hill, S.; Coronado, E. Molecular spins for quantum computation. Nat. Chem. 2019, 11, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Westerström, R.; Dreiser, J.; Piamonteze, C.; Muntwiler, M.; Weyeneth, S.; Brune, H.; Rusponi, S.; Nolting, F.; Popov, A.; Yang, S.; et al. An Endohedral Single-Molecule Magnet with Long Relaxation Times: DySc2N@C80. J. Am. Chem. Soc. 2012, 134, 9840–9843. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Anglès, X.; Alegret, N.; Clotet, A.; Rodríguez-Fortea, A.; Poblet, J.M. Endohedral Metallofullerenes Containing Lanthanides: A Robust Yet Simple Computational Approach. J. Phys. Chem. C 2013, 117, 12916–12921. [Google Scholar] [CrossRef]
- Nakanishi, R.; Satoh, J.; Katoh, K.; Zhang, H.; Breedlove, B.K.; Nishijima, M.; Nakanishi, Y.; Omachi, H.; Shinohara, H.; Yamashita, M. DySc2N@C80 Single-Molecule Magnetic Metallofullerene Encapsulated in a Single-Walled Carbon Nanotube. J. Am. Chem. Soc. 2018, 140, 10955–10959. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gu, X.; Jin, P. Overlooked Effects of La-4f Orbitals in Endohedral Metallofullerenes. Inorg. Chem. 2022, 61, 5891–5902. [Google Scholar] [CrossRef]
- Stevenson, S.; Phillips, J.P.; Reid, J.E.; Olmstead, M.M.; Rath, S.P.; Balch, A.L. Pyramidalization of Gd3N inside a C80 cage. The synthesis and structure of Gd3N@C80. Chem. Commun. 2004, 2814–2815. [Google Scholar] [CrossRef]
- Zuo, T.; Beavers, C.M.; Duchamp, J.C.; Campbell, A.; Dorn, H.C.; Olmstead, M.M.; Balch, A.L. Isolation and structural characterization of a family of endohedral fullerenes including the large, chiral cage fullerenes Tb3N@C88 and Tb3N@C86 as well as the Ih and D5h isomers of Tb3N@C80. J. Am. Chem. Soc. 2007, 129, 2035–2043. [Google Scholar] [CrossRef]
- Olmstead, M.M.; Zuo, T.; Dorn, H.C.; Li, T.; Balch, A.L. Metal ion size and the pyramidalization of trimetallic nitride units inside a fullerene cage: Comparisons of the crystal structures of M3N@Ih-C80 (M = Gd, Tb, Dy, Ho, Er, Tm, Lu, and Sc) and some mixed metal counterparts. Inorg. Chim. Acta 2017, 1, 321–326. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, P.; Shen, W.; Yu, P.; Huang, W.; Ehara, M.; Xie, Y.; Akasaka, T.; Lu, X. Crystallographic characterization of Er3N@C2n (2n = 80, 82, 84, 88): The importance of a planar Er3N cluster. Nanoscale 2019, 11, 13415–13422. [Google Scholar] [CrossRef]
- Shen, W.-Q.; Bao, L.-P.; Hu, S.-F.; Gao, X.-J.; Xie, Y.-P.; Gao, X.-F.; Huang, W.-H.; Lu, X. Isolation and crystallographic characterization of Lu3N@C2n (2n = 80 − 88): Cage selection by cluster size. Chem. Eur. J. 2018, 24, 16692–16698. [Google Scholar] [CrossRef] [PubMed]
- Schlesier, C.; Liu, F.; Dubrovin, V.; Spree, L.; Büchner, B.; Avdoshenko, S.M.; Popov, A.A. Mixed dysprosium-lanthanide nitride clusterfullerenes DyM2N@C80-Ih and Dy2MN@C80-Ih (M = Gd, Er, Tm, and Lu): Synthesis, molecular structure, and quantum motion of the endohedral nitrogen atom. Nanoscale 2019, 11, 13139–13153. [Google Scholar] [CrossRef] [PubMed]
- Basiuk, V.A.; Tahuilan-Anguiano, D.E. Complexation of free-base an 3d transition metal(II) phthalocyanines with endohedral fullerene Sc3N@C80. Chem. Phys. Lett. 2019, 722, 146–152. [Google Scholar] [CrossRef]
- Yamada, M.; Liu, M.T.H.; Nagase, S.; Akasaka, T. New Horizons in Chemical Functionalization of Endohedral Metallofullerenes. Molecules 2020, 25, 3626. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Troyanov, S.I.; Popov, A.A.; Krause, M.; Dunsch, L. Deviation from the Planarity-a Large Dy3N Cluster Encapsulated in an Ih-C80 Cage: An X-ray Crystallographic and Vibrational Spectroscopic Study. J. Am. Chem. Soc. 2006, 128, 16733–16739. [Google Scholar] [CrossRef]
- Zuo, T.; Olmstead, M.M.; Beavers, C.M.; Balch, A.L.; Wang, G.; Yee, G.T.; Shu, C.; Xu, L.; Elliott, B.; Echegoyen, L.; et al. Preparation and structural characterization of the Ih and the D5h isomers of the endohedral fullerenes Tm3N@C80: Icosahedral C80 cage encapsulation of a trimetallic nitride magnetic cluster with three uncoupled Tm3+ ions. Inorg. Chem. 2008, 47, 5234–5244. [Google Scholar] [CrossRef]
- Gan, L.-H.; Yuan, R. Influence of cluster size on the structures and stability of trimetallic nitride fullerenes M3N@C80. ChemPhysChem 2006, 7, 1306–1310. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Feng, L.; Akasaka, T.; Lu, X.; Adamowicz, L. Calculations of the Lu3N@C80 two-isomer equilibrium. Fuller. Nanotub. Carbon Nanostruct. 2019, 27, 382–386. [Google Scholar] [CrossRef]
- Krause, M.; Dunsch, L. Gadolinium nitride Gd3N in carbon cages. The influence of cluster size and bond strength. Angew. Chem. Int. Ed. 2005, 44, 1557–1560. [Google Scholar] [CrossRef]
- Lu, J.; Sabirianov, R.F.; Mei, W.N.; Gao, Y.; Duan, C.G.; Zeng, X.C. Structural and magnetic properties of Gd3N@C80. J. Phys. Chem. B 2006, 110, 23637–23640. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Liu, Y.; Han, P.D.; Zhuang, S.Y.; Wang, T.S.; Luo, S.C.; Xum, B.S. Metallofullerenes encaging mixed-metal clusters: Synthesis and structural studies of GdxHo3−xN@C80 and GdxLu3−xN@C80. ChemPhysChem 2015, 16, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Flores, C.; Bolivar-Pineda, L.M.; Basiuk, V.A. Lanthanide bisphthalocyanine single-molecule magnets: A DFT survey of their geometries and electronic properties from lanthanum to lutetium. Mater. Chem. Phys. 2022, 287, 126271. [Google Scholar] [CrossRef]
- Martínez-Flores, C.; Basiuk, V.A. Ln@C60 endohedral fullerenes: A DFT analysis for the complete series from lanthanum to lutetium. Comp. Theor. Chem. 2022, 1217, 113878. [Google Scholar] [CrossRef]
- Thomsen, C.; Reich, S. Raman Scattering in Carbon Nanotubes. In Light Scattering in Solid IX. Topics in Applied Physics; Cardona, M., Merlin, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 108. [Google Scholar] [CrossRef]
- Stevenson, S.; Lee, H.M.; Olmstead, M.M.; Kozikowski, C.; Stevenson, P.; Balch, A.L. Preparation and crystallographic characterization of a new endohedral, Lu3N@C80∙5 (o-xylene), and comparison with Sc3N@C80∙5 (o-xylene). Eur. J. Chem. 2002, 8, 4528–4535. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Yang, Z.Q. First-principles investigation on the electronic structures of intercalated fullerenes M3N@C80 (M = Sc, Y, and lanthanides). Chem. Phys. Lett. 2008, 4614, 285–289. [Google Scholar] [CrossRef]
- Koltover, V.K.; Bubnov, V.P.; Estrin, Y.I.; Lodygina, V.P.; Davydov, R.M.; Subramoni, M.; Manoharan, P.T. Spin-transfer complexes of endohedral metallofullerenes: ENDOR and NMR evidences. Phys. Chem. Chem. Phys. 2003, 5, 2774–2777. [Google Scholar] [CrossRef]
- Dunsch, L.; Krause, M.; Noack, J.; Georgi, P. Endohedral nitride cluster fullerenes: Formation and spectroscopic analysis of L3−xMxN@C2n (0 ≤ x ≤ 3; N = 39, 40). J. Phys. Chem. Solids 2004, 65, 309–315. [Google Scholar] [CrossRef]
- Popov, A.A.; Dunsch, L. Hindered cluster rotation and 45Sc hyperfine splitting constant in distonoid anion radical Sc3N@C80−, spatial spin−charge separation as a general principle for anions of endohedral fullerenes with metal-localized lowest unoccupied molecular orbitals. J. Am. Chem. Soc. 2008, 130, 17726–17742. [Google Scholar] [CrossRef]
- Valencia, R.; Rodríguez-Fortea, A.; Poblet, J.M. Understanding the stabilization of metal carbide endohedral fullerenes M2C2@C82 and related systems. J. Phys. Chem. A 2008, 112, 4550–4555. [Google Scholar] [CrossRef]
- Liu, D.; Hagelberg, F.; Park, S.S. Charge transfer and electron backdonation in metallofullerenes encapsulating NSc3. Chem. Phys. 2006, 330, 380–386. [Google Scholar] [CrossRef]
- Wu, J.; Hagelberg, F. Computational study on C80 enclosing mixed trimetallic nitride clusters of the form GdxM3−xN (M = Sc, Sm, Lu). J. Phys. Chem. C 2008, 112, 5770–5777. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, X.; Zhao, X. Relativistic electronic structure calculations on endohedral Gd@C60, La@C60, Gd@C74, and La@C74. App. Phys. A 2000, 70, 461–464. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Akasaka, T.; Lu, X.; Adamowicz, L. Computational modeling of the Ce@C82 metallofullerene isomeric composition. ECS J. Solid State Sci. Technol. 2019, 8, M118–M121. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Akasaka, T.; Lu, X.; Adamowicz, L. Calculated relative thermodynamic stabilities of the Gd@C82 isomers. ECS J. Solid State Sci. Technol. 2021, 10, 071013. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. Fast calculation of electrostatics in crystals and large molecules. J. Phys. Chem. 1996, 100, 6107–6110. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Delley, B.; Ellis, D.; Freeman, A.; Baerends, E.; Post, D. Binding energy and electronic structure of small copper particles. Phys. Rev. B 1983, 27, 2132–2144. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Basiuk, V.A.; Basiuk, E.V. Noncovalent complexes of Ih-C80 fullerene with phthalocyanines. Fuller. Nanotub. Carbon Nanostruct. 2018, 26, 69–75. [Google Scholar] [CrossRef]
- Basiuk, V.A. Electron smearing in DFT calculations: A case study of doxorubicin interaction with single-walled carbon nanotubes. Int. J. Quantum Chem. 2011, 111, 4197–4205. [Google Scholar] [CrossRef]
- Basiuk, V.A.; Prezhdo, O.V.; Basiuk, E.V. Thermal smearing in DFT calculations: How small is really small? A case of La and Lu atoms adsorbed on graphene. Mater. Today Commun. 2020, 25, 101595. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Flores, C.; Basiuk, V.A. DFT Analysis of the Electronic and Structural Properties of Lanthanide Nitride Cluster Fullerenes Ln3N@C80. Inorganics 2023, 11, 223. https://doi.org/10.3390/inorganics11050223
Martínez-Flores C, Basiuk VA. DFT Analysis of the Electronic and Structural Properties of Lanthanide Nitride Cluster Fullerenes Ln3N@C80. Inorganics. 2023; 11(5):223. https://doi.org/10.3390/inorganics11050223
Chicago/Turabian StyleMartínez-Flores, César, and Vladimir A. Basiuk. 2023. "DFT Analysis of the Electronic and Structural Properties of Lanthanide Nitride Cluster Fullerenes Ln3N@C80" Inorganics 11, no. 5: 223. https://doi.org/10.3390/inorganics11050223
APA StyleMartínez-Flores, C., & Basiuk, V. A. (2023). DFT Analysis of the Electronic and Structural Properties of Lanthanide Nitride Cluster Fullerenes Ln3N@C80. Inorganics, 11(5), 223. https://doi.org/10.3390/inorganics11050223