1. Introduction
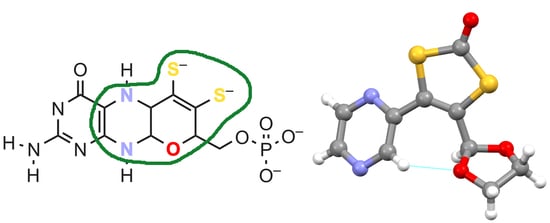
Molybdopterin (MPT;
Figure 1) is the natural ligand in the active sites of molybdenum and tungsten-dependent oxidoreductases [
1]. These enzymes catalyze transformations in the metabolic carbon, nitrogen, and sulfur cycles. Molybdopterin is expected to be a critically important component in the respective active sites due to its complex chemical structure and the non-innocence of the ene-dithiolate (or dithiolene) moiety with which it binds the central metal. Its exact role, however, has not been ultimately clarified yet. In order to better understand the influence of the various functional groups of molybdopterin, we are engaged in modeling distinct structural aspects of MPT alone and in combination with each other, excluding at the same time, as far as possible, moieties that are not present in natural MPT.
The central ring of MPT is a piperazine, which bears one C=C double bond where it is fused to the pyrimidine ring. It was found to be much easier to work with the oxidized than with the reduced N-heterocycles, though; therefore, we typically utilize pyrazines as analogs of the piperazine ring. In order to develop a more modular synthetic approach, which would allow the preparation of the ene-dithiolate chelating moiety in a large scale and its tethering to N-heterocyclic precursors of different kinds, a respective coupling strategy was investigated. It was previously attempted by others to link a pyrazine to the dithiolene moiety via a Stille reaction [
2]. The work of Joule and co-workers revealed that the cross-coupling between 2-chloroquinoxaline and 4-methyl-5-tri-
n-butylstannyl-1,3-dithiol-2-one catalyzed by various Pd(0) species and in different solvents does not work. Moreover, the use of iodobenzene did not lead to the formation of the desired product under these conditions.
In the 1990s, the positive influence of Cu(I) as a co-catalyst in slow-cycling Pd-catalyzed Stille reactions was described. Liebeskind and co-workers demonstrated copper(I) thiophene-2-carboxylate (CuTC) to be a suitable catalyst for this cross-coupling process. Respective optimized reactions with 1.5 equivalents of CuTC initiate very efficient and rapid intermolecular cross-couplings of aryl, heteroaryl, and vinyl stannanes with vinyl iodides and some aryl iodides in N-methyl-2-pyrrolidone (NMP) [
1,
3,
4]. Indeed, Joule and co-workers successfully obtained the cross-coupled product of 2-iodoquinoxaline and 4-methyl-5-tri-
n-butylstannyl-1,3-dithiol-2-one in a 44% yield when employing CuTC [
2,
5]. Notably, they did not observe the homocoupling product. Quinoxaline, however, bears an “extra” phenyl ring, which diminishes its suitability for MPT model compounds, and, to date, there is only one report available of a dithiocarbonate with a bare pyrazine attached to it [
6]. We here report a successful pathway towards the desired coupled products, as well as all the unsuccessful alternative approaches tested. The latter quite often do not receive the well-deserved attention, while their publication might help fellow researchers in avoiding experiments that are likely doomed to fail.
2. Results and Discussion
The considerable disadvantages of the Stille reaction are the toxicity of organo-tin compounds and the tedious and time-consuming isolation and purification of the products out of the reaction mixtures. Even after the application of various consecutive purification steps, the contamination by the tin compounds tends to persist, as evidenced by NMR spectroscopy. These side products might interfere with subsequent reactions, and therefore several alternative procedures for the coupling of the two synthons were tested in this study as alternative pathways. Joule and his co-workers showed that the formation of the boronic acid derivative from 4-methyl-1,3-dithiol-2-one, trimethyl or tri-iso-propyl borate, and lithium di-isopropyl amine (LDA) as a potentially suitable starting material for an alternative Suzuki coupling with 2-iodopyrazine was not possible [
2]. For this reason, we have halogenated the dithiolene instead and reacted it with the coupling partner, a boronic acid derivative, in a Suzuki reaction. Hence, 4-iodo-5-methyl-1,3-dithiol-2-one (
2) was prepared according to the literature procedure from 4-methyl-1,3-dithiol-2-one
1 (vide infra for synthesis of
1) in good yields (
Scheme 1) [
2,
7]. Compound
2 was then taken into a series of distinct trial reactions.
For an attempted proof of principle, two different Pd pre-catalysts were tested in Suzuki-type couplings to combine
2 with a simple phenyl boronic acid derivative as a first approach toward the targeted reactivity (
Scheme 2a). The reaction leads to the formation of homo-coupled boronic acids and the de-halogenation of
2 to compound
1. This indicates that the oxidative addition of
2 to Pd(0) was limited or not taking place at all due to the highly strained substituted five-membered ring, or because compound
2 might undergo ring opening under the applied alkaline conditions, resulting in Pd poisoning. The conversion of
1 to
2 proceeds through a zinc chloride intermediate, which inspired the next coupling attempt. The in-situ-prepared ZnCl-dithiocarbonate derivative
6 was employed together with 2-iodopyrazine (
5) in the presence of Pd pre-catalysts and under base-free conditions to avoid the potential ring opening/Pd poisoning (
Scheme 2b). Again, homo-coupling was observed exclusively and dithioketone
7 was identified as a product, along with the de-halogenated pyrazine species. The findings of these first two approaches were essentially reproduced in a third attempt when the in-situ-formed copper dithiocarbonate intermediate was reacted with
5 at a low temperature (
Scheme 2c). To drive the reaction towards the desired coupling product, intermediate
6, which has proven to be generally reactive in previous attempts, albeit with the wrong outcome, and copper iodide were reacted with 2-iodopyrazine (
5). This resulted again in the homo- coupled derivative as a major product. Most interestingly, however, in this case, the generation of the targeted coupling product
8 as a minor product was indeed observed (
Scheme 2d). These results support the importance of copper species as coupling mediators, as reported before, and emphasize the challenge that such coupling reactions constitute [
2,
3,
4,
5,
8]. In the case of the coupling partners used in this study, Pd catalysis completely failed, which was not anticipated, and the reasons (electronic, steric, catalyst substrate binding) for this are still under investigation.
Neither the Suzuki coupling nor any of the other described alternative routes led to the desired coupling product. For this reason, tin and the Stille coupling, which had been shown to work for related substrates before [
2], were reluctantly taken into consideration again, despite the disadvantages (vide supra). Consequently, 4-methyl-1,3-dithiol-2-one (
1) was synthesized again starting from colorless 1-bromopropan-2-one (
9) (
Scheme 3). For this purpose,
9 was freshly prepared starting from acetone according to a slightly modified literature procedure (acetic acid was added to the reaction mixture after the needed temperature was reached, instead of heating together with acetic acid) [
9]. The only moderately stable
9 was then reacted with the potassium
O-isopropylxanthate salt immediately after its synthesis [
10,
11]. The byproduct KBr was separated first by filtration and then by washing the organic phase with water. The ring-closing step leading to the formation of the 1,3-dithiol-2-one system was carried out with sulfuric acid in a Tschugajew reaction [
10,
12]. The synthesis of the tributyltin derivative
11 was achieved with freshly prepared LDA from di-isopropylamine and
n-BuLi in THF at −78 °C, which was added to a solution of 4-methyl-1,3-dithiol-2-one and tri-
n-butylstannyl chloride at −100 °C (
Scheme 3) [
2]. In the
1H-NMR spectrum, the disappearance of the signal at 6.31 ppm, which is assigned to the CH signal, confirmed the formation of the stannylated product. The exchange of the halogen, i.e., the transformation of 2-chloropyrazine to 2-iodopyrazine, was carried out in a so-called Finkelstein reaction by refluxing 2-chloropyrazine in a solution of sodium iodide in the presence of catalytic amounts of acid in acetonitrile [
13]. The coupling of 2-iodopyrazine to 4-methyl-5-tri-n-butylstannyl-1,3-dithiol-2-one (
11) mediated by 1.5 eq. CuTC in NMP gave 4-methyl-5-(pyrazin-2-yl)-1,3-dithiol-2-one (
12) (
Scheme 3).
The smallest amounts of residual tin alkyl contamination could be achieved when the obtained crystals were thoroughly washed with
n-hexane, followed by sublimation of the product. In fact,
12 is a novel compound and an interesting pro-ligand for molybdenum cofactor model complexes. One carbon of the ene function of the dithiocarbonate is substituted by pyrazine and the other by a methyl group. Attached to the carbon atom of MPT in this position is the oxygen atom of the pyrane ring. In order to account for the presence of this oxygen as well, the oxidation of the methyl substituent was attempted at various steps along the synthetic route to
12, as depicted in
Scheme 4. It was found that the transformation not only had to be carried out before any coupling attempts but also that the introduced oxygen (i.e., the aldehyde function) needed protection during the coupling reaction. The oxidation of
1 with SeO
2 led to the desired aldehyde substituted product
13 [
14]. Selenium dioxide (SeO
2) in a so-called Riley oxidation is considered to be a selective and mild agent for the oxidation of activated methylene and methyl functions next to a vinyl group to keto or aldehyde compounds. Note that the selenium dioxide used was freshly synthesized by the oxidation of elemental selenium with nitric acid and purified by sublimation. Moreover,
1 was refluxed with selenium dioxide for 22 h in freshly distilled 1,4-dioxane under an inert gas atmosphere. The resulting crude product was then sublimed to give fine X-ray-diffraction-quality colorless crystals in a needle shape.
The aldehyde oxygen of 4-formyl-1,3-dithiol-2-one (
13) was protected as an acetal functional group by refluxing in an inverse water separator with ethylene glycol and catalytic amounts of
p-toluene sulfonic acid in chloroform to obtain compound
14 [
15,
16]. Compound
14 was then stannylated as described for compound
1 to obtain the tributyltin derivative
15. In the
1H-NMR spectrum, the disappearance of the CH signal at 6.86 ppm and the formation of the stannylated product were observed. It was thus possible to couple a substituted/functionalized dithiolene unit with a “naked” pyrazine ring and obtain the targeted product 4-(1,3-dioxolan-2-yl)-5-(pyrazin-2-yl)-1,3-dithiol-2-one (
16), which could be unambiguously characterized by X-ray crystallography. To date, no dithiolene-substituted pyrazine-coupled compounds have been reported in the literature. It was, hence, shown that the Stille reaction is also applicable to more unstable non-annelated pyrazines and suitable for tolerating oxygen functionalizations on the dithiolene moiety. The products constitute direct precursors for models of MPT and are excellent starting materials for subsequent reactions or even for direct complexation reactions to obtain promising molybdenum cofactor models.
Crystallography
Compounds
12,
13,
14 and
16 gave X-ray-quality single crystals and their molecular structures were determined (
Figure 2). Compound
14 crystallized in the cold but melts at room temperature. It was therefore inevitable to measure the crystals at low temperatures (170 K). The crystal packing is determined by hydrogen-bonding interactions between the dithiocarbonate carbonyl oxygen O1 and C5 and acetal oxygen atom O3 and C2.
Table 1 lists the most important bond lengths to compare the effects of the different substituents. The –M and –I effects of the present aldehyde group in compound
13 lead to a resonance effect involving the lone pair of the S1 atom. Since this –M and –I effect of the substituent is most pronounced in compound
13, the C1-S1 bond is shortest here. Due to the presence of the pyrazine ring, which has an –M effect, the double bond character of the C1-S1 bond is weaker because the free electron pair of the S atom is no longer involved in the resonance. Therefore, the C1-S1 bonds in the compounds
12 and
16 with pyrazine are longer than for the compounds
13 and
14 without pyrazine. Since the C=C double bond can participate in resonance effects and might therefore have some mixed-in single-bond character, its bond length is an indicator of the extent of the resonance. The C=C double bond is slightly longer in
12,
13 and
16 than the average value for C=C double bonds of 1.331(9) Å [
17], which confirms its participation in resonance effects in those molecules bearing double/aromatic bonds that are only one single bond away from the ene. The short C=C distance in
14 also suggests that the resonant communication between dithiocarbonate C=O and the ene is limited. All metrical parameters are available in the
Supplementary Materials.
3. Materials and Methods
IR spectra were recorded on a Shimadzu IR Affinity-1 spectrophotometer.
1H and
13C NMR spectra were recorded on a Bruker Avance II 300 spectrometer (300 and 75 MHz, respectively). All chemical shifts are quoted with respect to TMS for the
1H signals and deuterated solvent for the
13C signals (CDCl
3 δ 77.0 ppm). The electron ionization (EI) mass spectrum was recorded on a single focusing sector-field Waters AMD40 spectrometer. Moreover, 1,4-dioxane and THF were dried over sodium and 0.4 nm molecular sieves. Syntheses, unless stated otherwise, were carried out in air. Compounds
1,
9,
10 and
11 were identified by comparison of their
1H NMR spectra with known data. Compound
13 was synthesized according to a literature procedure [
14].
3.1. X-ray Structural Analysis
Diffraction data were collected at r.t., or at a low temperature (−103.0 °C), using a STOE-IPDS 2T diffractometer with graphite-monochromatic molybdenum Kα radiation, λ = 0.71073 Å. The structures were solved by direct methods (SHELXS) [
18,
19] and refined by full-matrix least-squares techniques (SHELXL-2013, SHELXL-2016 and SHELXL-2018) using the WINGX GUI [
20]. All non-hydrogen atoms were refined with anisotropic displacement parameters. The two hydrogen atoms in
13 and the dithiolene hydrogen atom of
14 were located and refined freely. All other hydrogen atoms were refined isotropically on calculated positions using a riding model, with their
Uiso values constrained to 1.5
Ueq of their pivot atoms for terminal sp
3 carbon atoms and 1.2 times for the aromatic and methylene carbon atoms. Moreover,
13 crystallized as inversion twin and was refined with the respective applied twin law. The C
2H
4 moiety in
16 is disordered over two positions (51%:49% occupancy), and was modeled and refined without constraints or restraints. General crystallographic, crystal and refinement data along with metrical parameters for
12,
13,
14 and
16 are provided in the
Supplementary Materials. Crystallographic data were deposited with the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB21EZ, UK. These data can be obtained free of charge on quoting the depository numbers CCDC 1988723 (
13), 1988724 (
14), 1988725 (
16) and 1988726 (
12) by FAX (+44-1223-336-033), email (
deposit@ccdc.cam.ac.uk) or their web interface (at
http://www.ccdc.cam.ac.uk).
3.2. Pd-Mediated Cross-Coupling Trials
(a) An oven-dried 25 mL Schlenk tube was charged with dithioketone 2 (300 mg, 1.16 mmol), 1.5 eq. of 3,4-dimethoxy phenylboronic acid (317.3 mg, 1.74 mmol), 10 mol% PdCl2(PPh3)2 or 5 mol% of Pd/C (10 wt%) and K2CO3/Na2CO3 (2 eq.). The Schlenk tube was set under a N2 atmosphere and, at ambient conditions, dry DMF or DME/H2O (3–7 mL) was added. The resultant reaction mixture was stirred overnight at 90 °C and the reaction was monitored by TLC in regular intervals. The product spots on TLC were identified through TLC-MS as homo-coupled boronic acid derivatives and small fractions of de-halogenated dithioketone.
(b) The ZnCl intermediate 6 was synthesized by reacting compound 2 (500 mg, 3.78 mmol) with 2 eq. of t-BuLi (3.8 mL) and 1.5 eq. of ZnCl2 (5.7 mL) in dry THF at −78 °C, followed by continuous stirring for 1-2 h under an inert atmosphere. Then, the solution of 6 was added dropwise to a solution of 1.5 eq. 2-iodopyrazine (935 mg) and 5 mol% Pd(PPh3)4 or 2 mol% of Pd(dba)2 with 5 mol% of PPh3 in dry THF at −78 °C and allowed to warm up to room temperature. The reaction mixture was stirred for over 18 h and the reaction progress was monitored by TLC. The TLC-MS measurement identified the homo-coupling product of dithioketone 7 along with highly non-polar de-halogenated pyrazine.
3.3. Copper-Mediated Cross-Coupling Trials
(c) In an oven-dried 10 mL Schlenk tube, we added 1.3 eq. of t-BuLi (3.3 mL) in dry THF to 500 mg (3.782 mmol) of 2 under inert conditions at −78 °C, and the mixture was stirred for 1 h. To this solution, 0.6 eq. of CuCN·2LiCl (395.6 mg, 2.26 mmol) was added, followed by stirring for 1 h. To the resultant solution, 2-iodopyrazine in dry THF was added slowly and dropwise at 0 °C, and the reaction mixture slowly warmed to room temperature over the course of 2 h. The TLC showed the consumption of both starting materials but the products were confirmed as 7 and pyrazine.
(d) A freshly prepared ZnCl salt solution of 6 (2 mmol) in THF was added slowly to the suspension of 2-iodopyrazine (1.1 eq., 412 mg), 2 mol% CuI (7.7 mg, 0.04 mmol) and LiCl (1 eq., 84.8 mg, 2 mmol) in dry DMF at room temperature and the resultant reaction mixture was stirred at 100 °C for 3 h. The progress of the reaction was monitored by TLC. The TLC-MS analysis revealed the major fraction to be 7, with minor quantities of the desired product 8.
3.4. Synthesis of 4-Methyl-5-(pyrazin-2-yl)-1,3-dithiol-2-one (12)
To a solution of 2-iodopyrazine (364 mg, 1.77 mmol) in NMP (5.4 mL) cooled to 0 °C under an inert gas atmosphere was added CuTC (501 mg, 2.63 mmol). The mixture was stirred for 3 min, and, afterwards, 11 (0.97 g, 2.30 mmol) in DCM (4 mL) and NMP (4 mL) were added in two portions 10 min apart. After 1 h, the mixture was allowed to warm to room temperature and stirred for further 2 h. The solution was diluted with CH2Cl2 (30 mL) and then filtered through a pad of alumina and washed with 8% MeOH in CH2Cl2 (3 × 50 mL). The filtrate was washed with water (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried (over Na2SO4) and concentrated in vacuo to obtain a colorless solid, from which the product crystallized as colorless needles (yield: 0.58 mmol, 25%, 52.6 mg). 1H-NMR (300 MHz, CDCl3/TMS) δ: 2.53 (s, 3 H, CH3), 8.52 (d, 1 H, 3JH-5H-6 = 2.5, H-5), 8.6 (dd, 3JH-5H-6 = 2.5, 5JH-3H-6 = 1.6, 1 H, H-6), 8.77 (d, 5JH-3H-6 = 1.5, 1 H, H-3); 13C-NMR (75 MHz, CDCl3) δ: 16.4 (s,1C), 110.9 (s, 1C), 131.2 (s,1C), 143.2 (s, 2C), 143.6 (s,1C), 144.1 (s,1C), 190.0 (s,1C).
3.5. Synthesis of 4-(1,3-Dioxolan-2-yl)-1,3-dithiol-2-one (14)
The synthesis was carried out in a different method than already described in the literature [
15,
16]. Compound
13 (3.23 g, 22.1 mmol) and ethylene glycol (1.5 mL, 26.5 mmol) were mixed in CHCl
3 (175 mL) with a catalytic amount of
p-toluene sulfonic acid and refluxed in an inverse water trap for 3 h. After cooling to r.t., the resulting solution was washed with a saturated sodium chloride solution and dried over sodium sulfate, followed by removal of the solvent with a rotary evaporator. The product was obtained as a slightly yellowish oil, from which white crystals crystallized in the cold (3.17 g, 16.7 mmol, 76%). These could be characterized by X-ray diffraction structural analysis.
1H-NMR (300 MHz, CDCl
3, TMS) δ: 3.99-4.08 (m, 4 H, CH
2), 5.65 (d,
4J = 0.8, 1 H, CH), 6.86 (d,
4J = 0.9, 1 H, CH);
13C-NMR (75 MHz, CDCl
3): δ: 65.1 (s, 2C), 99.9 (s, 1C), 116.3 (s, 1C), 133.8 (s, 1C), 192.3 (s, 1C).
3.6. Synthesis of 4-(1,3-Dioxolan-2-yl)-5-tri-n-butylstannyl-1,3-dithiol-2-one (15)
The synthesis was carried out under an inert gas atmosphere of N2. Preparation of LDA: n-butyllithium (5.7 mL of a 2.7 M solution) was added dropwise over 15 min to a stirred solution of N-(propan-2-yl)propan-2-amine (2.17 mL, 15.4 mmol) in THF (15 mL) at –78 °C. The solution was then warmed to 0 °C and added to a stirred solution of 14 (2.0 g, 10.5 mmol) and tri-n-butylstannyl chloride (3.56 mL, 12.6 mmol) dissolved in THF (25 mL) at −78 °C. The resulting solution was stirred for 30 min, warmed to 0 °C and quenched by the addition of sat. aq. NH4Cl (50 mL) and EtOAc (50 mL). The layers were separated and the organic layer was washed with brine, dried (over Na2SO4) and the solvent evaporated in vacuo. Purification by column chromatography (EtOAc/n-hexane = 3:2) provided compound 15 as a light-yellow oil (3.94 g, 8.47 mmol, 81%). 1H-NMR (300 MHz, CDCl3/TMS) δ: 0.9–1.7 (m, 27 H, Sn(C4H9)3), 3-05-4.08 (m, 4 H, CH2), 5.52 (s, 1 H, CH); 13C-NMR (75 MHz, CDCl3) δ: 12.2–34.7 (m, 12C), 65.4 (s, 2C), 102.0 (s, 1C), 134.2 (s, 1C), 137.2 (s, 1C), 197.7 (s, 1C).
3.7. Synthesis of 4-(1,3-Dioxolan-2-yl)-5-(pyrazin-2-yl)-2H-1,3-dithiol-2-one (16)
CuTC (1.30 g, 6.82 mmol) was added to a solution of 2-iodopyrazine (940 mg, 4.54 mmol) in NMP (5 mL) cooled to 0 °C under an inert gas atmosphere. The mixture was stirred for 3 min, and, afterwards, 15 (2.33 g, 4.86 mmol) in DCM (4 mL) and NMP (4 mL) were added in two portions 10 min apart. After 1 h, the mixture was allowed to warm to room temperature and stirred for further 2 h. The solution was diluted with CH2Cl2 (30 mL) and then filtered through a pad of alumina and washed with 8% MeOH in CH2Cl2 (3 × 50 mL). The filtrate was washed with water (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried (Na2SO4) and concentrated. Purification by column chromatography (EtOAc/n-hexane = 4:1) provided compound 16 as a yellowish-white solid (26.3%, 1.28 mmol, 0.32 g). Single crystals were obtained from acetone, which could be characterized by X-ray diffraction. 1H-NMR (300 MHz, CDCl3/TMS) δ: 4.04-4.15 (m, 4 H, CH2), 6.06 (s, 1 H, CH), 8.58 (d, 3JH-5H-6 = 2.3, 1 H, H-5), 8.62 (dd, 3JH-5H-6 = 2.6, 5JH-3H-6 = 1.5, 1 H, H-6), 8.92 (d, 5JH-3H-6 = 1.5, 1 H, H-3); 13C-NMR (CDCl3) δ: 66.0 (s, 2C), 98.1 (s, 1C), 130.8 (s, 1C), 134.4 (s, 1C), 144.0 (s, 1C), 144.2 (s, 1C), 144.3 (s, 1C), 145.9 (s, 1C), 189.5 (s, 1C); IR spectrum (KBr): ν, cm−1: 805 (s), 856 (m), 944 (m), 986 (s), 1021 (s), 1059 (s), 1102 (vs), 1156 (vs), 1209 (s), 1244 (vs), 1260 (vs), 1311 (s), 1345 (m), 1407 (m), 1460 (m), 1609 (m), 1644 (vs), 1715 (m), 2856 (m), 2897 (m), 2926 (m), 2962 (m), 3420 (m, b). Mass (EI, 70 eV, 180 °C): m/z (%) = 268.6 (21) M+.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}