



How Metal Nuclearity Impacts Electrocatalytic H2 Production in Thiocarbohydrazone-Based Complexes

 , , , ,
, , , ,  and
and 
Abstract
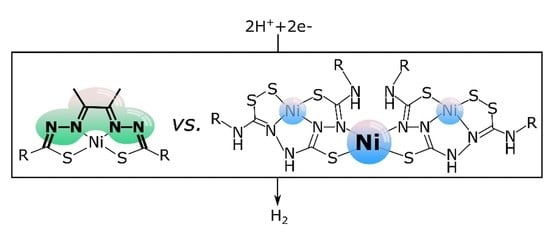
1. Introduction
2. Results and Discussions
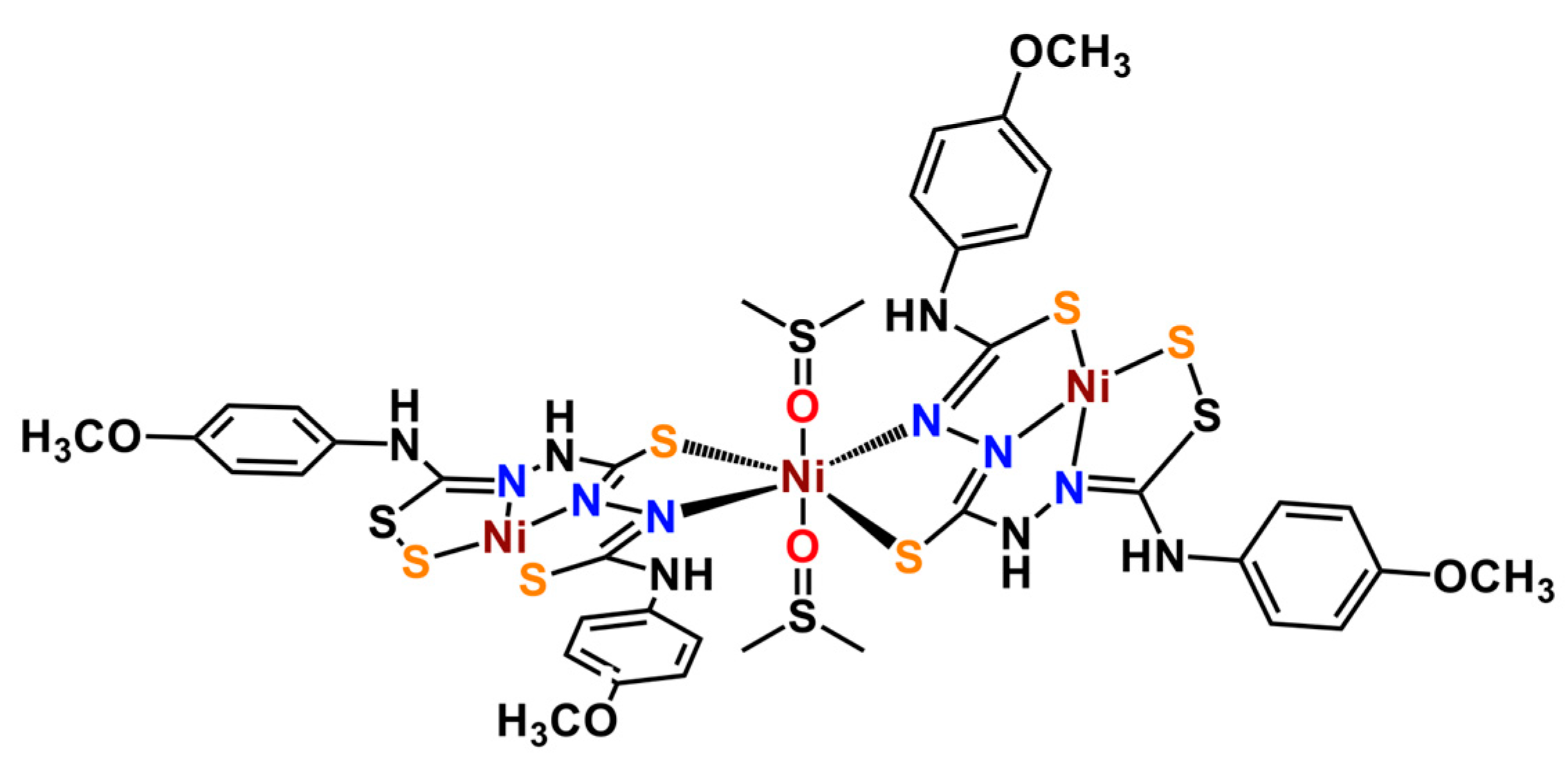
2.1. Synthesis and Single-Crystal X-ray Diffraction Analysis
2.2. Magnetic Studies
2.3. UV-Vis Spectroscopy
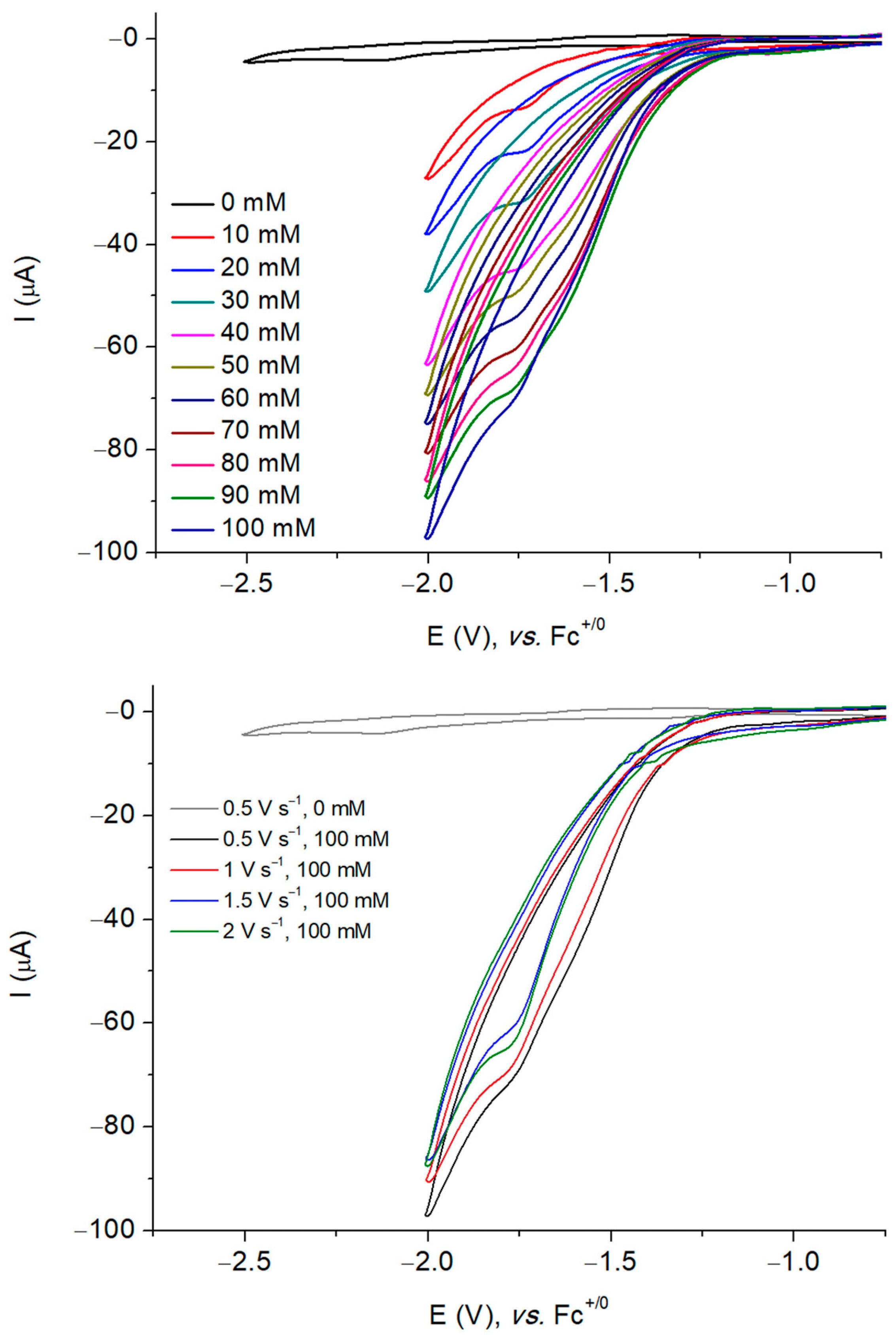
2.4. Electrochemistry
2.5. Electrocatalytic Production of H2
2.6. Benchmarking of Performances
2.7. Electronic Structure: Solvent Binding and Redox Processes
2.8. Mechanistic Considerations: Relative pKas and Reaction Pathways
3. Materials and Methods
3.1. Synthesis and Characterization
3.2. Synthesis of H6L
3.3. Synthesis of [Ni3(SH3L)2(DMSO)2] (Ni3L2)
3.4. X-ray Crystallography
3.5. SQUID Magnetometry
3.6. Electrochemistry
3.7. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turner, J.A. Sustainable Hydrogen Production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef]
- Edwards, P.P.; Kuznetsov, V.L.; David, W.I.F.; Brandon, N.P. Hydrogen and Fuel Cells: Towards a Sustainable Energy Future. Energy Policy 2008, 36, 4356–4362. [Google Scholar] [CrossRef]
- Kumar, S.S.; Himabintu, V. Hydrogen production by PEM water electrolysis—A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar] [CrossRef]
- Volbeda, A.; Charon, M.-H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal Structure of the Nickel–Iron Hydrogenase from Desulfovibrio Gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C.; Volbeda, A.; Cavazza, C.; Nicolet, Y. Structure/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases. Chem. Rev. 2007, 107, 4273–4303. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Garcin, E.; Piras, C.; de Lacey, A.L.; Fernandez, V.M.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Structure of the [NiFe] Hydrogenase Active Site: Evidence for Biologically Uncommon Fe Ligands. J. Am. Chem. Soc. 1996, 118, 12989–12996. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and Exploiting the Electrocatalytic Properties of Hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef]
- McKone, J.R.; Marinescu, S.C.; Brunschwig, B.S.; Winkler, J.R.; Gray, H.B. Earth-Abundant Hydrogen Evolution Electrocatalysts. Chem. Sci. 2014, 5, 865–878. [Google Scholar] [CrossRef]
- Dalle, K.E.; Warnan, J.; Leung, J.J.; Reuillard, B.; Karmel, I.S.; Reisner, E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. [Google Scholar] [CrossRef]
- Orio, M.; Pantazis, D.A. Successes, Challenges, and Opportunities for Quantum Chemistry in Understanding Metalloenzymes for Solar Fuels Research. Chem. Commun. 2021, 57, 3952–3974. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.L.; Stewart, M.P.; Bullock, R.M.; DuBois, M.R.; DuBois, D.L. A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s −1 for H2 Production. Science 2011, 333, 863–866. [Google Scholar] [CrossRef] [PubMed]
- DuBois, M.R.; DuBois, D.L. The Roles of the First and Second Coordination Spheres in the Design of Molecular Catalysts for H2 Production and Oxidation. Chem. Soc. Rev. 2009, 38, 62–72. [Google Scholar] [CrossRef]
- Razavet, M.; Artero, V.; Fontecave, M. Proton Electroreduction Catalyzed by Cobaloximes: Functional Models for Hydrogenases. Inorg. Chem. 2005, 44, 4786–4795. [Google Scholar] [CrossRef] [PubMed]
- Kaeffer, N.; Chavarot-Kerlidou, M.; Artero, V. Hydrogen Evolution Catalyzed by Cobalt Diimine–Dioxime Complexes. Acc. Chem. Res. 2015, 48, 1286–1295. [Google Scholar] [CrossRef]
- Drosou, M.; Kamatsos, F.; Mitsopoulou, C.A. Recent Advances in the Mechanisms of the Hydrogen Evolution Reaction by Non-Innocent Sulfur-Coordinating Metal Complexes. Inorg. Chem. Front. 2020, 7, 37–71. [Google Scholar] [CrossRef]
- Lyaskovskyy, V.; de Bruin, B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Straistari, T.; Fize, J.; Shova, S.; Réglier, M.; Artero, V.; Orio, M. A Thiosemicarbazone-Nickel(II) Complex as Efficient Electrocatalyst for Hydrogen Evolution. ChemCatChem 2017, 9, 2262–2268. [Google Scholar] [CrossRef]
- Haddad, A.Z.; Garabato, B.D.; Kozlowski, P.M.; Buchanan, R.M.; Grapperhaus, C.A. Beyond Metal-Hydrides: Non-Transition-Metal and Metal-Free Ligand-Centered Electrocatalytic Hydrogen Evolution and Hydrogen Oxidation. J. Am. Chem. Soc. 2016, 138, 7844–7847. [Google Scholar] [CrossRef] [PubMed]
- Straistari, T.; Hardré, R.; Massin, J.; Attolini, M.; Faure, B.; Giorgi, M.; Réglier, M.; Orio, M. Influence of the Metal Ion on the Electrocatalytic Hydrogen Production by a Thiosemicarbazone Palladium Complex. Eur. J. Inorg. Chem. 2018, 2018, 2259–2266. [Google Scholar] [CrossRef]
- Jain, R.; Mamun, A.A.; Buchanan, R.M.; Kozlowski, P.M.; Grapperhaus, C.A. Ligand-Assisted Metal-Centered Electrocatalytic Hydrogen Evolution upon Reduction of a Bis(Thiosemicarbazonato)Ni(II) Complex. Inorg. Chem. 2018, 57, 13486–13493. [Google Scholar] [CrossRef]
- Straistari, T.; Hardré, R.; Fize, J.; Shova, S.; Giorgi, M.; Réglier, M.; Artero, V.; Orio, M. Hydrogen Evolution Reactions Catalyzed by a Bis(Thiosemicarbazone) Cobalt Complex: An Experimental and Theoretical Study. Chem. Eur. J. 2018, 24, 8779–8786. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.P.; Mamun, A.A.; Toda, M.J.; Mashuta, M.S.; Losovyj, Y.; Kozlowski, P.M.; Buchanan, R.M.; Grapperhaus, C.A. Utilizing Charge Effects and Minimizing Intramolecular Proton Rearrangement to Improve the Overpotential of a Thiosemicarbazonato Zinc HER Catalyst. Inorg. Chem. 2019, 58, 12986–12997. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, M.; Barrozo, A.; Straistari, T.; Queyriaux, N.; Putri, A.; Fize, J.; Giorgi, M.; Réglier, M.; Massin, J.; Hardré, R.; et al. Ligand-Based Electronic Effects on the Electrocatalytic Hydrogen Production by Thiosemicarbazone Nickel Complexes. Dalton Trans. 2020, 49, 5064–5073. [Google Scholar] [CrossRef]
- Coutard, N.; Kaeffer, N.; Artero, V. Molecular Engineered Nanomaterials for Catalytic Hydrogen Evolution and Oxidation. Chem. Commun. 2016, 52, 13728–13748. [Google Scholar] [CrossRef]
- DuBois, D.L. Development of Molecular Electrocatalysts for Energy Storage. Inorg. Chem. 2014, 53, 3935–3960. [Google Scholar] [CrossRef]
- Das, A.; Hessin, C.; Ren, Y.; Desage-El Murr, M. Biological Concepts for Catalysis and Reactivity: Empowering Bioinspiration. Chem. Soc. Rev. 2020, 49, 8840–8867. [Google Scholar] [CrossRef]
- Ladomenou, K.; Papadakis, M.; Landrou, G.; Giorgi, M.; Drivas, C.; Kennou, S.; Hardré, R.; Massin, J.; Coutsolelos, A.G.; Orio, M. Nickel Complexes and Carbon Dots for Efficient Light-Driven Hydrogen Production. Eur. J. Inorg. Chem. 2021, 2021, 3097–3103. [Google Scholar] [CrossRef]
- Barrozo, A.; Orio, M. Unraveling the Catalytic Mechanisms of H2 Production with Thiosemicarbazone Nickel Complexes. RSC Adv. 2021, 11, 5232–5238. [Google Scholar] [CrossRef]
- Pieri, C.; Bhattacharjee, A.; Barrozo, A.; Faure, B.; Giorgi, M.; Fize, J.; Réglier, M.; Field, M.; Orio, M.; Artero, V.; et al. Hydrogen evolution reaction mediated by an all-sulfur trinuclear nickel complex. Chem. Commun. 2020, 56, 11106–11109. [Google Scholar] [CrossRef]
- Fourmond, V.; Canaguier, S.; Golly, B.; Field, M.J.; Fontecave, M.; Artero, V. A Nickel–Manganese Catalyst as a Biomimic of the Active Site of NiFe Hydrogenases: A Combined Electrocatalytical and DFT Mechanistic Study. Energy Environ. Sci. 2011, 4, 2417–2427. [Google Scholar] [CrossRef]
- Fourmond, V.; Jacques, P.-A.; Fontecave, M.; Artero, V. H2 Evolution and Molecular Electrocatalysts: Determination of Overpotentials and Effect of Homoconjugation. Inorg. Chem. 2010, 49, 10338–10347. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Sharma, B.; Pécaut, J.; Simon, P.; Fontecave, M.; Tran, P.D.; Derat, E.; Artero, V. Molecular Cobalt Complexes with Pendant Amines for Selective Electrocatalytic Reduction of Carbon Dioxide to Formic Acid. J. Am. Chem. Soc. 2017, 139, 3685–3696. [Google Scholar] [CrossRef]
- Artero, V.; Saveant, J.-M. Toward the Rational Benchmarking of Homogeneous H2-Evolving Catalysts. Energy Environ. Sci. 2014, 7, 3808–3814. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Homogeneous Catalysis of Electrochemical Hydrogen Evolution by Iron(0) Porphyrins. J. Am. Chem. Soc. 1996, 118, 3982–3983. [Google Scholar] [CrossRef]
- Galan, B.R.; Schöffel, J.; Linehan, J.C.; Seu, C.; Appel, A.M.; Roberts, J.A.S.; Helm, M.L.; Kilgore, U.J.; Yang, J.Y.; DuBois, D.L.; et al. Electrocatalytic Oxidation of Formate by [Ni(PR2NR′2)2(CH3CN)]2+ Complexes. J. Am. Chem. Soc. 2011, 133, 12767–12779. [Google Scholar] [CrossRef]
- Wilson, A.D.; Newell, R.H.; McNevin, M.J.; Muckerman, J.T.; Rakowski DuBois, M.; DuBois, D.L. Hydrogen Oxidation and Production Using Nickel-Based Molecular Catalysts with Positioned Proton Relays. J. Am. Chem. Soc. 2006, 128, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Wiedner, E.S.; Appel, A.M.; DuBois, D.L.; Bullock, R.M. Thermochemical and Mechanistic Studies of Electrocatalytic Hydrogen Production by Cobalt Complexes Containing Pendant Amines. Inorg. Chem. 2013, 52, 14391–14403. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Ghosh, P.; Lunsford, A.M.; Wang, N.; Bhuvanesh, N.; Hall, M.B.; Darensbourg, M.Y. Hemilabile Bridging Thiolates as Proton Shuttles in Bioinspired H2 Production Electrocatalysts. J. Am. Chem. Soc. 2016, 138, 12920–12927. [Google Scholar] [CrossRef]
- Tang, H.; Hall, M.B. Biomimetics of [NiFe]-Hydrogenase: Nickel- or Iron-Centered Proton Reduction Catalysis? J. Am. Chem. Soc. 2017, 139, 18065–18070. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis Pro. Available online: https://www.rigaku.com/products/crystallography/crysalis (accessed on 1 March 2023).
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Cobo, S.; Heidkamp, J.; Jacques, P.-A.; Fize, J.; Fourmond, V.; Guetaz, L.; Jousselme, B.; Ivanova, V.; Dau, H.; Palacin, S.; et al. A Janus Cobalt-Based Catalytic Material for Electro-Splitting of Water. Nat. Mater. 2012, 11, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 1986, 33, 8822–8824, Erratum in Phys. Rev. B 1986, 34, 7406–7406. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Neese, F. An Improvement of the Resolution of the Identity Approximation for the Formation of the Coulomb Matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Noodleman, L. Valence Bond Description of Antiferromagnetic Coupling in Transition Metal Dimers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Noodleman, L.; Case, D.A. Density-Functional Theory of Spin Polarization and Spin Coupling in Iron—Sulfur Clusters. In Advances in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 1992; Volume 38, pp. 423–470. [Google Scholar]
- Noodleman, L.; Davidson, E.R. Ligand Spin Polarization and Antiferromagnetic Coupling in Transition Metal Dimers. Chem. Phys. 1986, 109, 131–143. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Lester, W.A. Recent Advances in Quantum Monte Carlo Methods; Recent Advances in Computational Chemistry; World Scientific: Singapore, 1997; Volume 2, ISBN 978-981-02-3009-8. [Google Scholar]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An Efficient Implementation of Time-Dependent Density-Functional Theory for the Calculation of Excitation Energies of Large Molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-Dependent Density Functional Theory within the Tamm–Dancoff Approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-Dependent Density Functional Theory for Radicals An Improved Description of Excited States with Substantial Double Excitation Character. Chem. Phys. Lett. 1999, 302, 375–382. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Chemcraft. Available online: https://www.chemcraftprog.com (accessed on 1 March 2023).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proton Site | Ni3L20 | Ni3L2− |
|---|---|---|
| Nic | 0.0 | 0.0 |
| Nis | −1.3 | −5.3 |
| Sc | −1.3 | −7.8 |
| Ssc | −4.6 | X |
| Ss | X | X |
| Sd | −13.6 | X |
| Nc | X | X |
| Nsc | −6.4 | −8.3 |
| Ns | −5.5 | X |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadakis, M.; Barrozo, A.; Delmotte, L.; Straistari, T.; Shova, S.; Réglier, M.; Krewald, V.; Bertaina, S.; Hardré, R.; Orio, M. How Metal Nuclearity Impacts Electrocatalytic H2 Production in Thiocarbohydrazone-Based Complexes. Inorganics 2023, 11, 149. https://doi.org/10.3390/inorganics11040149
Papadakis M, Barrozo A, Delmotte L, Straistari T, Shova S, Réglier M, Krewald V, Bertaina S, Hardré R, Orio M. How Metal Nuclearity Impacts Electrocatalytic H2 Production in Thiocarbohydrazone-Based Complexes. Inorganics. 2023; 11(4):149. https://doi.org/10.3390/inorganics11040149
Chicago/Turabian StylePapadakis, Michael, Alexandre Barrozo, Léa Delmotte, Tatiana Straistari, Sergiu Shova, Marius Réglier, Vera Krewald, Sylvain Bertaina, Renaud Hardré, and Maylis Orio. 2023. "How Metal Nuclearity Impacts Electrocatalytic H2 Production in Thiocarbohydrazone-Based Complexes" Inorganics 11, no. 4: 149. https://doi.org/10.3390/inorganics11040149
APA StylePapadakis, M., Barrozo, A., Delmotte, L., Straistari, T., Shova, S., Réglier, M., Krewald, V., Bertaina, S., Hardré, R., & Orio, M. (2023). How Metal Nuclearity Impacts Electrocatalytic H2 Production in Thiocarbohydrazone-Based Complexes. Inorganics, 11(4), 149. https://doi.org/10.3390/inorganics11040149